
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTurn-folding in fluorescent anthracene-substituted cyclopenta[d]isoxazoline short peptides†‡
Marco Leusciatti a,
Barbara Mannuccib,
Teresa Reccab and
Paolo Quadrelli*a
a,
Barbara Mannuccib,
Teresa Reccab and
Paolo Quadrelli*a
aDepartment of Chemistry, University of Pavia, Viale Taramelli 12, 27100 – Pavia, Italy. E-mail: paolo.quadrelli@unipv.it
bCentro Grandi Strumenti (CGS), University of Pavia, Via Bassi 21, 27100 – Pavia, Italy
First published on 1st June 2021
Abstract
Cyclopenta[d]isoxazoline aminols were used for the synthesis of β-turn mimics. The peptide chain choice ascertained the influence of their structural features on the applicability/reliability/robustness of these scaffolds as β-turn inducers and their limitations. The amino acid selection as well as steric demands can favor or disfavor the structure folding and the correct design of the peptide chains deeply influences the potential use of these nitrosocarbonyl-based compounds as turn-inducers.
Introduction
Reverse turns are a major class of protein secondary structures and represent chain reversal sites; thus they are places where the globular character of a protein is created.1 Turns are local features, pivotal motifs of peptide strands and helices2 and it is therefore not surprising that their structural properties are extensively studied using different models. Turns occur within protein binding sites, at protein–protein interfaces and in bioactive peptides, playing a central role in recognition.3 Hence, the efforts in this area4 are directed to the designing and developing novel structures for specific applications5 and using classical and uncommon scaffolds for promising new synthetic targets.6β-Turn motifs, commonly observed in small peptides and proteins,7 take advantage of conformationally constrained molecules. The key point is the correspondence among conformations in solution and those determined in silico.8 For these reasons, three-dimensional interactions, spatial orientation determination derived from a conformational analysis, based on spectroscopic techniques and molecular modeling, are essential for the design of analogues in the drug discovery process.7 β-Turns criteria rely upon intramolecular hydrogen bonds between the carbonyl oxygen of the first residue (i) and the amide NH proton of the fourth residue (i + 3), within a pseudo-ten-membered ring.2,9 Critical distances between the Cα carbon atoms (dcrit < 7 Å) must be respected as well as the one between the carbonyl oxygen of the first amino acid and the amide hydrogen of the fourth one (<4 Å) (Fig. 1). Turns are capable of initiating productive structure formation in proteins without a large loss in chain entropy since the interactions involved in turn formation are largely local.10 The turn becomes a nucleation point that facilitates cooperative formation of neighboring interactions. Turns can also act passively in folding, serving only as flexible structural connectors. In this case, other folding events can promote structure formation, and turns form only as a consequence of structure consolidation from other regions of the protein. Fig. 1 shows the β-turn formation initiated at the turn corner residues (red box). Turn formation can promote cooperative favorable interactions in the other regions of peptide sequence (green or pink circles).11
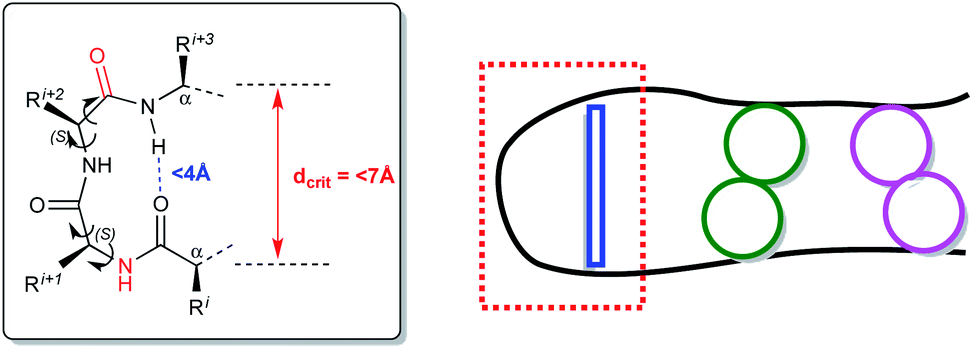 | ||
| Fig. 1 β-Turn criteria (left inset). β-Turn formation at the turn corner (red box, connection in blue). Cooperative favorable interactions in other regions of peptide sequence (green or pink circles) (right). | ||
Recently, we presented and developed our original approach towards the design of non-peptidic turn-inducers12 based on easily synthesized cyclopenta[d]isoxazoline aminols through the nitrosocarbonyl intermediates (RCONO, 3) chemistry (Scheme 1).13 The mild oxidation of nitrile oxides 1 with tertiary amine N-oxides is a valuable alternative to the periodate oxidation of hydroxamic acids 2 for the generation of the fleeting intermediates 3; the efficient trapping with cyclopentadiene (or other dienes), affords the hetero Diels–Alder (HDA) cycloadducts 4 that are synthetically elaborated to furnish the conformationally restricted carbocyclic aminols 6 through amide hydrolysis and reductive N–O bond cleavage of the cycloadducts 5.14,15 Cycloadducts 5 were prepared by using nitrile oxides of type 1 and in particular benzonitrile oxide (R = Ph), isolated as regioisomers in comparable amounts.
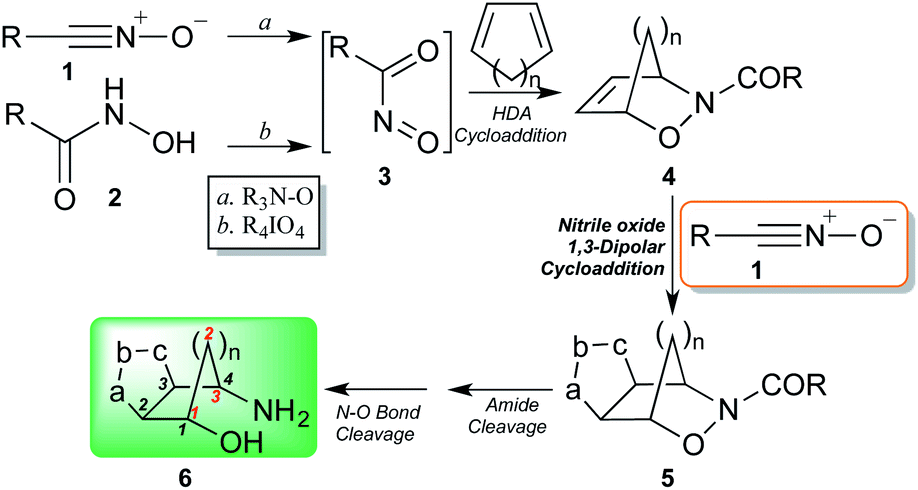 | ||
| Scheme 1 Synthetic pathway of cyclopenta[d]isoxazoline aminols through nitrosocarbonyl intermediates chemistry. | ||
On pursuing our research work on the synthesis of novel turn motifs, we developed this topic through further investigations in order to verify the influence of the two side chains on the type of turn, gaining a better understanding of applicability/reliability/robustness of aminol scaffolds of type 6 as β-turn inducers. We wish to present here the peptide chain effect on the turn formation through the characterizations of the diasteroisomeric turn structures through NMR and circular dichroism (CD) techniques. Furthermore, we took advantage of the stability and fluorescence properties of the anthracenenitrile oxide (R = anthryl in Scheme 1) to investigate the spectroscopic features of the synthesized turns, aiming to finalize them as fluorescent tags, for imaging studies at the protein turn points.
Results and discussion
The regioisomeric aminols 6a,b were prepared according to the previously reported methodology,13–17 using anthracenenitrile oxide in the 1,3-dipolar cycloaddition step. The 1,3-dipole was prepared from the corresponding commercially available oxime upon oxidation with a NCS/Py/DCM protocol.18 The hydroxy group protection was performed under standard procedure affording the amines 7a,b (85%) that were fully characterized (Scheme 2).12,16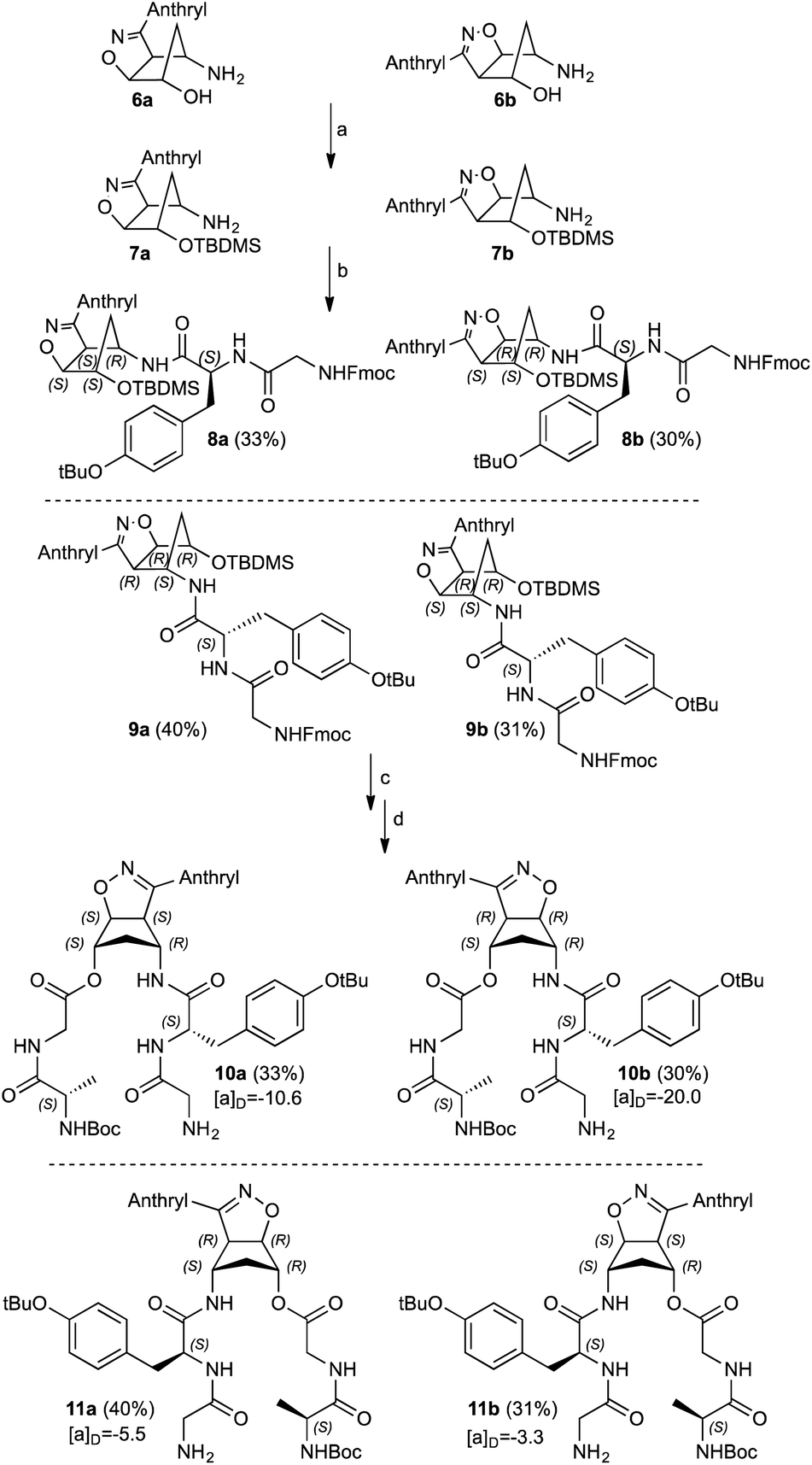 | ||
| Scheme 2 Synthesis of compounds 10a,b and 11a,b: (a) TBDMSiCl, imidazole, DCM, r.t., 18 h. (b) Fmoc–Gly–Tyr(tBu)–OH, HBTU, DIEA, DCM, r.t., 48 h. (c) n-Bu4NF, THF, r.t., 3 h. (d) Boc–Ala–Gly–OH, DIC, DMAP, DCM, r.t., 48 h. Yields (%) and [α]D (MeOH) (see also Experimental section). | ||
Compounds 7a,b were coupled with the commercially available Fmoc–Gly–Tyr(tBu)–OH and the diastereoisomeric adducts 8a and 9a were isolated in good yields as well as compounds 8b and 9b. The hydroxy functionalities deprotection was secured by standard n-Bu4NF treatment; during this step the Fmoc protection is also removed without detrimental effects in the subsequent synthetic step. The corresponding alcohols were not isolated but immediately coupled with Boc–Ala–Gly–OH affording the diastereoisomeric double strand peptides 10a/11a and 10b/11b. All the compounds reported were separated from unreacted starting materials or decomposition products and fully characterized (Scheme 2).
Structurally similar to previous synthetized β-turn mimics,12,16 compounds 10a,b and 11a,b remarkably differs from two relevant aspects: the presence of the fluorescent anthracene moiety on the isoxazoline ring moiety and the two peptide side arms bearing aminoacid residues with different steric demands, namely a tyrosine and alanine residues coupled with simple glycine.
The UV-vis and fluorescent spectra revealed an interesting behavior for all the analyzed compounds and referred to the presence of the anthracene moiety. Fig. 2 (top) shows UV-vis spectra recorded in methanol with the typical absorbance of the phenyl ring of the tyrosine moiety at 253 nm and the aromatic vibrational transition pattern between 300–400 nm. The fluorescence at 355 nm is shown in Fig. 2 (middle) and reports similar maxima around 418 and 438 nm for all the compounds but with different intensities. The fluorescence intensities in fact are remarkably increased when the irradiation was done at the tyrosine phenyl ring band at 253 nm indicating a charge transfer from the phenyl ring to the anthracene moiety.
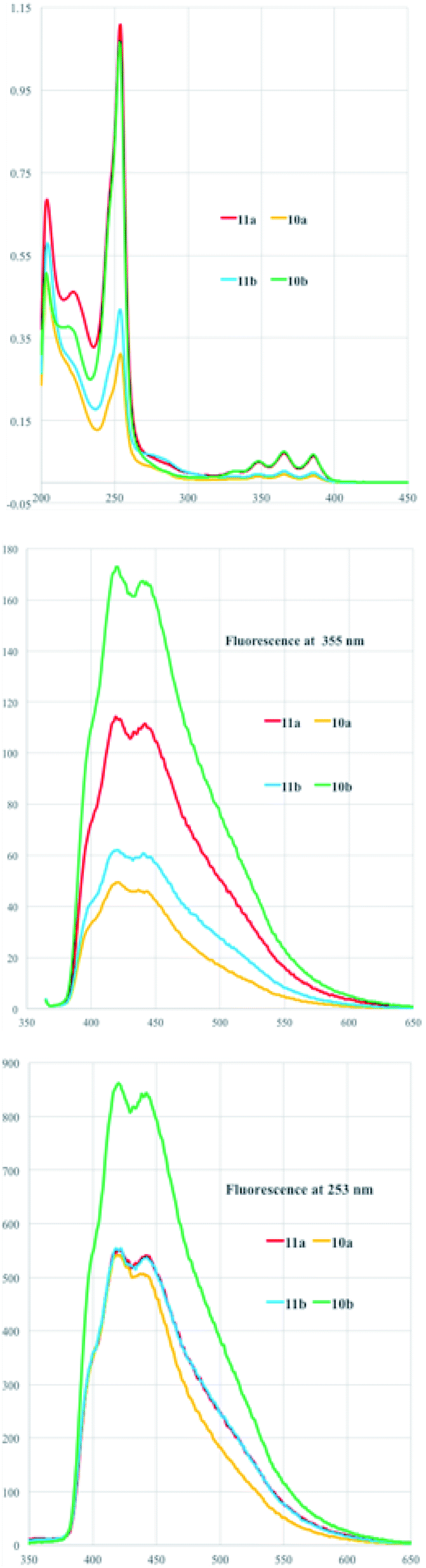 | ||
| Fig. 2 UV-vis (top) and fluorescent spectra (bottom) in MeOH as solvent upon irradiation at 355 nm (middle) and at 253 nm (bottom) of diastereoisomers 10a,b and 11a,b. | ||
The conformational analysis of both the series of compounds19 relies upon CD analyses coupled with NMR experiments.20,21
Collection of the CD spectra was obtained from MeOH 10−5 M solutions of diastereoisomers 10a/11a and 10b/11b within a wavelength range of 200–300 nm (Fig. 3). Compound 10a shows a CD profile with a positive absorption band at 202 nm and two negative bands centered at 220 and 254 nm, indicating a left β-turn in accordance with the previously data reported in literature.22 The CD spectrum of 11a shows a comparable and specular trend with a negative absorption band at 202 nm and positive bands centered at 230 and 248 nm, consistent for a right β-turn. A further negative band is also found at 276 nm.
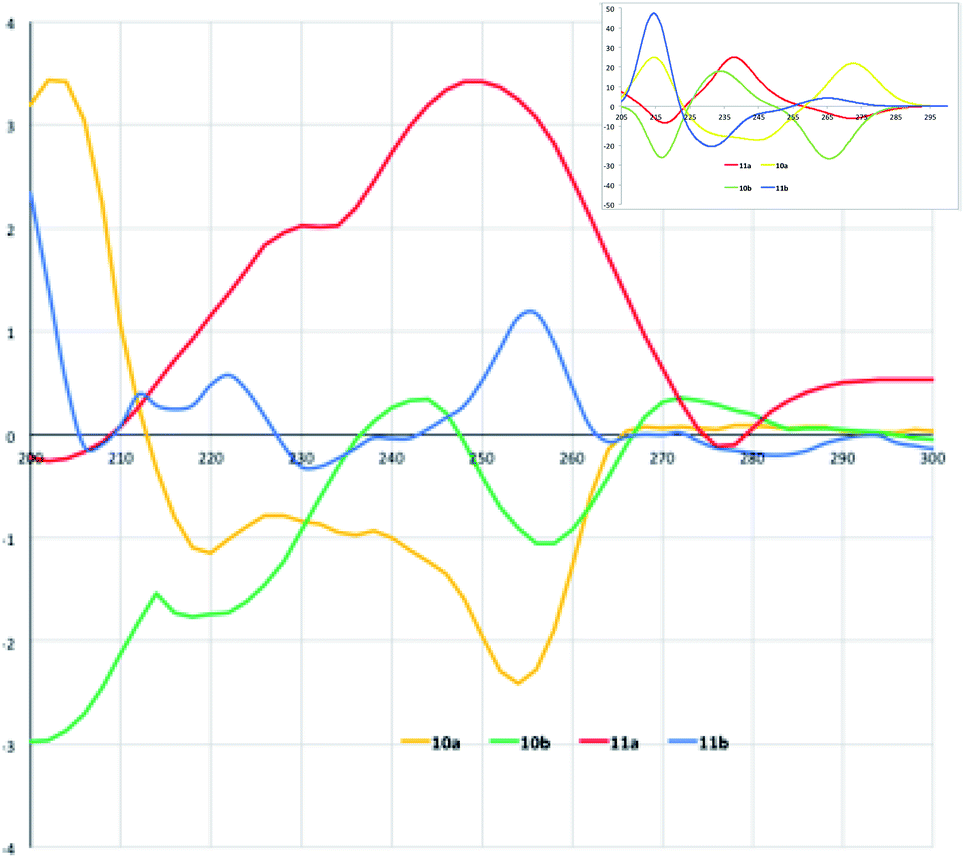 | ||
| Fig. 3 Experimental CD spectra (MeOH as solvent) of diastereoisomers 10a,b and 11a,b. In the inset: calculated CD of the model molecules reported in the cited ref. 12 for comparison with experimental CD spectra of molecules in the present work. | ||
In a similar way, compound 10b CD profile has a negative absorption band at 214 nm followed by a second positive band at 244 nm. Negative and positive absorption bands also follow at 258 and 272 nm. This profile can be referred to a right β-turn.19 Finally, the CD spectrum of 11b, although more “corrugated” compared with the others, shows two positive absorptions at 212 and 222 nm; the absorption turns into negative with a maximum at 232 nm to become strongly positive at 256 nm. Noteworthy, these values are remarkably close to those observed for the model malonic derivatives (see calculated CD of model molecules12 in the inset of Fig. 3) and in general with previous observations.16 Previously reported simulations of the CD absorption profiles of the model malonic derivatives12 done by TD-DFT calculations23 at the B3LYP/6-31g(d,p) level in methanolic solution nicely fitted with the experimental profiles indicating that the configuration inversions at the isoxazoline-norbornane moieties correspond to an inversion of the chiroptical properties of the products at hand.
This fitting allowed assigning the products conformations in spite of the interference effect in the CD spectra of the anthracene moiety due to its large UV absorption and fluorescence. The assigned conformations are the same for all the new synthetized diastereoisomers because of a good overlapping of CD spectra.12 However, here the CD profiles indicate that when the aminoacid residues are somewhat sterically demanding and the strands are structurally elaborated the turn mimic shows difficulties in maintaining the correct shape and perfect folding.
H-bonding in diastereoisomers 10a/11a and 10b/11b in CDCl3 and DMSO-d6 was evaluated through temperature coefficients (tc) of the amide, NH2 and NHBoc protons of the aminoacidic residues at 298.15 and 318.15 K for CDCl3 and up to 348.15 K for DMSO-d6 as well as DMSO-d6-titration experiments.22 Typically, low tc values in CDCl3 ≤ 2.4 ppb K−1 are related not only to shielded protons but also to accessible ones and only values significantly larger than 2.4 ppb K−1 in CDCl3 can be unambiguously assigned to NH protons initially shielded, which become exposed to the solvent upon increasing temperature.3,24 Conversely, low tc in DMSO-d6 (<5 ppb K−1) are related to inaccessible protons to the solvent.
Solutions (10−3 M) of the compounds 10a/11a and 10b/11b in the deuterated solvent of choice were used to record the 1H NMR spectra. Table 1 reports the tc values expressed in Δppb/ΔK highlighted in different colors for CDCl3 and DMSO-d6 for the related diastereoisomers. The results indicate that in CDCl3 diastereoisomers 10a/11a and 10b/11b display somewhat a uniform borderline situation; the values of the NH protons indicated as 1, and corresponding to the aminolic NH protons, are in the range 0.0–4.0 Δppb/ΔK and similarly happens for the NH protons 2-Tyr found in the range 0.5–3.5 Δppb/ΔK. Marked higher values are found for protons 3-Gly, NH2 and 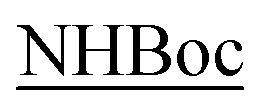 (4.0–11.0 Δppb/ΔK) denoting that these protons are totally accessible, as it derives from their structural location.
(4.0–11.0 Δppb/ΔK) denoting that these protons are totally accessible, as it derives from their structural location.
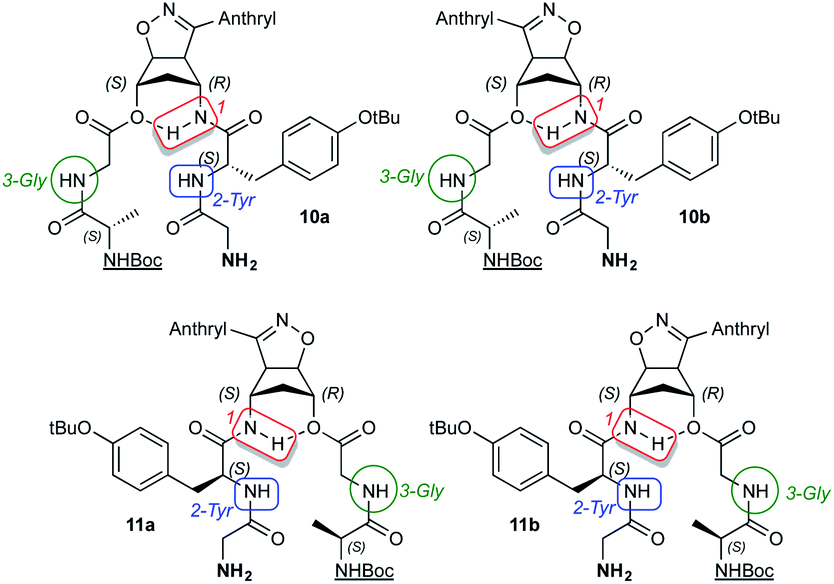
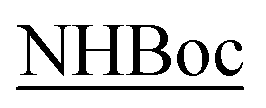
On the other hand, the tc values in DMSO-d6 indicate that the NH protons 1 have values ranging at 0.8–3.2 Δppb/ΔK for the four substrates and are clearly inaccessible to the solvent. Same behavior is shown in the NH protons 2-Tyr whose tc values range from 4.0 to 5.6 Δppb/ΔK, a borderline situation. Higher threshold values (up to 8.2 Δppb/ΔK) are found for the protons 3-Gly, NH2 and 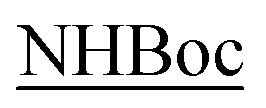 indicating the accessibility of those protons to the solvent. The main novelty here is the low accessibility of the 2-Tyr protons along with the expected behavior of the aminolic NH protons 1.
indicating the accessibility of those protons to the solvent. The main novelty here is the low accessibility of the 2-Tyr protons along with the expected behavior of the aminolic NH protons 1.
Furthermore, we performed DMSO-d6-titration experiments with gradual DMSO-d6 addition to CDCl3 solutions (∼3 × 10−2 M) of the four diastereoisomers 10a/11a and 10b/11b (Fig. 4). A first set of results concerns the NH protons indicated as 1, 2-Tyr and 3-Gly whose structural correspondence can be seen in the scheme of Table 1. The protons of type 1 maintain their chemical shifts almost unchanged or slightly waving around a narrow range of chemical shifts over the addition of increasing amounts of DMSO-d6 to the CDCl3 solutions for compounds 10a,b and 11a. Larger variations of chemical shifts are found in compound 11b. Variation ΔNH within 0.50 ppm indicate a very low accessibility of the amide proton 1, engaged in H-bonding with the ester oxygen atom and that is the case for all the titrated compounds.
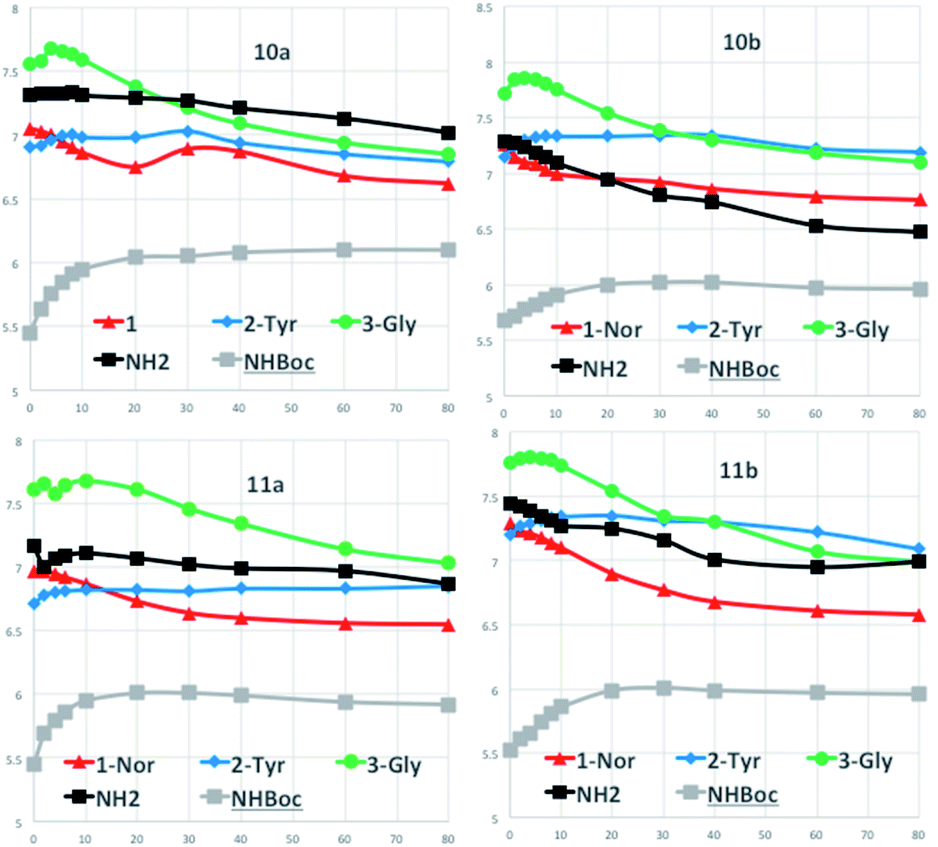 | ||
| Fig. 4 DMSO-d6-titration of compounds 10a/11a and 10b/11b. | ||
A similar trend is nicely observed for 2-Tyr protons but not for the 3-Gly protons that are somewhat involved in H-bonding with the added DMSO-d6. This can be observed for compounds 10a,b and 11a,b, where a contained chemical shift variation for the 2-Tyr proton is the opposite trend with respect to the 3-Gly protons.
The NH2 and 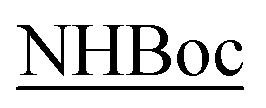 protons, located at the end of the peptide chains, are free to be involved in H-bonding with DMSO-d6 and the titration data confirm these behaviors, correctly showing large chemical shift variations.
protons, located at the end of the peptide chains, are free to be involved in H-bonding with DMSO-d6 and the titration data confirm these behaviors, correctly showing large chemical shift variations.
To further confirm the secondary structures of the four diastereoisomers 10a, 10b, 11a and 11b, 2D proton NMR experiments were performed. Complete sequence-specific assignments of resonances of the five residues in each compound was achieved using a combination of TOCSY experiments to identify the spin systems and ROESY experiments to identify the near-neighbour connectivities. In fact, because of their short rotational correlation times, these products gave extremely weak cross peaks in a NOESY spectrum.25,26 Hence, to investigate the connectivities characteristic of the residues involved in each turn structure, ROESY spectra were collected.
Fig. 5 reports the region of the ROESY spectrum relative to compound 11b, best representative of the general behavior of all the synthesized turns. As can be seen, in the HalphaHN and in the HNHN region of the ROESY spectrum of the compound 11b, a sequential ROE connection HN–HN(i,i + 1) as well as a HalphaHN(i,i + 2) ROE cross peaks were observed, involving the tyrosine residue.
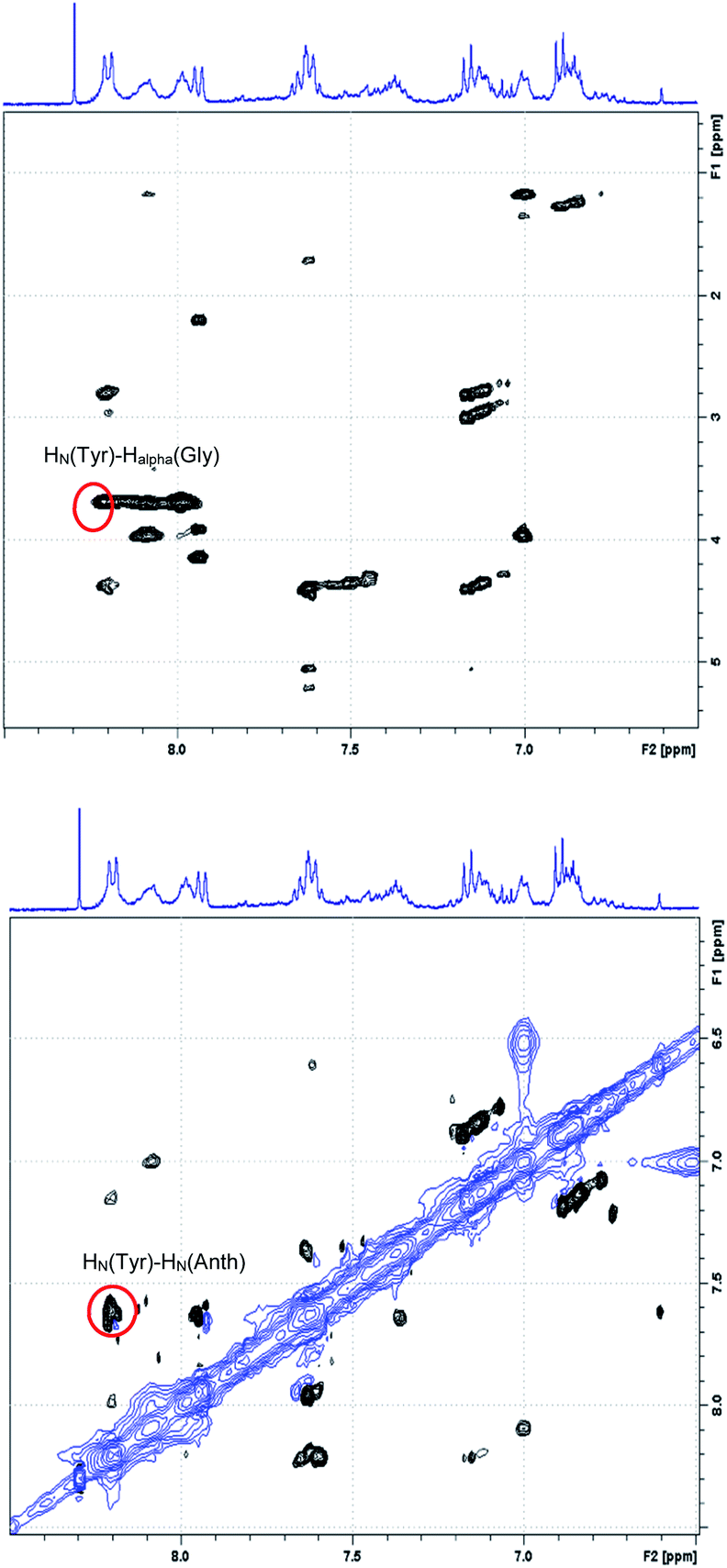 | ||
| Fig. 5 HalphaHN region (top) and HNHN region (bottom) of the ROESY spectrum of the 11b sample: red circles highlight the tyrosine cross peaks characteristic of the turn-like structure. | ||
Similar cross peaks were observed for the other three compounds, with the different intensities of the cross peaks; diastereoisomers belonging to the b series showed more intense peaks than those of the a series. The complete spectra can be seen in the ESI.‡
The presence of these dipolar couplings, together with the results from CD analysis, the values of temperature coefficients and the NMR titration data, suggest that the entire cyclopenta[d]isoxazoline peptide functionalized-derivatives adopt a turn-like conformation in a significative fraction of the time in the NMR scale.
The reported results further enforce the indication that the oxazanorbornenes chemistry gives the possibility to synthesize stereo-ordinated constrained aminols holding the geometrical features to be used as turn inducers, transferring these constraints to the entire molecules they belong to. The driving force that makes these products induce β-turns very efficiently is strictly related to the presence of a fused isoxazolinic ring to the cyclopentane moiety (Fig. 6). This latter usually adopts an envelope conformation with the flap directed toward the isoxazoline ring, thus showing a boat like appearance to the bicyclic system.12,16
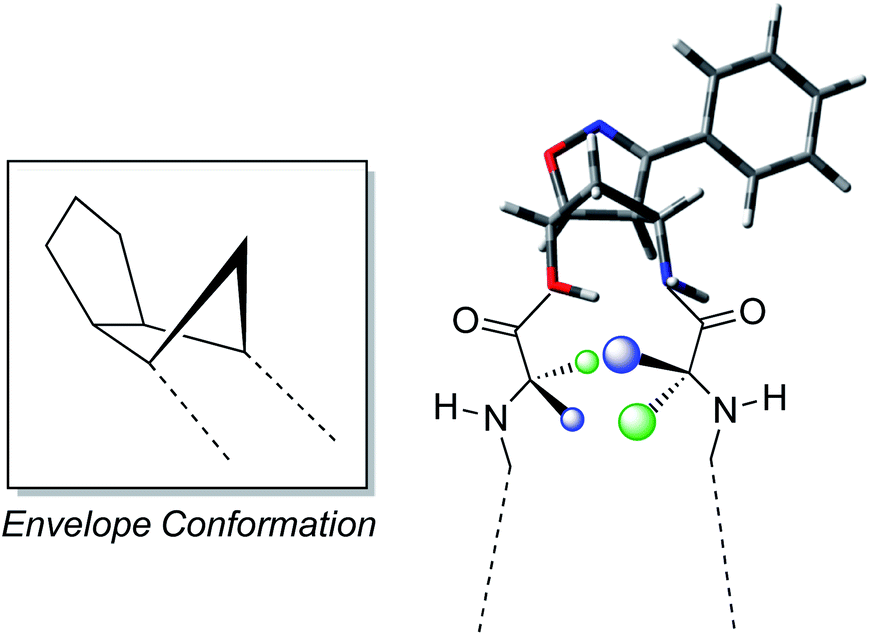 | ||
| Fig. 6 Envelope conformation of the isoxazoline aminol structure and development of the turn conformation. | ||
In the present work the insertion of the peptide chains and the introduction of aminoacids with different steric demand shine further light on the use of isoxazoline-cyclopentane aminols of type 6a,b as key structures for the synthesis of turn inducers. In the previous papers,12,16 we outlined the methodology to synthetize and to study the turn conformations of this type of systems and in the present work our results are an evolution of the set-up protocol.
From the synthetic point of view the methodology is again found to be robust and reliable. The attachment of the Fmoc–Gly–Tyr(tBu)–OH fragment to the protected amines 7a,b gave the expected products in good yields. The subsequent deprotection step liberates the OH group that is coupled with the Boc–Ala–Gly–OH to afford the expected diasteroisomeric products 10a,b and 11a,b in very good yields.
Another valuable feature of these new compounds relies upon the change of the nitrile oxide portion in the 1,3-dipolar cycloaddition reaction conducted to prepare the aminol 6a,b. We used the anthracenenitrile oxide, known to be a stable as well as fluorescent 1,3-dipole (Fig. 7).25 The fluorescence properties of the 1,3-dipole were easily transferred to the isoxazoline derivatives as the UV-vis studies coupled with the fluorescence spectra clearly demonstrate. The interesting and potentially valuable fluorescence activation studies are that irradiation at the tyrosine phenyl group enhance the fluorescence of the compounds. In this way, turn mimics assume the characteristic of fluorescent tags (Fig. 7).
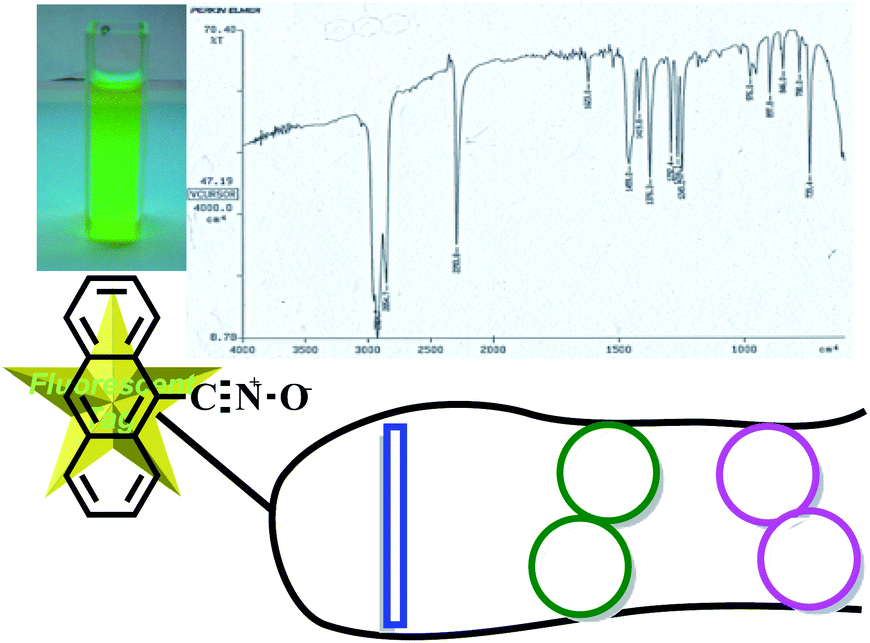 | ||
| Fig. 7 Folded compounds structure with fluorescent tag. | ||
The CD spectra somewhat differ from previous observation in reported works due to the presence/interference of the anthracene moieties.27 The Cotton effects are much weaker for compounds 10a,b and 11a,b with respect to the products previously described12,16 bearing simpler aminoacidic residues.28 Therefore, it seems that compounds 10a,b and 11a have some population of folded conformers because their Cotton effects in the range 248–258 nm are significantly large. However, the population of unfolded conformers is the majority and in one case, 11b, the CD profile seems to indicate a more critical folding behaviour. For this reasons NMR ROESY experiments positively corroborated the compounds turn arrangements.
The behavior and trend depicted by the CD is confirmed by the NMR titration experiments where the expected behavior of the NH 1 and 2-Tyr protons indicated the existence of strong H-bonding for compounds 10a,b and 11a,b.
From the structurally point of view the existence of a second important H-bonding at the tyrosine level of the peptide chain, together with the ROE cross peak HN(2-Tyr)–HN(1-anth), configure a structure of the folded compounds as shown in Fig. 8. All the other protons in this model are free or eventually involved in intramolecular H-bonding or intermolecularly linked when in solution.
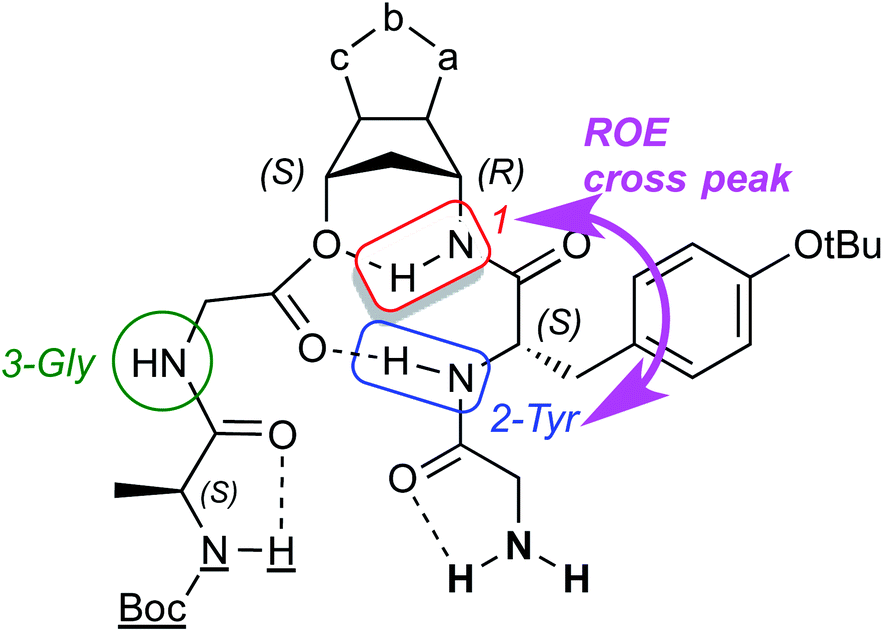 | ||
| Fig. 8 Folded compounds structure. | ||
In summary, the introduction of aminoacids in the side chains of cyclopenta[d]isoxazoline aminol structures produce some changes in the abilities of the isoxazoline ring to maintain the turn conformation in the synthesized compounds, mainly because of steric effects. Nevertheless, additional H-bonding interactions and the results collected by NMR and CD experiments somewhat enforce the use of this aminolic model to produce folding processes in this type of peptide motifs. Elongation of the peptide chains with a larger number of aminoacids (>4 units) is in our synthetic plans, extending the length of their beta strand mimics and presenting more beta strand like hydrogen bonds that can be observed in regions far from that of the engineered β-turn. In this way we will offer a clearer picture of the turn inducing ability of isoxazoline-aminol structures.
Conclusions
In conclusion, the synthetized diastereoisomeric compounds represent the reliable “proof” for the use of aminols, simply and easily prepared, for the synthesis of non-peptidic turn-inducers. We confirmed the crucial role of the cyclopenta[d]isoxazoline aminol structures in the formation of the β-turn arrangement. The performed experiments ascertained the influence of the two side chains features of on the applicability/reliability/robustness of these scaffolds as β-turn inducers and the limitations in terms of amino acid choice determined by the strong conformational preference of the bi-dimensional heterocyclic aminols. The choice of aminoacid can favor or disfavor the structure folding and the correct design of the peptide chains deeply influence the potential use of these nitrosocarbonyl-based compounds as turn-inducers. A decisive step forward in the evolution of this study on the ability of isoxazoline aminol structures to induce turns will be given by replacing the cyclopentane ring based compounds with the larger cyclohexane derivatives obtained from the same nitrosocarbonyl chemistry, thoroughly investigated by our group both from the theoretical and synthetic grounds.29,30 In this latter work, we prepared the same type of aminols that are ready to be proposed as turn-inducers. Indeed, the results here presented call for the introduction of variations on the region of engineered beta turn and these compounds, ready available from out consolidated chemistry, will be promptly used as potential turn inducers. Finally, the spectroscopic features of the synthesized turns will be finalized to design proper fluorescent tags, for imaging studies at the protein turn points and activity-based protein profiling (ABPP) investigations.31,32Experimental section
Melting points (mp) are uncorrected and were determined by the capillary method. HRMS were done on a X500D QTOF system (ABSciex) available at the CGS of the University of Pavia. 1H and 13C NMR spectra were recorded at 298 K on a AVIII 400 MHz Bruker NMR spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with a z-gradient coil probe, available at the CGS of the University of Pavia. All 1D and 2D NMR spectra were acquired using the standard pulse sequences available with Bruker Topspin 3.6. Solvents are specified in the text. Chemical shifts (δ) are expressed in ppm and referred to the solvent signal (δH 2.50 ppm for DMSO-d6 and 7.28 ppm for CDCl3), and coupling constants (J) are in hertz (Hz): b, broad; s, singlet; bs, broad singlet; d, doublet; t, triplet; m, multiplet. ROESY spinlock was set to 200 ms. IR spectra (nujol mulls) were recorded on a Perkin-Elmer RX-1 spectrophotometer and absorptions (ν) are in cm−1. The UV-vis spectra and Fluorescence spectra were recorded on a Perkin-Elmer LS-55 spectrophotometer. CD spectra were recorded on a Jasco J1500 spectropolarimeter at the Centro Grandi Strumenti (CGS) of the University of Pavia equipped with Spectra Analysis program. Column chromatography, TLC and MPLC: silica gel H60 and GF254, respectively; eluants: from cyclohexane/ethyl acetate 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to pure ethyl acetate or chloroform/methanol when needed.
:1 to pure ethyl acetate or chloroform/methanol when needed.
Starting and reference materials
Aminols 6a,b were prepared according to the established procedures.14,15 TBDMSiCl, Imidazole, Fmoc–Gly–Tyr(tBu)–OH, HBTU, DIEA, n-Bu4NF, Boc–Ala–Gly–OH, DIC, DMAP were purchased from chemical suppliers. Solvents and other reagents were also purchased and used as they were, without any further purification. The protected amines 7a,b were prepared according to the well-established procedure reported in literature and were found identical to authentic samples available in our laboratories.12,16Synthesis of compounds 7a,b
In a 100 mL flask, 956 mg (3 mmol) of aminols 6a,b were dissolved in 50 mL of anhydrous DCM along with 450 mg (6.61 mmol) of imidazole. TBDMSiCl (498 mg, 3.30 mmol) was then added under stirring at room temperature and the solution was left to react for 48 h under open air. After this period of time, the solution was diluted with DCM and the washed with brine (3 × 20 mL). The organic phase was dried over anhydrous Na2SO4 and filtered. Upon evaporation of the solvent, the pale yellow oils relative to the protected compounds 7a,b were obtained and fully characterized.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 0.16 (s, 3H, CH3), 0.21 (s, 3H, CH3), 0.92 (s, 9H, tBu), 1.67 and 2.16 (m, 1H + 1H, CH2), 3.08 (m, 1H, CH–N), 4.21 (d, 1H, J = 8 Hz, H4isox.), 4.54 (bs, 1H, CH–O), 5.32 (d, 1H, J = 8 Hz, H5isox.), 7.62 (m, 5H, anthr. and NH), 7.99 (d, 2H, anthr.), 8.19 (d, 3H, anthr. and NH), 8.78 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −4.5, −4.4, 18.2, 26.2, 41.8, 56.9, 68.3, 79.5, 92.4, 123.5, 125.3, 126.2, 127.6, 129.3, 130.3, 131.3, 156.7. C26H32N2O2Si (432.64): HRMS: calcd (2 MW + H) 865.454; found 865.4515.N) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = −1.18 (s, 3H, CH3), −0.92 (s, 3H, CH3), 0.11 (s, 9H, tBu), 1.39 and 1.78 (m, 1H + 1H, CH2), 3.31 (s, 1H, CH–N), 3.76 (s, 1H, CH–O), 4.05 (d, 1H, J = 8 Hz, H4isox.), 5.05 (d, 1H, J = 8 Hz, H5isox.), 7.41 (m, 5H, anthr. and NH), 7.80 (d, 2H, anthr.), 7.97 (d, 3H, anthr. and NH), 8.67 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −6.0, −5.5, 17.5, 25.3, 31.1, 41.5, 59.7, 67.4, 75.9, 93.6, 123.1, 126.1, 127.6, 129.2, 129.4, 131.3, 155.3. C26H32N2O2Si (432.64): HRMS: calcd (2 MW + H) 865.454; found 865.4515.
N) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 0.16 (s, 3H, CH3), 0.21 (s, 3H, CH3), 0.92 (s, 9H, tBu), 1.67 and 2.16 (m, 1H + 1H, CH2), 3.08 (m, 1H, CH–N), 4.21 (d, 1H, J = 8 Hz, H4isox.), 4.54 (bs, 1H, CH–O), 5.32 (d, 1H, J = 8 Hz, H5isox.), 7.62 (m, 5H, anthr. and NH), 7.99 (d, 2H, anthr.), 8.19 (d, 3H, anthr. and NH), 8.78 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −4.5, −4.4, 18.2, 26.2, 41.8, 56.9, 68.3, 79.5, 92.4, 123.5, 125.3, 126.2, 127.6, 129.3, 130.3, 131.3, 156.7. C26H32N2O2Si (432.64): HRMS: calcd (2 MW + H) 865.454; found 865.4515.N) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = −1.18 (s, 3H, CH3), −0.92 (s, 3H, CH3), 0.11 (s, 9H, tBu), 1.39 and 1.78 (m, 1H + 1H, CH2), 3.31 (s, 1H, CH–N), 3.76 (s, 1H, CH–O), 4.05 (d, 1H, J = 8 Hz, H4isox.), 5.05 (d, 1H, J = 8 Hz, H5isox.), 7.41 (m, 5H, anthr. and NH), 7.80 (d, 2H, anthr.), 7.97 (d, 3H, anthr. and NH), 8.67 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −6.0, −5.5, 17.5, 25.3, 31.1, 41.5, 59.7, 67.4, 75.9, 93.6, 123.1, 126.1, 127.6, 129.2, 129.4, 131.3, 155.3. C26H32N2O2Si (432.64): HRMS: calcd (2 MW + H) 865.454; found 865.4515.Coupling of amines 7a,b with Fmoc–Gly–Tyr(tBu)–OH
In a 100 mL flask, 222 mg (0.51 mmol) of amines 7a,b were dissolved in 50 mL of anhydrous DCM along with 214 mg (0.56 mmol) HBTU and 360 μL of DIPEA. Fmoc–Gly–Tyr(tBu)–OH (273 mg, 0.51 mmol) were then added under stirring at room temperature and the solution was left to react for 48 h under inert atmosphere. After this period of time, the organic phase is washed with brine and dried over anhydrous Na2SO4 and filtered. Upon evaporation of the solvent, oily residues were collected and the products 8a,b and 9a,b were isolated from column chromatography and fully characterized.O), 1668 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = −0.03 (s, 3H, CH3), −0.01 (s, 3H, CH3), 0.73 (s, 9H, tBu), 1.08 (s, 9H, tBuO), 1.51 and 1.89 (m, 1H + 1H, CH2), 1.74 and 2.02 (m, 1H + 1H, CH2), 3.29 (m, 2H, CH2), 3.84 (bs, 1H, CH–N), 3.75 (m, 1H, CO–CH–N), 4.03 (m, 1H + 2H, CH2 and H4isox.), 4.35 (bs, 1H, CH–O), 4.99 (d, 1H, J = 5 Hz, H5isox.), 6.41 (m, 4H, arom. Tyr), 7.11 (m, 2H, Fmoc), 7.21 (m, 2H, Fmoc), 7.40 (m, 4H, anthr.), 7.46 (m, 5H, Fmoc., NHFmoc, NHTyr and NH), 7.68 (d, 2H, Fmoc), 7.77 (d, 2H, anthr.), 7.97 (d, 2H, anthr.), 8.56 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −4.6, −4.4, 18.3, 26.3, 29.0, 37.0, 43.5, 47.0, 66.2, 78.0, 79.0, 91.9, 120.6, 123.6, 125.2, 125.7, 126.1, 127.5, 128.1, 129.3, 129.7, 131.3, 132.0, 141.1, 144.2, 156.3, 156.7, 169.6, 174.2, 174.9, 177.3. C56H62N4O7Si (931.22): HRMS: calcd 931.446; found 931.4433.O), 1680 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = −1.36 (s, 3H, CH3), −0.94 (s, 3H, CH3), 0.01 (s, 9H, tBu), 1.01 (s, 9H, tBuO), 1.88 (m, 1H, CH), 2.61 and 2.71 (m, 1H + 1H, CH2), 3.20 (m, 2H, CH2), 3.46 (m, 2H, CH2), 3.77 (m, 1H, CH–N), 4.07 (m, 4H, CH2, CO–CH–N and H4isox.), 4.22 (m, 1H, CH–O), 5.11 (dd, 1H, J = 5, 1 Hz, H5isox.), 6.60 (AA′BB′ syst., 2H, arom. Tyr), 6.89 (AA′BB′ syst., 2H, arom. Tyr), 7.12 (m, 2H, Fmoc), 7.21 (m, 2H, Fmoc), 7.38 (m, 5H, anthr., Fmoc and NHTyr), 7.52 (m, 2H, Fmoc), 7.69 (d, 2H, J = 8 Hz, anthr.), 7.79 (d, 2H, J = 8 Hz, anthr.), 7.97 (m, 3H, anthr. and NHFmoc), 7.99 (d, 1H, NH), 8.57 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −6.4, −5.4, 17.3, 25.3, 29.0, 38.7, 43.8, 47.1, 54.9, 55.9, 60.3, 63.4, 66.3, 66.6, 73.9, 78.0, 88.6, 120.6, 122.5, 123.8, 125.7, 126.1, 126.9, 127.1, 127.6, 128.1, 128.5, 129.2, 129.5, 130.2, 131.3, 132.3, 141.2, 144.3, 154.0, 155.9, 157.0, 169.4, 170.8. C56H62N4O7Si (931.22): HRMS: calcd 931.446; found 931.4445.O), 1652 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = −0.01 (s, 3H, CH3), −0.04 (s, 3H, CH3), 0.78 (s, 9H, tBu), 1.00 (m, 1H, CH), 1.07 (s, 9H, tBuO), 1.56 and 2.05 (m, 1H + 1H, CH2), 2.25 and 2.40 (m, 1H + 1H, CH2), 3.29 (m, 2H, CH2), 3.84 (bs, 1H, CH–N), 3.91 (m, 1H, CO–CH–N), 4.09 (m, 2H, CH2), 4.30 (dd, 1H, J = 5, 1 Hz, H4isox.), 4.41 (bs, 1H, CH–O), 5.18 (d, 1H, J = 5 Hz, H5isox.), 6.57 (AA′BB′ syst., 2H, arom. Tyr), 6.72 (AA′BB′ syst., 2H, arom. Tyr), 7.17 (m, 2H, Fmoc), 7.29 (m, 2H, Fmoc), 7.46 (m, 5H, anthr., Fmoc and NHFmoc), 7.55 (m, 2H, Fmoc), 7.64 (d, 1H, J = 7 Hz, NH), 7.76 (d, 3H, J = 8 Hz, anthr. and NHTyr), 7.84 (d, 2H, J = 8 Hz, anthr.), 8.03 (d, 2H, J = 8 Hz, anthr.), 8.63 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −4.5, 18.4, 26.3, 29.0, 29.5, 37.1, 43.5, 47.0, 52.9, 54.2, 63.5, 66.2, 78.0, 78.9, 92.3, 120.6, 122.7, 123.6, 125.1, 125.7, 126.2, 127.5, 127.7, 128.1, 129.3, 129.6, 129.9, 130.3, 131.2, 132.2, 141.2, 144.3, 153.8, 156.0, 156.8, 169.0, 169.6. C56H62N4O7Si (931.22): HRMS: calcd 931.446; found 931.4454.O), 1652 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = −1.28 (s, 3H, CH3), −0.90 (s, 3H, CH3), 0.01 (s, 9H, tBu), 1.01 (m, 1H, CH), 1.05 (s, 9H, tBuO), 1.43 and 1.97 (m, 1H + 1H, CH2), 2.63 and 2.78 (m, 1H + 1H, CH2), 3.43 (m, 2H, CH2), 3.78 (m, 1H, CH–N), 3.96 (m, 1H, CO–CH–N), 4.05 (m, 3H, CH2 and H4isox.), 4.21 (m, 1H, CH–O), 4.89 (d, 1H, J = 5 Hz, H5isox.), 6.67 (AA′BB′ syst., 2H, arom. Tyr), 6.93 (AA′BB′ syst., 2H, arom. Tyr), 7.11 (m, 2H, Fmoc), 7.20 (m, 2H, Fmoc), 7.41 (m, 6H, anthr., Fmoc and NHFmoc), 7.50 (d, 2H, J = 8 Hz, anthr.), 7.55 (d, 1H, J = 7 Hz, NH), 7.67 (d, 2H, J = 8 Hz, anthr.), 7.97 (d, 2H, J = 8 Hz, anthr. and NHTyr), 8.57 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = −6.2, −5.3, 17.4, 25.4, 27.3, 27.9, 29.0, 43.8, 47.1, 54.8, 55.3, 56.4, 63.4, 66.3, 66.8, 74.7, 78.1, 89.3, 120.6, 122.5, 123.9, 124.0, 125.0, 125.7, 126.1, 126.9, 127.1, 127.6, 128.1, 128.5, 129.3, 129.5, 130.2, 131.3, 132.4, 141.2, 142.9, 144.3, 154.1, 155.6, 157.0, 169.4, 170.7. C56H62N4O7Si (931.22): HRMS: calcd 931.446; found 931.4446.Coupling of compounds 8a,b/9a,b with Boc–Ala–Gly–OH
In a 100 mL flask, 100 mg (0.12 mmol) of compounds 8a,b/9a,b were dissolved in 50 mL of anhydrous DCM. DIC (31 mg, 0.24 mmol) were added along with a catalytic amount of DMAP and Boc–Ala–Gly–OH (33.2 mg, 0.13 mmol) under stirring at room temperature and the solution was left to react for 48 h under inert atmosphere. After this period of time, the organic phase is washed with a saturated solution NaHCO3 and brine and dried over anhydrous Na2SO4 and filtered. Upon evaporation of the solvent, oily residues were collected and the products 10a,b and 11a,b were isolated from column chromatography and fully characterized.O), 1530 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.18 (d, 3H, CH3),1.35 (s, 9H, tBu), 1.26 (s, 9H, tBuO), 1.72 and 2.18 (m, 1H + 1H, CH2), 2.36 and 2.22 (m, 1H + 1H, CH2Tyr), 3.51 (m, 2H, CH2Gly), 3.67 (m, 2H, CH2Gly), 3.97 (m, 3H, CH–N, CHAla and CHTyr), 4.14 (m, 1H, H4isox.), 4.43 (bs, 1H, CH–O), 5.20 (d, 1H, J = 9 Hz, H5isox.), 6.70 (m, 4H, arom. Tyr), 6.97 (d, 1H, J = 7 Hz, NHAla), 7.58 (1H, NH), 7.60 (m, 4H, anthr.), 7.78 (d, 1H, J = 8 Hz, NHTyr), 7.83 (t, 2H, J = 6 Hz, NH2), 7.94 (d, 2H, anthr.), 8.02 (t, 1H, J = 6 Hz, NHGly), 8.17 (d, 2H, anthr.), 8.77 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 18.4, 24.2, 24.8, 28.6, 29.0, 29.5, 36.8, 38.1, 42.0, 42.4, 48.1, 49.7, 50.2, 52.6, 54.1, 64.6, 77.3, 78.0, 78.3, 78.7, 91.7, 99.8, 123.1, 123.7, 125.2, 126.1, 127.5, 129.3, 129.4, 129.8, 130.4, 131.3, 132.4, 153.8, 155.7, 156.1, 168.6, 169.5, 173.5, 185.1, 192.5. C45H54N6O9 (822.96): HRMS: calcd 845.384 (MW + Na); found 845.3853.O), 1506 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.18 (d, 3H, CH3),1.27 (s, 9H, tBu), 1.36 (s, 9H, tBuO), 1.58 and 2.16 (m, 1H + 1H, CH2), 2.80 and 2.98 (m, 1H + 1H, CH2Tyr), 3.72 (m, 2H, CH2Gly), 3.73 (m, 2H, CH2Gly), 3.92 (m, 1H, CH–N), 3.99 (m, 1H, CHAla),4.27 (m, 1H, CH–O), 4.38 (m, 1H, CHTyr), 4.43 (m, 1H, H4isox.), 5.16 (d, 1H, J = 5 Hz, H5isox.), 6.86 (m, 2H, arom. Tyr), 6.97 (d, 1H, J = 7 Hz, NHAla), 7.14 (m, 2H, arom. Tyr), 7.62 (m, 4H, anthr.), 7.71 (s, 1H, J = 8 Hz, NH), 7.97 (m, 4H, anthr. and NH2), 8.08 (t, 1H, J = 5 Hz, NHGly), 8.20 (m, 3H, anthr. and NHTyr), 8.80 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 18.4, 28.7, 29.0, 29.5, 42.3, 42.6, 50.2, 55.0, 56.8, 78.1, 78.7, 123.9, 125.2, 126.2, 127.7, 129.3, 129.4, 130.1, 131.3, 132.7, 153.9, 155.7, 168.9, 169.6, 170.1, 170.3, 170.6, 173.6, 173.7. C45H54N6O9 (822.96): HRMS: calcd 823.403 (MW + H); found 823.4025.O), 1506 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.16 (d, 3H, CH3), 1.35 (s, 9H, tBu), 1.23 (s, 9H, tBuO), 1.75 and 2.16 (m, 1H + 1H, CH2), 2.41 and 2.59 (m, 1H + 1H, CH2Tyr), 3.53 (m, 2H, CH2Gly), 3.67 (m, 2H, CH2Gly), 3.96 (m, 1H, CH–N), 4.09 (m, 1H, CHTyr), 4.33 (m, 1H, H4isox.), 4.44 (bs, 1H, CH–O), 5.34 (d, 1H, J = 5 Hz, H5isox.), 6.74 (d, 2H, arom. Tyr), 6.91 (d, 2H, arom. Tyr), 6.97 (d, 1H, J = 7 Hz, NHAla), 7.61 (m, 5H, anthr. and NH), 7.84 (m, 3H, NH2 and NHTyr), 7.96 (d, 2H, anthr.), 8.02 (t, 1H, J = 5 Hz, NHGly), 8.19 (d, 2H, anthr.), 8.78 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 18.4, 21.5, 24.2, 28.6, 29.0, 36.9, 38.2, 42.1, 42.5, 48.1, 50.2, 53.3, 54.3, 64.3, 77.1, 78.1, 78.7, 92.0, 123.1, 123.8, 125.1, 126.2, 127.7, 129.3, 129.5, 129.9, 130.4, 131.2, 132.6, 153.8, 155.7, 155.9, 168.6, 169.5, 169.6, 172.5, 173.6. C45H54N6O9 (822.96): HRMS: calcd 845.384 (MW + Na); found 845.3837.O), 1508 (CN) cm−1.1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.18 (d, 3H, CH3), 1.37 (s, 18H, tBu), 1.72 and 2.21 (m, 1H + 1H, CH2), 2.79 and 2.95 (m, 1H + 1H, CH2Tyr), 3.70 (m, 2H, CH2Gly), 3.71 (m, 2H, CH2Gly), 3.93 (m, 1H, CH–N), 3.98 (m, 1H, CHTyr), 4.15 (m, 1H, H4isox.), 4.43 (bs, 1H, CH–O), 5.07 (d, 1H, J = 5 Hz, H5isox.), 6.91 (d, 2H, arom. Tyr), 7.00 (m, 1H, NHAla), 7.17 (d, 2H, arom. Tyr), 7.63 (m, 5H, anthr. and NH), 7.95 (d, 4H, anthr. and NH2), 8.06 (m, 1H, NHGly), 8.20 (m, 3H, anthr. and NHTyr), 8.80 (s, 1H, anthr.). 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 18.4, 19.3, 28.7, 29.0, 29.5, 37.3, 38.0, 42.2, 42.5, 50.3, 54.9, 56.8, 70.0, 74.4, 78.1, 78.7, 90.5, 123.1, 123.9, 125.1, 126.2, 127.7, 129.3, 130.1, 130.2, 131.3, 132.7, 135.1, 154.0, 155.7, 168.9, 169.6, 173.6, 173.7. C45H54N6O9 (822.96): HRMS: calcd 823.403 (MW + H); found 823.4023.Author contributions
Authors declare equal contribution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
University of Pavia is gratefully acknowledged for supporting the research activities that allowed obtaining the reported results. Financial support: We thank “VIPCAT – Value Added Innovative Protocols for Catalytic Transformations” project (CUP: E46D17000110009) for valuable financial support. We also thank for the financial support the project “Scent of Lombardy” (CUP: E31B19000700007).References
- A. M. C. Marcelino and L. M. Gierasch, Biopolymers, 2008, 889, 380 CrossRef PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed.
- C. André, B. Legrand, C. Deng, C. Didierjean, G. Pickaert, J. Martinez, M. C. Averlant-Petit, M. Amblard and M. Calmes, Org. Lett., 2012, 14, 960 CrossRef PubMed.
- D. S. Kemp, Trends Biotechnol., 1990, 8, 249 CrossRef CAS PubMed.
- Y. Shi, P. Teng, P. Sang, F. She, L. Wei and J. Cai, Acc. Chem. Res., 2016, 49, 428 CrossRef CAS PubMed.
- R. F. Hirschmann, K. C. Nicolaou, A. R. Angeles, J. S. Chen and A. B. Smith III, Acc. Chem. Res., 2009, 42, 1511 CrossRef CAS PubMed.
- G. D. Rose, L. M. Gierash and J. A. Smith, Adv. Protein Chem., 1985, 37, 1 CrossRef CAS PubMed.
- J. B. Ball and P. F. Alewood, J. Mol. Recognit., 1990, 3, 55 CrossRef CAS PubMed.
- K. S. Rotondi and L. M. Gierasch, Biopolym, 2006, 84, 13 CrossRef CAS PubMed.
- H. J. Dyson and P. E. Wright, Curr. Opin. Struct. Biol., 1993, 3, 60 CrossRef CAS.
- D. E. Kim, C. Fisher and D. Baker, J. Mol. Biol., 2000, 298, 971 CrossRef CAS PubMed.
- M. G. Memeo, M. Mella, V. Montagna and P. Quadrelli, Chem.–Eur. J., 2015, 21, 16374 CrossRef CAS PubMed.
- M. G. Memeo and P. Quadrelli, Chem. Rev., 2017, 117, 2108 CrossRef CAS PubMed.
- P. Quadrelli, M. Mella, A. Gamba Invernizzi and P. Caramella, Tetrahedron, 1999, 55, 10497 CrossRef CAS.
- P. Quadrelli, M. Mella, P. Paganoni and P. Caramella, Eur. J. Org. Chem., 2000, 2613 CrossRef CAS.
- M. G. Memeo, M. Bruschi, L. Bergonzi, G. Desimoni, G. Faita and P. Quadrelli, ACS Omega, 2018, 3, 13551 CrossRef CAS PubMed.
- Y. Moggio, L. Legnani, B. Bovio, M. G. Memeo and P. Quadrelli, Tetrahedron, 2012, 68, 1384 CrossRef CAS.
- C. Grundmann and J. M. Dean, J. Org. Chem., 1965, 30, 2809 CrossRef CAS.
- R. A. Brown, T. Marcelli, M. De Poli, J. Solà and J. Clayden, Angew. Chem., Int. Ed., 2012, 51, 1395 CrossRef CAS PubMed.
- (a) N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC; (b) D. A. Lightner and J. E. Gurst, Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy, Wiley, J. & Sons, New York, 2000 Search PubMed.
- (a) M. C. Manning and R. W. Woody, Biopolymers, 1991, 31, 569 CrossRef CAS PubMed; (b) C. Toniolo, A. Polese, F. Formaggio, M. Crisma and J. Kamphuis, J. Am. Chem. Soc., 1996, 118, 2744 CrossRef CAS.
- (a) K.-Y. Jung, K. Vanommeslaeghe, M. E. Lanning, J. L. Yap, C. Gordon, P. T. Wilder, A. D. MacKerell and S. Fletcher, Org. Lett., 2013, 15, 3234 CrossRef CAS PubMed; (b) V. Semetey, D. Rognasn, C. Hemmerlin, R. Graff, J.-P. Briand, M. Marraud and G. Guichard, Angew. Chem., Int. Ed., 2002, 114, 1973 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. S. Stevens, N. Sugawara, G. M. Bonora and C. Toniolo, J. Am. Chem. Soc., 1980, 102, 7048 CrossRef CAS.
- A. Bax and S. Grzesiek, Encyclopedia of Magnetic Resonance, J. Wiley & Sons, Ltd., New York, 2007 Search PubMed.
- Y. Wang and D. Shortle, Folding Des., 1997, 2, 93 CrossRef CAS PubMed.
- L. F. Minuti, M. G. Memeo, S. Crespi and P. Quadrelli, Eur. J. Org. Chem., 2016, 821 CrossRef CAS.
- K. Sato, N. Taki, U. Nagai and T. Higashijima, Bull. Chem. Soc. Jpn., 1983, 56, 2476 CrossRef CAS.
- P. Quadrelli, S. Romano and P. Caramella, Eur. J. Org. Chem., 2010, 6600 CrossRef CAS.
- P. Quadrelli, M. Mella, S. Carosso, B. Bovio and P. Caramella, Eur. J. Org. Chem., 2007, 6003 CrossRef CAS.
- M. Moiola, S. Crespi, M. G. Memeo, S. Collina, H. Overkleeft, B. Florea and P. Quadrelli, ACS Omega, 2019, 4, 7766 CrossRef CAS PubMed.
- M. Moiola, A. Bova, S. Crespi, M. G. Memeo, M. Mella, H. S. Overkleeft, B. I. Florea and P. Quadrelli, ChemistryOpen, 2019, 8, 770 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Professor Rolf Huisgen, “Maestro” of pericyclic reactions. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03685h |
| This journal is © The Royal Society of Chemistry 2021 |