
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of macroscopic monolithic metal–organic gels for ultra-fast destruction of chemical warfare agents†
Chuan Zhou ab,
Shouxin Zhangab,
Hongjie Panb,
Guang Yangb,
Lingyun Wangb,
Cheng-an Taoc and
Heguo Li*b
ab,
Shouxin Zhangab,
Hongjie Panb,
Guang Yangb,
Lingyun Wangb,
Cheng-an Taoc and
Heguo Li*b
aState Key Laboratory of NBC Protection for Civilians, Beijing 102205, PR China
bResearch Institute of Chemical Defense, Beijing 102205, PR China
cCollege of Liberal Arts and Science, National University of Defense Technology, Changsha 410073, China
First published on 23rd June 2021
Abstract
The potential threat that has originated from chemical warfare agents (CWAs) has promoted the development of advanced materials to enhance the protection of civilian and military personnel. Zr-based metal–organic frameworks (Zr-MOFs) have recently been demonstrated as excellent catalysts for decomposing CWAs, but challenges of integrating the microcrystalline powders of Zr-MOFs into monoliths still remain. Herein, we report hierarchically porous monolithic UiO-66-X xerogels for the destruction of CWAs. We found that the UiO-66-NH2 xerogel with a larger pore size and a higher surface area than the UiO-66-NH2 powder possessed better degradability of 2-chloroethyl ethyl sulfide (2-CEES), which is a sulfur mustard simulant. These UiO-66-X xerogels exhibit outstanding performance for decomposing CWAs. The half-lives of vesicant agent sulfur mustard (HD) and nerve agent O-ethyl S-[2-(diisopropylamino)ethyl] methylphosphonothioate (VX) are as short as 14.4 min and 1.5 min, respectively. This work is, to the best of our knowledge, the first report on macroscopic monolithic UiO-66-X xerogels for ultrafast decomposition of CWAs.
1. Introduction
Chemical warfare agents (CWAs) such as sulfur mustard (HD) and VX are lethal chemicals that pose an extreme risk to national security and public health, due to occasional or deliberate emissions.1 Detoxification of CWAs is an increasing research field that has generated great interest from the scientific and industrial communities. Current state-of-the-art materials for the capture of CWAs mainly rely on impregnated active carbons, which suffer from problems such as slow degradation kinetics, low capacity, and/or a lack of tailorability.2 Thus, there is an urgent need to explore novel self-detoxifying materials to efficiently protect civilian and military personnel.Recently, resourceful studies have been carried out on the catalytic performance of the decontamination of CWAs. Among them, metal–organic frameworks (MOFs) that are assembled by metal ions or clusters with organic linkers through coordination bonds have been extensively studied and have shown intriguing performances in CWA removal, due to their exceptionally large surface area, high porosity, tunable textural and chemical properties, and their abundant catalytic sites.3–5 In particular, Zr-based metal–organic frameworks (Zr-MOFs) have attracted great research attention as potential heterogeneous catalysts, due to their easy and numerous synthetic methods,6 and their exceptionally high thermal, chemical and mechanical stabilities.7 Meanwhile, it has been demonstrated that Zr-MOFs such as MOF-808, PCN-777, NU-1000 and UiO-66-NH2 have unprecedented catalytic degradation rates for nerve agents with half-lives of less than 0.5 min.8–10 However, the crystallization of Zr-MOFs often leads to microcrystalline powders. The application of Zr-MOF powders is greatly hampered, due to their inherent problems such as particle aggregation, poor processability and handling, mass transfer limitations, and significant pressure drop in an adsorption bed.11–15 To overcome the issues of Zr-MOF powders, many methods have been reported to construct Zr-MOF macroscale structures by either integrating Zr-MOFs into support materials such as fibers,16–18 polymeric monoliths19,20 and foams,21–23 or pelletizing Zr-MOF powders via mechanical compression or extrusion.24–26 However, both strategies still have issues such as reduced adsorption capacities, due to the use of Zr-MOF as a secondary component, and pressure-induced losses of crystallinity and porosity. Therefore, the development of pure monolithic Zr-MOFs has become a novel research field in recent years.
Metal–organic gels (MOGs) linked by metal–ligand coordination associated with hydrogen bonding, hydrophobic effects, π–π stacking and van der Waals interactions are a class of fabricable, hierarchically porous, pure MOF materials.27,28 Preparation of MOGs mainly depends on mismatched growth of the MOF particles by regulating key factors such as synthesis temperature and time,27 reactant concentration,11 organic-linker functionality and solvent29 in the gelation process. Therefore, the combination of Zr-MOFs with a suitable gelation process could shed light on Zr-MOF applications in the detoxification of CWAs. Up to now, only sporadic examples involving Zr-based metal–organic gels (Zr-MOGs) have been reported. For example, the synthesis of Zr-MOGs from ZrCl4 in a mixture of solvent/water proceeds through a rapid method at room temperature.29 Bueken et al. described a series of Zr-MOGs synthesized in DMF using ZrOCl2·8H2O as a precursor and a mixture of HCl/AA as a modulator.11 However, there has been almost no systematic or detailed investigation on the post-treatment of gel synthesis.
In this contribution, we systematically investigate the effect that post-treatment conditions have on porosity for the optimization and preparation of macroscopic monolithic UiO-66-X xerogels (UiO-66 and UiO-66-NH2 xerogels). As far as we know, no report regarding UiO-66-X xerogels for the catalytic destruction of real CWAs has appeared in the literature. Herein, we have first investigated the catalytic degradation performance of the UiO-66-NH2 xerogel and the UiO-66-NH2 powder for use with 2-chloroethyl ethyl sulfide (2-CEES), which is a sulfur mustard simulant. Then, we attempted to identify the sulfur mustard (HD) and VX decomposition abilities of neat UiO-66-X xerogels at room temperature. This is also the first demonstration of ultra-fast destruction of CWAs using Zr-MOFs xerogels.
2. Materials and methods
2.1. Chemicals and characterization methods
Unless otherwise noted, ACS reagent grade chemicals and solvents were purchased from commercial vendors and were used without further purification. Terephthalic acid (H2BDC), 2-aminoterephthalic acid (H2N-BDC), zirconyl chloride octahydrate (ZrOCl2·8H2O) and 2-chloroethyl ethyl sulfide (2-CEES) were obtained from Shanghai Macklin Biochemical Co., Ltd. Methanol, alcohol, acetone, N,N-dimethylformamide (DMF), hydrochloric acid (37%) and acetic acid were obtained from Shanghai Aladdin Chemicals Co., Ltd.Powder X-ray diffraction (PXRD) data were collected at room temperature on a Bruker D2 Phaser diffractometer, with Cu K-alpha radiation (λ = 1.54178 Å). Thermogravimetric analysis (TGA) was performed in air using a Mettler TGA/DSC 1 instrument equipped with an alumina crucible and heated at a rate of 5 °C min−1 up to 800 °C. N2 physisorption measurements were measured at 77 K on a micromeritics ASAP 2020 instrument. Prior to the measurements, the samples (20–40 mg) were degassed for 8 h at 110 °C and under 0.1 mbar vacuum. Transmission electron microscopy (TEM) was performed on a JEM-2100 microscope, operated in scanning mode (accelerating voltage of 200 kV). Scanning electron microscope (SEM) images were recorded on a JEOL JSM-IT300 microscope and were processed using the ImageJ software. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) and C, H and N elemental analyses were carried out using a Leeman Prodigy 7 ICP-OES analyser and a Vario EL elemental analyser, respectively.
2.2. Synthesis of the UiO-66 xerogel
The UiO-66 xerogel was prepared under optimum conditions by dissolving H2BDC (5 mmol) and ZrOCl2·8H2O (7.25 mmol) in DMF (30 mL) under sonication for 15 min. Afterwards, HCl (1.5 mL) and acetic acid (2.0 mL) as modulators were added to the above solution, and the mixture was left for 5 min under sonication. The resulting homogeneous solution was then allowed to stand at 373 K for 2 h in a closed container to yield a thick white UiO-66 gel. Subsequently, fresh DMF was added to the above UiO-66 gel and was mixed well. The diluted gel suspension was centrifuged (5000 rpm, 5 min) and the supernatant was decanted. The gel was then washed three times with ethanol and dried in air at 30 °C. The obtained xerogel was immersed in acetone and methanol for 12 h and dried in air at 30 °C to produce the macroscopic monolithic UiO-66 xerogel, as shown in Fig. 1.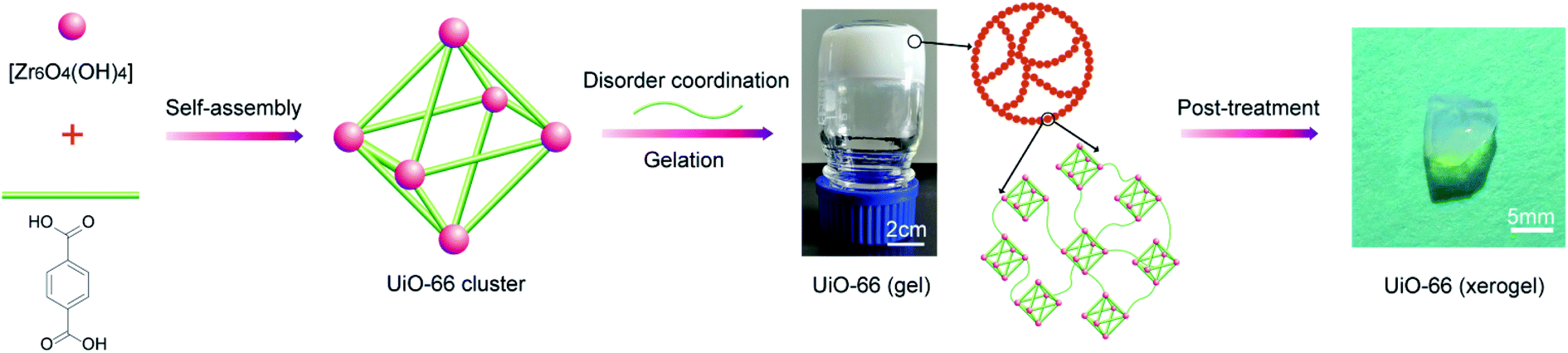 | ||
| Fig. 1 Schematic of the synthesis of the UiO-66 xerogel. | ||
2.3. Synthesis of the UiO-66-NH2 xerogel
The UiO-66-NH2 xerogel was prepared by replacing H2BDC in the UiO-66 xerogel synthesis procedure with an equimolar amount of H2N-BDC.2.4. Synthesis of the UiO-66-NH2 powder33
ZrOCl2·8H2O (0.54 mmol), H2N-BDC (0.75 mmol), 1 mL of HCl and 15 mL of DMF were added into a 100 mL Pyrex Schott bottle before being sonicated for 10 min until fully dissolved. The Pyrex Schott bottle was sealed and heated to 80 °C for 12 h. The resulting pale yellow solid was centrifuged and washed three times with DMF and alcohol. The sample was activated for 8 h in a vacuum oven at 110 °C.2.5. Degradation experiments
Important! CWAs (HD, VX) are known to be lethal if inhaled or in contact with skin. Experiments should be performed under/by trained personnel in adequate facilities.The catalytic degradation of 2-CEES, HD and VX was evaluated. The degradation of 2-CEES and HD was carried out by extraction: 25 μL CHCl3 solution containing 2.5 μL 2-CEES or HD was added to 25 mg xerogel in each glass vial, which were vortexed and left standing for various periods of time before extracting with 3 mL ethanol for 3 times and analyzing for residual 2-CEES or HD by GC-MS. 20 μL Pentane solution containing 0.4 μL VX was added to 20 mg xerogel in each glass vial. Afterwards, the vials containing the reaction mixtures were vortexed and were left standing at room temperature. Each vial was taken out for analysis at regular times. The residual VX was extracted in 2 mL of acetonitrile and the solution was subjected to gas chromatography with an FID detector.
3. Results and discussion
3.1. Xerogel optimization
Based on the essential principle of optimising the porosity of the xerogel, we selected UiO-66 as a prototype to perform a screening of several post-treatment parameters such as drying temperature, washing procedure, centrifugation procedure and metal-to-ligand ratio, in order to determine their influence on the porosity of the xerogel (Table S1†). Tian et al. noted that a low temperature of drying is beneficial for the formation of the monolithic HKUST-1 gel.12 Subsequently, we investigated the effect of drying temperature on the porosity of the UiO-66 xerogel. The result showed that the Brunauer–Emmett–Teller surface area (SBET) and the total pore volume (Vtot) of the xerogel could be enhanced by appropriately increasing the drying temperature (Table S1,† #1–#3). This may be due to the more rapid solvent evaporation rate at a higher temperature, resulting in an increase in mesoporous content in the xerogel. We further evaluated the influence of the washing procedure (washing solvents and washing times) on the porosity of the UiO-66 xerogel by washing the gel before drying at 30 °C. The result showed that the use of ethanol instead of DMF to wash the UiO-66 gel helps to enhance the SBET and Vtot values of the xerogel (Table S1,† #1 vs. #5, #4 vs. #6). In addition, we found that SBET and Vtot of the xerogel could be significantly increased by increasing the number of ethanol washing times (Table S1,† #5 vs. #6), whereas the number of DMF washing times had little significant effect (Table S1,† #1 vs. #4). Furthermore, we examined the effect of centrifugation time and found that a prolonged centrifugation time had no significant effect on the porosity of the xerogel (Table S1,† #1 vs. #7). A defect density would also greatly affect the porosity of the material.30,31 Finally, a series of UiO-66 xerogels (Table S1,† UiO-66-G, UiO-66-H, UiO-66-I) were synthesized by modulating different metal-to-ligand ratios, and these were characterized by TGA. All the synthesized samples exhibited excellent thermal stability in air up to 500 °C. The numbers of BDC linker in each unit of synthesized UiO-66 xerogel could be calculated according to the amount of mass lost (%) between 300 °C and 600 °C.32 The mass loss (%) of the BDC linkers for UiO-66-G, UiO-66-H and UiO-66-I was 40.6%, 40.3% and 37.5%, respectively (Fig. S1†). The calculated BDC ligands per unit of UiO-66-G, UiO-66-H and UiO-66-I were 4.5, 4.4 and 4.1, respectively. Less BDC ligands means more missing-linker defects, indicating that more defects exist in the structural units of UiO-66-I, and this results in a higher specific surface area and pore volume.3.2. Structural and morphological characterizations
Fig. 2a shows the XRD patterns of the monolithic UiO-66 xerogel and the UiO-66-NH2 xerogel. The apparent peaks at around 7° and 8.5° correspond well with the (111) and (200) reflections of the UiO-66 structure.33 Furthermore, broad diffraction peaks primarily originate from Scherrer broadening and nanoparticles. The morphologies of the monolithic UiO-66 xerogel and the UiO-66-NH2 xerogel are characterized by SEM and TEM. The SEM images show a rough surface with closely packed nanoparticles, as seen in Fig. 3e–h. The TEM results further confirm that the monolith consists of compact nanoparticles with particle interstitial space (Fig. 3a–d). Elemental analysis shows that the experimental compositions of the UiO-66-X xerogels are significantly different from their theoretical compositions (Table S2†), indicating the presence of defects, as illustrated by the lower experimentally observed carbon and nitrogen content relative to the theoretical content. In addition, the increase in hydrogen content of the experimental samples may indicate the existence of water in the crystal structure. The TGA curve of the monolithic UiO-66-X xerogels is illustrated in Fig. 2b. The high thermal stability of the monolith is consistent with the previously reported literature.34 As shown in Fig. 2c, the N2 adsorption–desorption isotherms are of Type IV, and this confirms the existence of a micro–mesoporous structure of the monolithic UiO-66 xerogel and of the UiO-66-NH2 xerogel. The Brunauer–Emmett–Teller surface areas (SBET), micro pore volumes (Wo) and total pore volumes (Vtot) are shown in Table S3.† Interestingly, these experimental values are higher than the theoretical maxima, and this may be related to the presence of extensive mesoporosity in the xerogels. The NLDFT analysis confirms the microporosity (Fig. 2d), while the BJH pore size distribution significantly highlights the extensive mesoporosity of between 5 and 20 nm (Fig. 2e). The mesoporosity of the xerogels is not generated by crystalline defects, but corresponds to interparticle voids.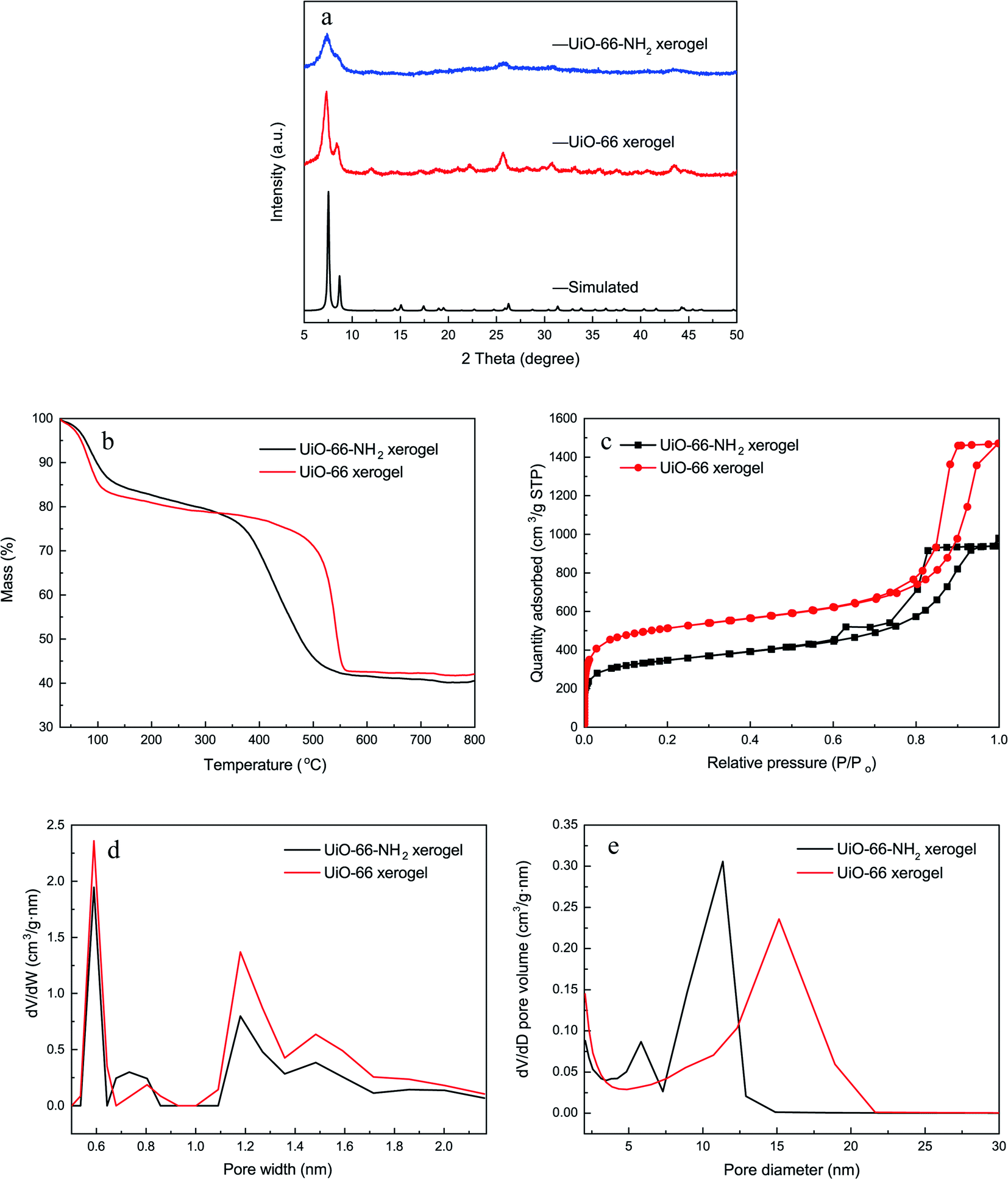 | ||
| Fig. 2 Characterization of the xerogels. (a) XRD patterns; (b) TGA curves; (c) N2 adsorption–desorption isotherms; (d) NLDFT pore size distributions; (e) BJH pore size distributions. | ||
 | ||
| Fig. 3 Electron microscopy (top: low magnification; down: high magnification) images for the UiO-66-X xerogels. TEM images of the UiO-66 xerogel (a and b) and UiO-66-NH2 xerogel (c and d); SEM images of the UiO-66 xerogel (e and f) and UiO-66-NH2 (g and h) xerogel. | ||
3.3. Degradation properties
In order to understand the effect of state on the decomposition of CWAs, a decomposition experiment for 2-CEES was implemented at room temperature. Data for the structural characterization of the UiO-66-NH2 powder are given in Fig. S2.† Fig. 4 shows the decomposition rates of 2-CEES over time on the UiO-66-NH2 xerogel and UiO-66-NH2 powder. The UiO-66-NH2 xerogel exhibits a faster reaction rate, with a half-life of 8.2 min, than the UiO-66-NH2 powder that exhibits a t1/2 value of 29 min. This finding can be attributed to the larger pore size and higher surface area of the UiO-66-NH2 xerogel, allowing for 2-CEES molecules to diffuse more rapidly and/or access more active sites.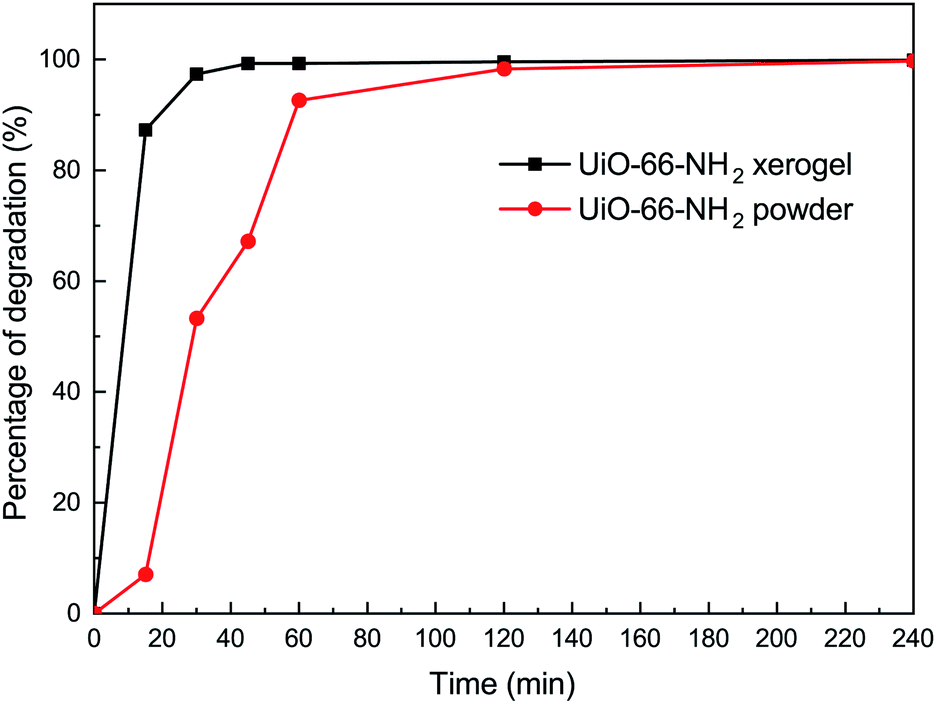 | ||
| Fig. 4 Degradation of 2-CEES on the UiO-66-NH2 xerogel and on the UiO-66-NH2 powder. | ||
In addition to the simulant 2-CEES, we further tested our monolithic UiO-66-X xerogels on the hydrolytic degradation of HD, a real CWA (Fig. 5). As is well-known, Zr-based metal–organic frameworks (Zr-MOFs) are highly active in the decomposition of P–X bonds (X = F, Cl, S, O) of nerve agents, but they are poorly active in the destruction of C–Cl of vesicant agents. This limits the practical application of Zr-MOFs for the degradation of unknown chemical threats. In this regard, we have found that the incorporation of an amino-group leads to significant improvement of the degradation of C–Cl bonds found in HD. For the UiO-66-NH2 xerogel, we observed 100% HD conversion in 240 min of reaction. The UiO-66-NH2 xerogel (t1/2 = 14.4 min) shows a faster destruction rate compared to the pristine UiO-66 xerogel (t1/2 = 24.8 min).
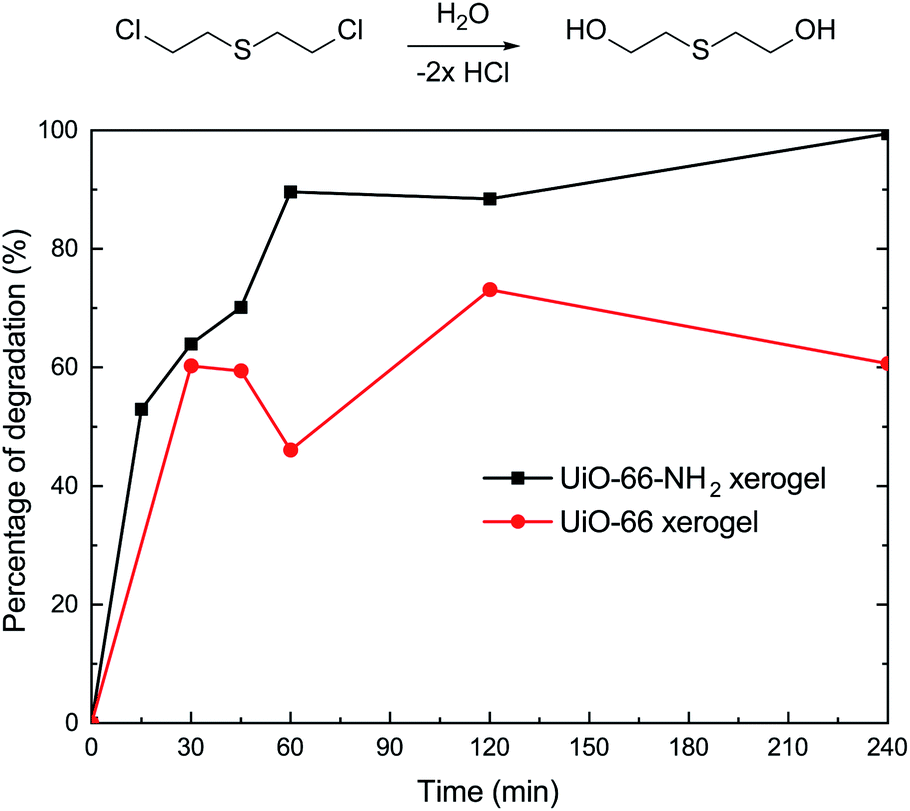 | ||
| Fig. 5 Degradation of HD on the UiO-66 xerogel and on the UiO-66-NH2 xerogel. | ||
To further investigate the catalytic performance of our materials, we also evaluated the behavior of the xerogels for the hydrolytic degradation of nerve agent VX. In this regard, it should be noted that both of the xerogels exhibited rapid VX degradation (t1/2 ≤ 1.5 min) and 100% conversion within 3 min (Fig. 6). There was little difference in the VX decomposing ability for the UiO-66 and UiO-66-NH2 xerogels. This discovery is in accordance with the previous report of Ryu et al.10 who studied the detoxification of GD on UiO-66 and UiO-66-NH2 powders.
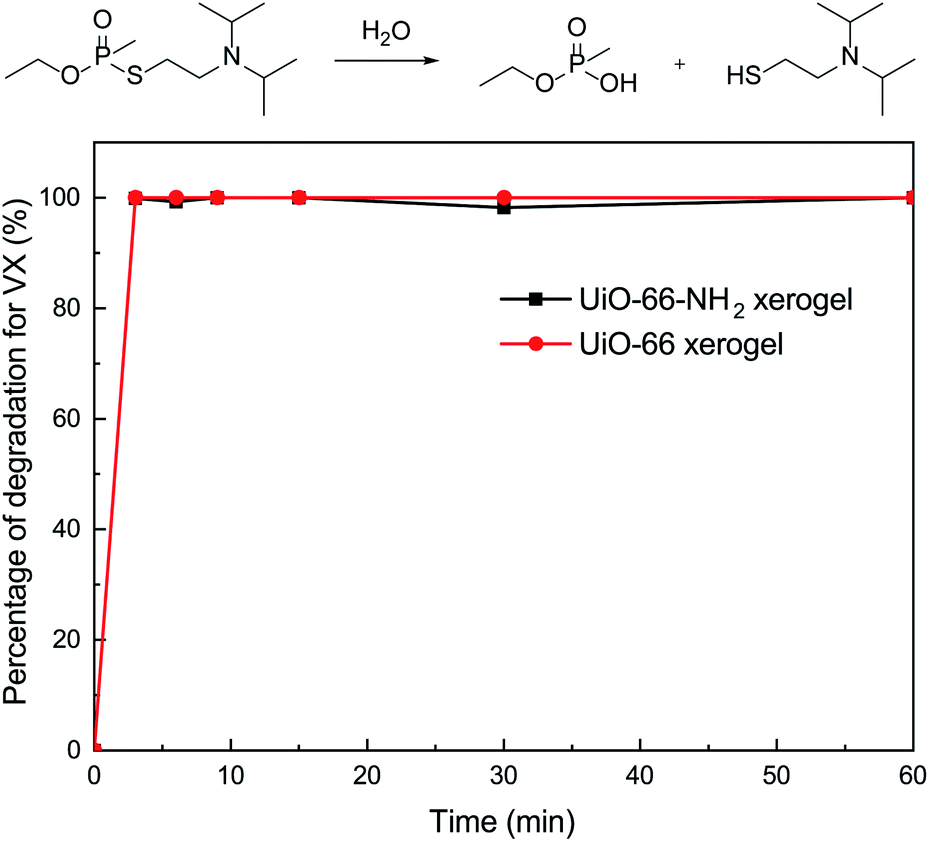 | ||
| Fig. 6 Degradation of VX on the UiO-66 xerogel and on the UiO-66-NH2 xerogel. | ||
4. Conclusions
In summary, UiO-66-X xerogels have been prepared and studied for the degradation of CWAs. The results indicate that the UiO-66-NH2 xerogel (t1/2 = 8.2 min) displays excellent catalytic performance for the degradation of 2-CEES compared to the UiO-66-NH2 powder (t1/2 = 29 min). Using these UiO-66-X xerogels shortens the half-lives of HD and VX to 14.4 min and 1.5 min, respectively. We also confirmed that there is little obvious degradability difference between the UiO-66 and UiO-66-NH2 xerogels for nerve agents in actual situations. This contribution shows that Zr-MOFs xerogels have remarkable prospects for use in future CWA protective applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the State Key Laboratory of NBC Protection for Civilian Foundation of China (SKLNBC201807) and by the National Natural Science Foundation of China (No. 22075319). The authors also acknowledge the Analytical Instrumentation Center of Peking University.References
- M. Zhang, Y. Liu, J. Chen, H. Liu, X. Lu, J. Wu, Y. Zhang, Y. Lin, Q. Liu, H. Wang, L. Guo, R. Gao, B. Xu and J. Xie, Anal. Chem., 2020, 92, 10578–10587 CrossRef CAS.
- S. S. Mondal and H. J. Holdt, Angew. Chem., Int. Ed., 2016, 55, 42–44 CrossRef CAS PubMed.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed.
- I. Stassen, B. Bueken, H. Reinsch, J. F. M. Oudenhoven, D. Wouters, J. Hajek, V. Van Speybroeck, N. Stock, P. M. Vereecken, R. Van Schaijk, D. De Vos and R. Ameloot, Chem. Sci., 2016, 7, 5827–5832 RSC.
- T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120, 8130–8160 CrossRef CAS.
- C. Zhou, B. Yuan, S. Zhang, X. Yang and J. Zhong, Chem. Ind. Eng. Prog., 2019, 38, 4614–4622 Search PubMed.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- M. C. de Koning, M. van Grol and T. Breijaert, Inorg. Chem., 2017, 56, 11804–11809 CrossRef CAS PubMed.
- J. M. Palomba, C. V. Credille, M. Kalaj, J. B. DeCoste, G. W. Peterson, T. M. Tovar and S. M. Cohen, Chem. Commun., 2018, 54, 5768–5771 RSC.
- S. G. Ryu, M.-K. Kim, M. Park, S. O. Jang, S. H. Kim and H. Jung, Microporous Mesoporous Mater., 2019, 274, 9–16 CrossRef CAS.
- B. Bueken, N. Van Velthoven, T. Willhammar, T. Stassin, I. Stassen, D. A. Keen, G. V. Baron, J. F. M. Denayer, R. Ameloot, S. Bals, D. De Vos and T. D. Bennett, Chem. Sci., 2017, 8, 3939–3948 RSC.
- T. Tian, Z. Zeng, D. Vulpe, M. E. Casco, G. Divitini, P. A. Midgley, J. Silvestre-Albero, J. C. Tan, P. Z. Moghadam and D. Fairen-Jimenez, Nat. Mater., 2018, 17, 174–179 CrossRef CAS PubMed.
- S. M. F. Vilela, P. Salcedo-Abraira, L. Micheron, E. L. Solla, P. G. Yot and P. Horcajada, Chem. Commun., 2018, 54, 13088–13091 RSC.
- W. Cui, X. Kang, X. Zhang and X. Cui, J. Phys. Chem. Solids, 2019, 134, 165–175 CrossRef CAS.
- J. Hou, A. F. Sapnik and T. D. Bennett, Chem. Sci., 2020, 11, 310–323 RSC.
- D. B. Dwyer, N. Dugan, N. Hoffman, D. J. Cooke, M. G. Hall, T. M. Tovar, W. E. Bernier, J. DeCoste, N. L. Pomerantz and W. E. Jones, Jr, ACS Appl. Mater. Interfaces, 2018, 10, 34585–34591 CrossRef CAS PubMed.
- H. Liang, A. Yao, X. Jiao, C. Li and D. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 20396–20403 CrossRef CAS PubMed.
- D. L. McCarthy, J. Liu, D. B. Dwyer, J. L. Troiano, S. M. Boyer, J. B. DeCoste, W. E. Bernier and W. E. Jones, Jr, New J. Chem., 2017, 41, 8748–8753 RSC.
- Y. Hara, K. Kanamori and K. Nakanishi, Angew. Chem., Int. Ed., 2019, 58, 19047–19053 CrossRef CAS PubMed.
- Y. Dong, L. Cao, J. Li, Y. Yang and J. Wang, RSC Adv., 2018, 8, 32358–32367 RSC.
- J. Zhao, L. Xu, Y. Su, H. Yu, H. Liu, S. Qian, W. Zheng and Y. Zhao, J. Environ. Sci., 2021, 101, 177–188 CrossRef PubMed.
- L. Wen, X. Chen, C. Chen, R. Yang, M. Gong, Y. Zhang and Q. Fu, Arabian J. Chem., 2020, 13, 5669–5678 CrossRef CAS.
- D. Xie, Y. Gu, H. Wang, Y. Wang, W. Qin, G. Wang, H. Zhang and Y. Zhang, J. Colloid Interface Sci., 2019, 542, 269–280 CrossRef CAS PubMed.
- Y. Khabzina, J. Dhainaut, M. Ahlhelm, H. J. Richter, H. Reinsch, N. Stock and D. Farrusseng, Ind. Eng. Chem. Res., 2018, 57, 8200–8208 CrossRef CAS.
- L. R. Redfern, L. Robison, M. C. Wasson, S. Goswami, J. Lyu, T. Islamoglu, K. W. Chapman and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 4365–4371 CrossRef CAS PubMed.
- D. W. Lee, T. Didriksen, U. Olsbye, R. Blom and C. A. Grande, Sep. Purif. Technol., 2020, 235, 116182–116193 CrossRef CAS.
- L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C. Y. Su, Nat. Commun., 2013, 4, 1774–1782 CrossRef PubMed.
- A. Carne-Sanchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506–2513 CrossRef PubMed.
- J. Santos-Lorenzo, R. San José-Velado, J. Albo, G. Beobide, P. Castaño, O. Castillo, A. Luque and S. Pérez-Yáñez, Microporous Mesoporous Mater., 2019, 284, 128–132 CrossRef CAS.
- X. Zhang, Y. Yang, L. Song, J. Chen, Y. Yang and Y. Wang, J. Hazard. Mater., 2019, 365, 597–605 CrossRef CAS PubMed.
- C. Yin, Q. Liu, R. Chen, J. Liu, J. Yu, D. Song and J. Wang, Ind. Eng. Chem. Res., 2019, 58, 1159–1166 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- J. Ren, H. W. Langmi, B. C. North, M. Mathe and D. Bessarabov, Int. J. Hydrogen Energy, 2014, 39, 890–895 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01703a |
| This journal is © The Royal Society of Chemistry 2021 |