
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT insight into the effect of Cu atoms on adsorption and dissociation of CO2 over a Pd8/ TiO2(101) surface†
Li Liu *ab and
Pingli Lvbc
*ab and
Pingli Lvbc
aState Key Laboratory of Biobased Material and Green Papermaking, Qilu University of Technology (Shandong Academy of Sciences), Jinan, 250353, China. E-mail: liuli_1636@qlu.edu.cn; Fax: +86 0531 89631630; Tel: +86 0531 89631168
bSchool of Light Industry and Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, China
cSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
First published on 12th May 2021
Abstract
In order to improve the photocatalytic activity of a bimetallic cocatalyst, understanding its mechanism is very important for the development of a CO2 photocatalyst. In this study, density functional theory (DFT) calculations were performed to investigate CO2 adsorption and dissociation over Pd–Cu bimetallic clusters loaded on a TiO2(101) surface, aiming at understanding the origin of the effect caused by the presence of Cu. The results demonstrated that the introduction of a Cu atom has a dual effect on the adsorption and dissociation of CO2: (1) it provides the positive polarization charge center to enhance CO2 adsorption, and (2) it up-shifts the d-band center of the Cu atom to improve the activation of CO2. Thus, the activity of the Pd7Cu1/TiO2(101) surface, as compared with that of the Pd8/TiO2(101) surface, can be significantly improved, and the active center is the introduced Cu atom. This result is not only helpful for the development of effective CO2 photocatalysts but also crucial to understand the basic mechanism of bimetallic catalysis.
1. Introduction
In recent decades, the significant increase in the amount of carbon dioxide (CO2) in the atmosphere has been widely regarded as a global environmental problem.1 Therefore, extensive research has been carried out to reduce CO2 levels. In particular, the photocatalytic reduction of CO2 into hydrocarbon fuels and chemicals is considered an effective method of controlling and utilizing CO2.2–4 Photocatalytic CO2 reduction by H2O was first reported more than 30 years ago and is currently gaining more attention, because sunlight can be used as the primary energy source for this reduction. There have been many studies on CO2 photoreduction, and various materials (especially semiconductors) have been evaluated for this purpose.2–7 Among all types of semiconductors, TiO2 has been the most commonly used photocatalyst because of its many advantages, including chemical and thermal stability, abundance, low toxicity, low cost, and high UV photoactivity.8–10Although TiO2 is the most explored semiconductor for the photocatalytic reduction of CO2, its efficiency is still far from optimal due to the lack of visible light photoreaction, rapid electron–hole recombination of photogenerated charges, and inefficient CO2 capture.11,12 The combination of TiO2 and other active ingredients is expected to overcome these limitations. In the past few decades, noble metals, such as Au, Pt, Ag, Cu, and Pd, have been studied as cocatalysts to promote the capability of TiO2 semiconductors in photocatalytic CO2 activation and conversion.11–18 In addition, bimetallic cocatalysts (such as PdCu, AuCu, CuPt, and AgPd, etc.) have higher CO2 conversion activity than single metal nanoparticles (NPs).11,12,16,19–25 For example, Garcia's group reported that Au and Cu loaded TiO2 photocatalyst in the appropriate Au/Cu ratio is an extremely efficient material for the solar-light reduction of CO2 to CH4.11 Tan et al. reported that the Ag/Pd bimetals supported on N-doped TiO2 nanosheet exhibit high selectivity for CO2 conversion.12 Recently, Huang et al. reported that the efficiency of CO2 hydrogenation to C2H5OH can be optimized by adjusting the composition of Pd–Cu NPs and catalyst support.25 Long et al.16 also indicated that isolated Cu atoms in a Pd lattice can form highly selective active sites for photocatalytic conversion of CO2 to CH4.
It has been established that bimetallic cocatalysts often exhibit better catalytic performance than their corresponding elemental metal counterparts due to their composition and synergic effects on the catalytic properties.26 However, the mechanism for improving photocatalytic activity and selectivity of bimetallic cocatalysts remains unclear and needs further study. Furthermore, the photoreduction of CO2 is a complex reaction process, mainly encompasses the following elementary steps: (i) photon absorption and excited carrier generation; (ii) activation of CO2 to form CO2δ−; (iii) dissociation of the C–O bond; and (iv) desorption of reduced products from the active sites.4 Among them, some of the main challenges have not been completely resolved, such as the complicated activation and adsorption mechanisms of CO2, the mechanisms and pathways of photocatalytic reaction, and low efficiency and selectivity of different products.13 Therefore, it is essential to use theoretical calculations to explain the role of bimetallic cocatalysts in enhancing the activity, selectivity and stability of semiconductor photocatalysts, and to provide theoretical basis for design of new, high-performance CO2 reduction cocatalysts. In this study, we chose TiO2(101) loaded with Pd–Cu bimetallic NPs, which is reported to be an effective catalyst for the photocatalytic reduction of CO2,16 to explore the catalysis of bimetallic nanoparticles, and to clarify how the presence of Cu atom improves the catalytic activity of Pd-loaded TiO2(101) surface. The (101) surface of anatase was selected because this surface is the most stable. The calculation results indicates that the electronic structure of supported Pd–Cu clusters is very important to the catalytic performance. The strong positive polarization potential and the elevated Cu d-band center make Pd7Cu1 have higher activity on TiO2(101), which promotes adsorption and dissociation of CO2.
2. Computational methods
All of the DFT27 calculations were carried out with the GGA-PBE28–31 functional using the CASTEP package.32 Electronic wave functions were expanded in a plane-wave basis set, and ultrasoft pseudopotentials were used to describe the ionic cores.33 The cutoff energy was set to 400 eV, and the Monkhorst–Pack34 k-points sampling was generated with a 2 × 2 × 1 grid. In order to determine the activation barriers of CO2 dissociation, the complete LST/QST method was used to search for the transition states.35 To confirm the transition states, we performed nudged elastic band (NEB) calculations using TS confirmation.It is well-known that the standard DFT calculation will greatly underestimate the band gap due to the insufficient cancellation of the self-interaction energy inherent in the DFT functional, which may affect the calculation results.36–38 To investigate this possibility, we did the test calculations by using the DFT+U method for Pd8 cluster on TiO2(101) surface and CO2 on Pd8/TiO2 (101) surface. U = 3.5 eV was chosen according to the literature.36,39 It can be seen from Table S1 in ESI† that the charge distribution on the Pd8 cluster calculated by DFT+U is similar to that calculated by DFT, and the adsorption stability trend of Pd8/TiO2(101) to CO2 is consistent (see Fig. S1 in ESI†). Because we only focus on qualitative trend analysis, the DFT is used in the following calculation.
A six layer slab with a (3 × 3) supercell was adopted to simulate the TiO2(101) surface. The Pd8 cluster has been studied for a long time and widely reported in the literature. Previous DFT calculations have shown that the lowest energy structure of the free Pd8 cluster has a bicapped octahedral geometry with D2d symmetry.40,41 Therefore, Pd8 clusters with D2d symmetry were used in this work, as shown in Fig. 1a. During the calculation, the three top layers and the adsorbates relaxed. A vacuum region of 15 Å was chosen for TiO2(101). The free CO2 molecule was optimized in a 10 Å × 10 Å × 10 Å unit cell. The optimized C–O bond length and O–C–O angle are 1.178 Å and 180.0°, respectively, which are consistent with the experimental42 and theoretically43 values reported previously.
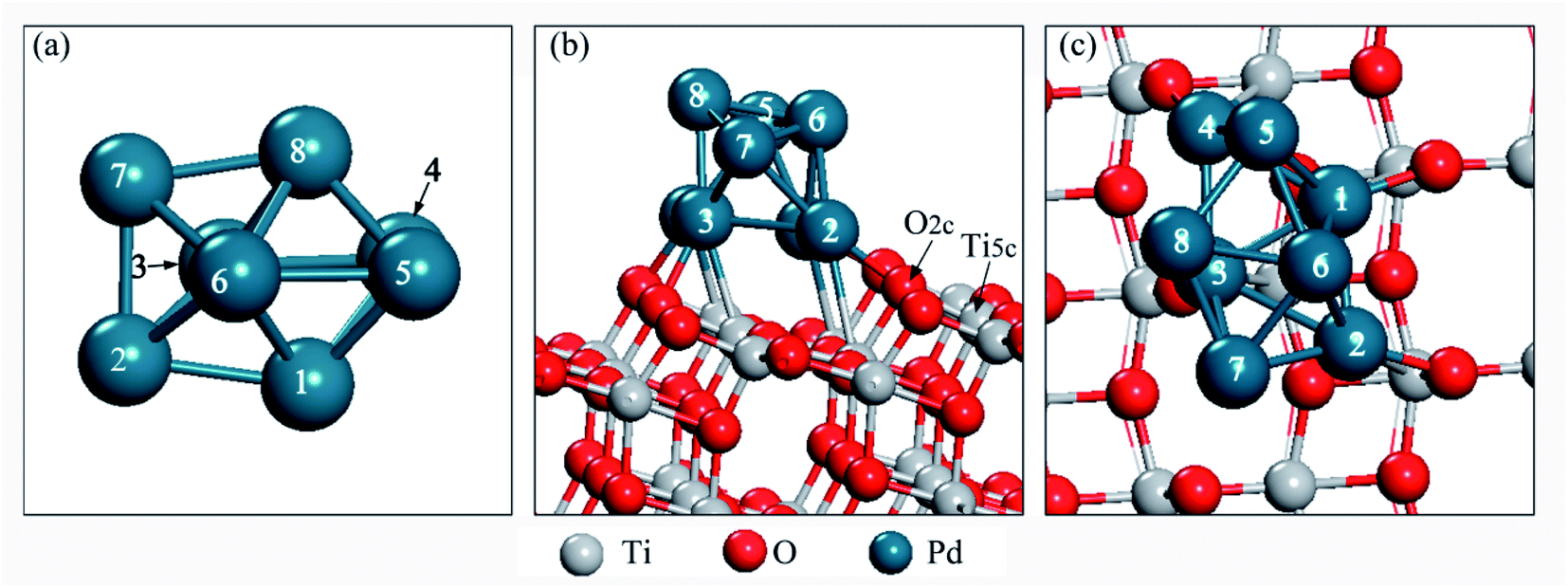 | ||
| Fig. 1 (a) Pd8 cluster, (b) side view of the Pd8/TiO2(101) surface, and (c) top view of the Pd8/TiO2(101) surface. | ||
The adsorption energy of CO2 is calculated as follows:
| Eads = ECO2 + surface − [ECO2 + Esurface] |
3. Results and discussion
3.1. Pd8 and Pd7Cu1 clusters on TiO2(101)
In order to find the most stable configuration of Pd8 on TiO2(101) surface, we constructed various possible configurations (see Fig. S2 in ESI†). Through optimization, we found that the structure shown in Fig. 1 is the most stable. As for Pd7Cu1 on TiO2(101) surface, we tested eight possible replacing modes in order to find the most stable configuration, as shown in Fig. S3.† We chose the most stable configuration for further research, as shown in Fig. 2. A comparison to the optimized Pd8 cluster shows that the Pd–TiO2(101) interactions result in a completely different structure. On TiO2(101), the cluster adopts a bilayer structure. This is due to the fluxionality of the metal cluster.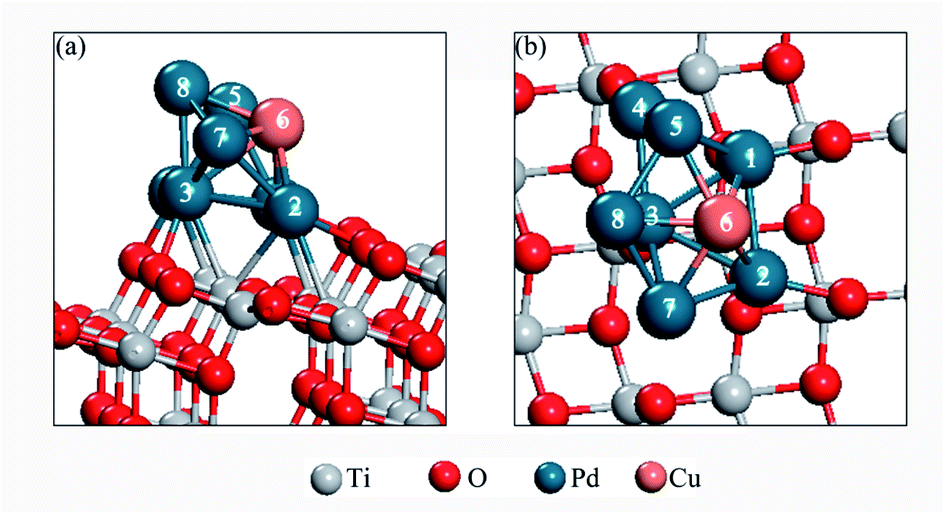 | ||
| Fig. 2 The side (a) and top view (b) of the most stable model of the Pd7Cu1 cluster on the TiO2(101) surface. | ||
The charge density difference and Mulliken charge analysis for the Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces were calculated to further understand the interaction between the Pd8 (or Pd7Cu1) cluster and the TiO2 surface from the perspective of electronic structure. As shown in Fig. 3a, when Pd8 or Pd7Cu1 clusters supported on the surface of TiO2(101), obvious charge transfer occurs at the interface, which is a long-range interaction that affects the charge distribution of the Pd or Cu atoms in the upper layer, and can lead to charge polarization between the clusters and the TiO2(101) surface. It can be seen from the electrostatic potential of Pd8/TiO2(101) and Pd7Cu1/TiO2(101) in Fig. S4† that the polarization potentials of Pd8 and Pd7Cu1 are positive, while the polarization potentials of TiO2(101) surface are negative. Therefore, when the interface forms and reaches an equilibrium, the final polarized electric field is from the clusters to the TiO2(101) surface.
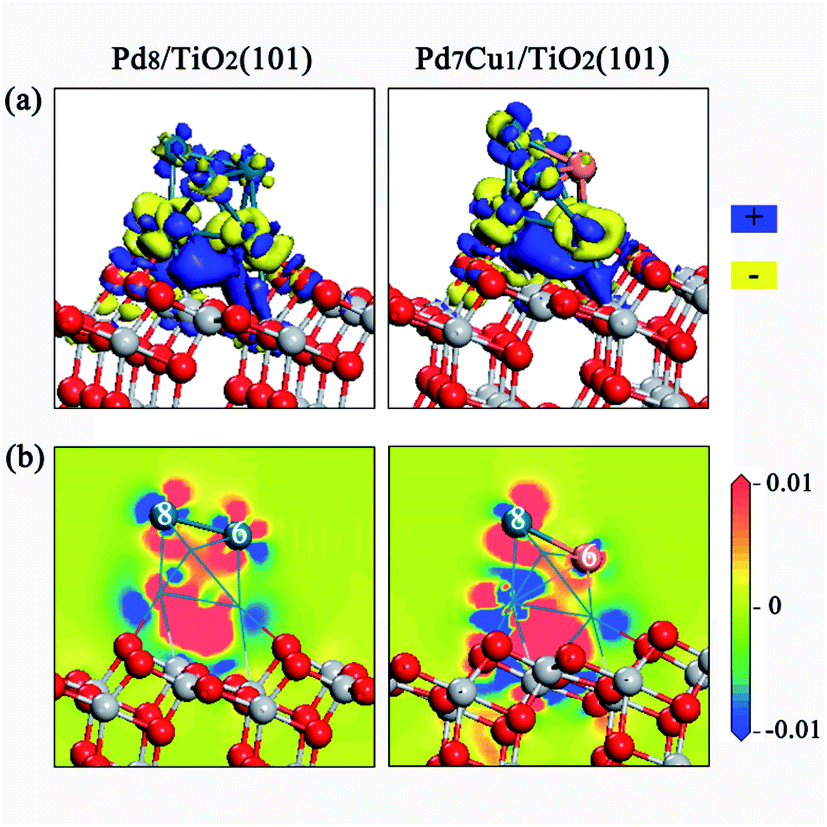 | ||
| Fig. 3 Charge density difference for the Pd8/TiO2(101) (left) and Pd7Cu1/TiO2(101) (right) surfaces. | ||
As shown in Fig. 3b, although both the Pd8 and Pd7Cu1 clusters can provide electrons to the TiO2(101) surface, the charge transfer of each atom is different. Mulliken charge analysis also confirmed this conclusion. The results in Table 1 show that all interfacial Pd atoms (from Pd-1 to Pd-4) of the Pd8 cluster are positively charged. However, all of the top-layered Pd atoms (from Pd-5 to Pd-8) are negatively charged. For the Pd7Cu1 cluster, because the electronegativity of Cu is lower than that of Pd, the Cu-6 can provide its electrons to the neighboring Pd atoms and become positively charged.
| Pd8/TiO2(101) | Pd-1 | Pd-2 | Pd-3 | Pd-4 | Pd-5 | Pd-6 | Pd-7 | Pd-8 | Total |
| Charge/|e| | 0.19 | 0.13 | 0.08 | 0.09 | −0.10 | −0.01 | −0.08 | −0.01 | 0.29 |
| Pd7Cu1/TiO2(101) | Pd-1 | Pd-2 | Pd-3 | Pd-4 | Pd-5 | Cu-6 | Pd-7 | Pd-8 | Total |
| Charge/|e| | 0.17 | 0.16 | 0.14 | 0.04 | −0.18 | 0.14 | −0.10 | −0.05 | 0.32 |
3.2. Adsorption and activation of CO2 over Pd8/TiO2(101)
For the Pd8/TiO2(101) surface, the most stable CO2 adsorption configuration is shown in Fig. 4a, labeled as Pd8–CO2. The other unstable adsorption models are displayed in Fig. S5.† We summarized our calculated results in Table 2. In order to describe conveniently, the two oxygen atoms of the CO2 are labeled Oa and Ob, respectively.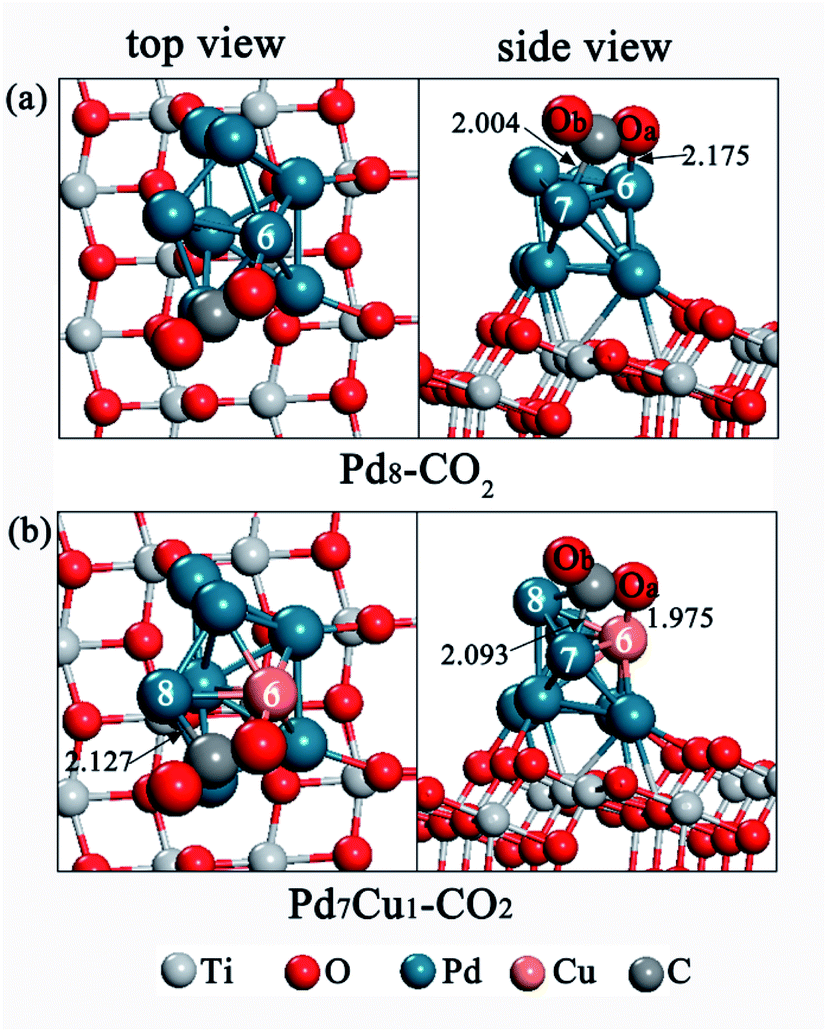 | ||
| Fig. 4 Top and side views of CO2 adsorption configuration on (a) the Pd8/TiO2(101) surface and (b) the Pd7Cu1/TiO2(101) surface. Distances are in Å. | ||
| Configurations | C–Oa bond (Å) | C–Ob bond (Å) | Oa–C–Ob angle (°) | Eads (eV) |
|---|---|---|---|---|
| Pd8–CO2 | 1.249 | 1.233 | 141.7 | −0.58 |
| Pd7Cu1–CO2 | 1.281 | 1.249 | 135.9 | −1.06 |
| CO2 molecule | 1.180 | 1.180 | 180.0 |
In configuration Pd8–CO2 (Fig. 4a), CO2 is adsorbed on Pd-6 and Pd-7 atoms in form of a bidentate carbonate, with an adsorption energy of −0.58 eV. The Oa bonds to the Pd-6 atom, and the C atom bonds to the Pd-7 atom. The bond lengths of Oa–Pd-6 and C–Pd-7 bonds are 2.175 Å and 2.004 Å, respectively. As shown in Table 2, compared with the free CO2 molecule, the adsorbed CO2 in configuration Pd8–CO2 has been heavily distorted. The length of the C–Oa and C–Ob bonds are both elongated, and the Oa–C–Ob angle is reduced to 141.7°, which indicates that CO2 is activated. In addition, it can be seen from Table S2† that the adsorbed CO2 has a negative charge of −0.43|e|, as a consequence of electron transfer from surface to CO2, and leading to a bent anionic CO2δ− species. The CO2δ− can increase the reactivity of CO2, including the breaking of C–O bond. Local density of states (LDOS) analysis was also conducted to further understand the interaction between the Pd8/TiO2(101) surface and adsorbed CO2. Fig. S6a† shows the LDOS of free CO2; Fig. S6b† shows the LDOS of Pd8–CO2 configuration. From Fig. S6b,† for C and Pd-7 (or Oa and Pd-6) atom in configuration Pd8–CO2, it can be clearly seen that the orbital overlap is more obvious in the range of −10 to 0 eV. This indicates that the interaction between the CO2 and the Pd8/TiO2(101) is strong.
3.3. Adsorption and activation of CO2 over Pd7Cu1/TiO2(101)
For CO2 adsorption on Pd7Cu1/TiO2(101), the most stable adsorption configuration is shown in Fig. 4b, labeled as Pd7Cu1–CO2. Other unstable adsorption configurations are shown in Fig. S5.† The most stable adsorption configuration Pd7Cu1–CO2 appears at the Cu-6 site, which is assisted by nearby Pd-7 and Pd-8 atoms. In configuration Pd7Cu1–CO2, the C atom of CO2 bridges two Pd atoms (Pd-7 and Pd-8), and the Oa atom bonds to the Cu-6 atom. The bond lengths of the Oa–Cu-6, C–Pd-7, and C–Pd-8 bonds are 1.975 Å, 2.093 Å, and 2.127 Å, respectively. This configuration has a high exothermic adsorption energy of −1.06 eV. According to Table 2, the bond lengths of C–Oa and C–Ob bonds are elongated to 1.281 Å and 1.249 Å, respectively, which are longer than those of gas-phase CO2 molecule, and the Oa–C–Ob angles are reduced to 135.9°, indicating CO2 is activated upon adsorption on Pd7Cu1/TiO2(101) surface. The Mulliken charge analysis in Table S2† shows that the adsorbed CO2 on Pd7Cu1/TiO2(101) surface has a negative charge of −0.61|e|. This indicates that electron transfer occurs from surface to CO2, and forms a negatively charged CO2δ− species. The LDOS in Fig. S6c† shows that the resonant peaks lie mainly in the range of −10 to 0 eV, indicating that the adsorbed CO2 has a strong tendency to hybridize with the Pd7Cu1/TiO2(101) surface at lower energy levels. Moreover, it can be seen from Table 2 that the presence of monatomic Cu significantly increases the adsorption stability of CO2. Structurally, Pd7Cu1–CO2 is similar to Pd8–CO2 on the Pd8/TiO2(101) surface without Cu (Fig. 4a), but its adsorption energy is more negative (the adsorption energy increases by 0.48 eV). In addition, when CO2 adsorbs on the Pd8/TiO2(101) surface, the Oa–Pd-6 bond distance is 2.175 Å. However, when CO2 adsorbs on the Pd7Cu1/TiO2(101) surface, the bond distance of the Oa–Cu-6 is shortened to 1.975 Å. This indicates that the interaction between CO2 and surface becomes stronger. The Mulliken charge analysis of CO2 on Pd7Cu1/TiO2(101) (Table S2†) showed the largest gain of electrons, suggesting stronger formation of CO2δ− anions. This suggests that the introduction of Cu atom enhances the activation of CO2. Thus, Pd7Cu1/TiO2(101) is more favorable for CO2 adsorption and activation compared to Pd8/TiO2(101). This conclusion is similar to our previous work, where we also found the presence of Cu atoms to promote adsorption and activation of CO2 on Au/TiO2(101).43 In addition, this result is in good agreement with a recent experimental report by Long et al.16 who studied photocatalytic CO2 reduction by H2O on clean TiO2, PdxCu1–TiO2 (x = 1, 3, 5, 7, 9, 11), and Pd–TiO2. They found that the PdxCu1 alloys with isolated Cu atoms in Pd lattice can provide highly active sites to enhance the adsorption and activation of CO2.3.4. Dissociation of CO2 over Pd8/TiO2(101) and Pd7Cu1/TiO2(101)
Having elucidated the adsorption and activation of CO2, we now discuss the effect of bimetallic cocatalyst Pd–Cu on the dissociation of CO2. There are two possible pathways for CO2 dissociation: direct dissociation or H-assisted dissociation. Here, H comes from the dissociation of H2O in the system, see Fig. S7.† We first study the direct dissociation path. The potential energy profiles are displayed in Fig. 5. Optimized structures of the transition states and products involved in this pathway are also included in Fig. 5. Starting from Pd8–CO2, CO2 subsequently dissociates into adsorbed CO* and O* on the Pd8/TiO2(101) surface through the transition state TS1. Here the asterisk (*) indicates an adsorbed species. As represented by the black line in Fig. 5, this process is endothermic by 1.44 eV and has an activation barrier of 2.80 eV. Finally, the CO* can be hydrogenated to HCO* via a C–H bond formation (see Fig. S8†) or desorbs from the surface into the gas phase with a heat of 0.28 eV needed to fulfill this process (see Fig. 5). And at the same time, the O* may migrate to surface oxygen vacancy (one of the most common defects in metal oxides) and becomes lattice oxygen or combine with H* in the system to form surface hydroxyl groups (OH*). For CO2 dissociation on the Pd7Cu1/TiO2(101) surface (the red line in Fig. 5), beginning with Pd7Cu1–CO2, the adsorbed CO2 dissociated into CO* and O* crosses a barrier of 2.18 eV and is endothermic by 0.59 eV. Then, CO* desorption or further hydrogenation to generate HCO*. Therefore, from the viewpoint of activation barrier and reaction energy, the Pd7Cu1/TiO2(101) surface is more favorable for CO2 direct dissociation than the Pd8/TiO2(101) surface, both kinetically and thermodynamically.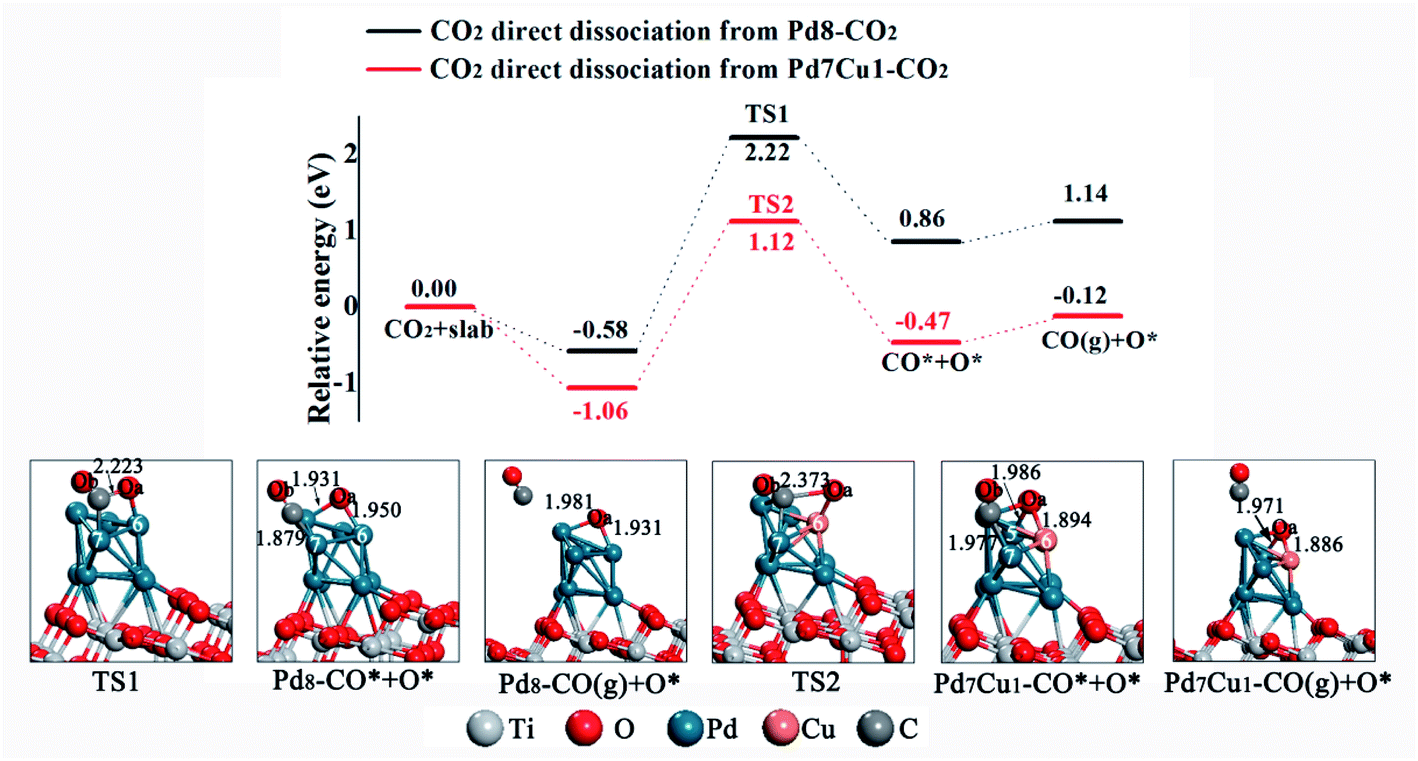 | ||
| Fig. 5 Potential energy profile for the direct dissociation of CO2 on the Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces. Distances are in Å. | ||
Next, we study the H-assisted CO2 dissociation. The existence of the H atom may activate CO2 via formation of COOH* or HCOO* species, which will alter the potential of CO2 dissociation. We started with the most stable adsorption configurations Pd8–CO2 and Pd7Cu1–CO2 and placed an H atom on the Pd-5 and Pd-8 site. For COOH* formation, the structural tends to form the cis-COOH* (the formation of trans-COOH* is shown in Fig. S9†), in which the H atom attack the Oa atom of CO2 to form an Oa–H bond, as shown in Fig. 6. On Pd8/TiO2(101) surface, the potential energy profile in Fig. 6 (black line) shows that the energy barrier for the first step of the cis-COOH* formation is 1.62 eV (TS3). This process is endothermic by 0.82 eV. Then the C–Oa band breaks and forms adsorbed CO* and OH* through the transition state TS4, with an activation barrier of 0.78 eV. This process is exothermic by 0.21 eV. Thus, the rate-determining step of this route is the process of forming cis-COOH*. On the Pd7Cu1/TiO2(101) surface, the energy profile in Fig. 6 (red line) shows that the first step of the cis-COOH* formation is endothermic by 0.34 eV and has an activation barrier of 1.49 eV (TS5). Then the C–Oa band breaks and forms CO* and OH* through the transition state TS6, with an activation barrier of 0.30 eV. This process is exothermic by 0.60 eV. The activation barrier of TS5 is higher than TS6, indicating that the formation of cis-COOH* is also the rate-determining step in this reaction. The above results for H-assisted CO2 dissociation via the cis-COOH* path on Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces show that the activation barrier of the rate-determining step (TS5, 1.49 eV) on the Pd7Cu1/TiO2(101) surface is lower than on the Pd8/TiO2(101) surface (TS3, 1.62 eV). Moreover, the corresponding reaction energies are 0.97 and 0.34 eV, indicating that Pd7Cu1/TiO2(101) surface is more favorable for CO2 dissociation than Pd8/TiO2(101) surface. Therefore, compared to the Pd8/TiO2(101) surface, the H-assisted CO2 dissociation via the cis-COOH* path on the Pd7Cu1/TiO2(101) surface is more favorable both kinetically and thermodynamically. This result indicates that the presence of Cu-6 atom promotes the H-assisted CO2 dissociation process.
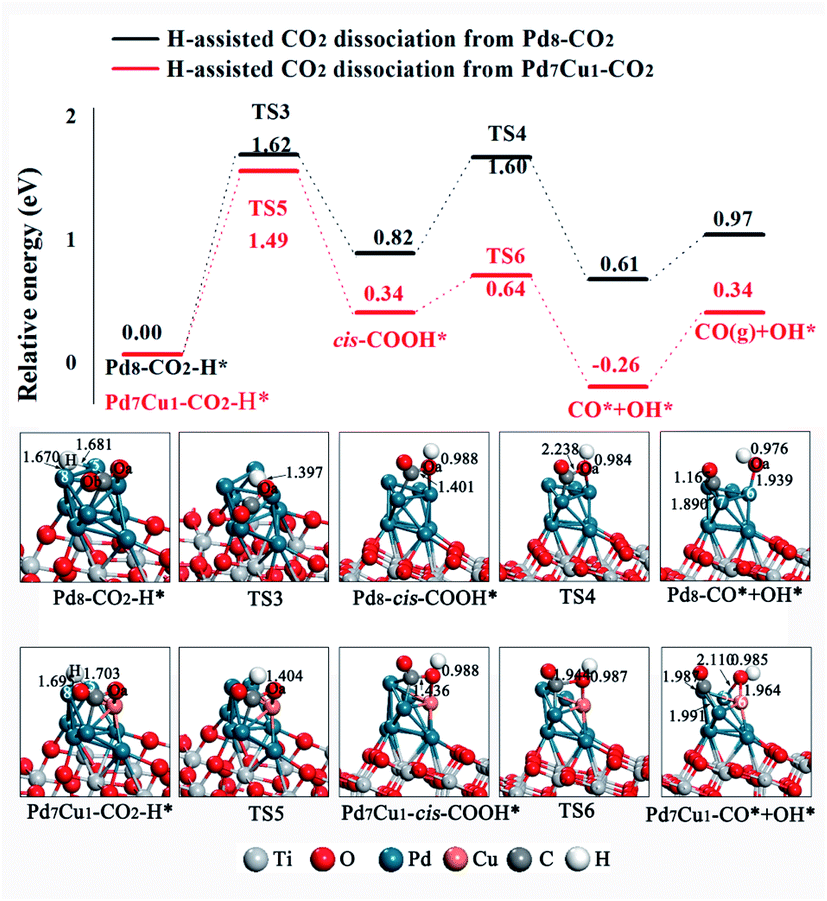 | ||
| Fig. 6 Potential energy profile for the H-assisted dissociation of CO2 via the cis-COOH* path on the Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces. Distances are in Å. | ||
As for HCOO* formation, the H atom attack the C atom of CO2 to form an C–H bond, as shown in Fig. 7. On Pd8/TiO2(101) surface, a stable bidentate HCOO* configuration was found, in which the two O atoms bond to surface Pd-6 and Pd-7, respectively. CO2 hydrogenation to HCOO* requires surmounting a barrier of 1.45 eV, and the reaction is 0.13 eV endothermic. Next, the HCOO* cleaves to form HCO* and O* in a 1.27 eV endothermic step with 2.84 eV energy barrier, limiting the rate of reaction. Then the H atom of HCO* combine with O* and C–H band breaks to form CO* and OH* with an energy barrier of 1.85 eV and reaction energy of 0.99 eV. Finally, CO* desorption or further hydrogenation to generate HCO*. On the Pd7Cu1/TiO2(101) surface, the energy profile in Fig. 7 (red line) shows that the first step of the HCOO* formation is endothermic by 0.08 eV and has an activation barrier of 1.24 eV. Subsequently, the C–Oa band breaks and forms HCO* and O* through the transition state TS11, with an activation barrier of 2.50 eV. This process is endothermic by 1.13 eV. Then the H atom of HCO* combine with O* and C–H band breaks to form CO* and OH* with an energy barrier of 1.57 eV and reaction energy of 0.92 eV. Finally, CO* desorption or further hydrogenation to generate HCO*. This result for H-assisted CO2 dissociation via the HCOO* path on Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces show that the activation barrier of the rate-determining step on the Pd7Cu1/TiO2(101) surface is lower than on the Pd8/TiO2(101) surface (2.50 vs. 2.84 eV). Moreover, the corresponding reaction energies are 1.09 and 0.82 eV, indicating that Pd7Cu1/TiO2(101) surface is more favorable for CO2 dissociation than Pd8/TiO2(101) surface. Therefore, compared to the Pd8/TiO2(101) surface, the H-assisted CO2 dissociation via the HCOO* path on the Pd7Cu1/TiO2(101) surface is also more favorable both kinetically and thermodynamically, indicating that the presence of Cu-6 atom promotes the H-assisted CO2 dissociation process. In addition, compared to the COOH*-mediated route, HCOO*-mediated path is less competitive because of its higher barrier (2.84 and 2.50 vs. 1.62 and 1.49 eV). Combined with CO2 adsorption and activation over Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces, we can conclude that the catalytic activity of the Pd7Cu1/TiO2(101) is higher than that of Pd8/TiO2(101).
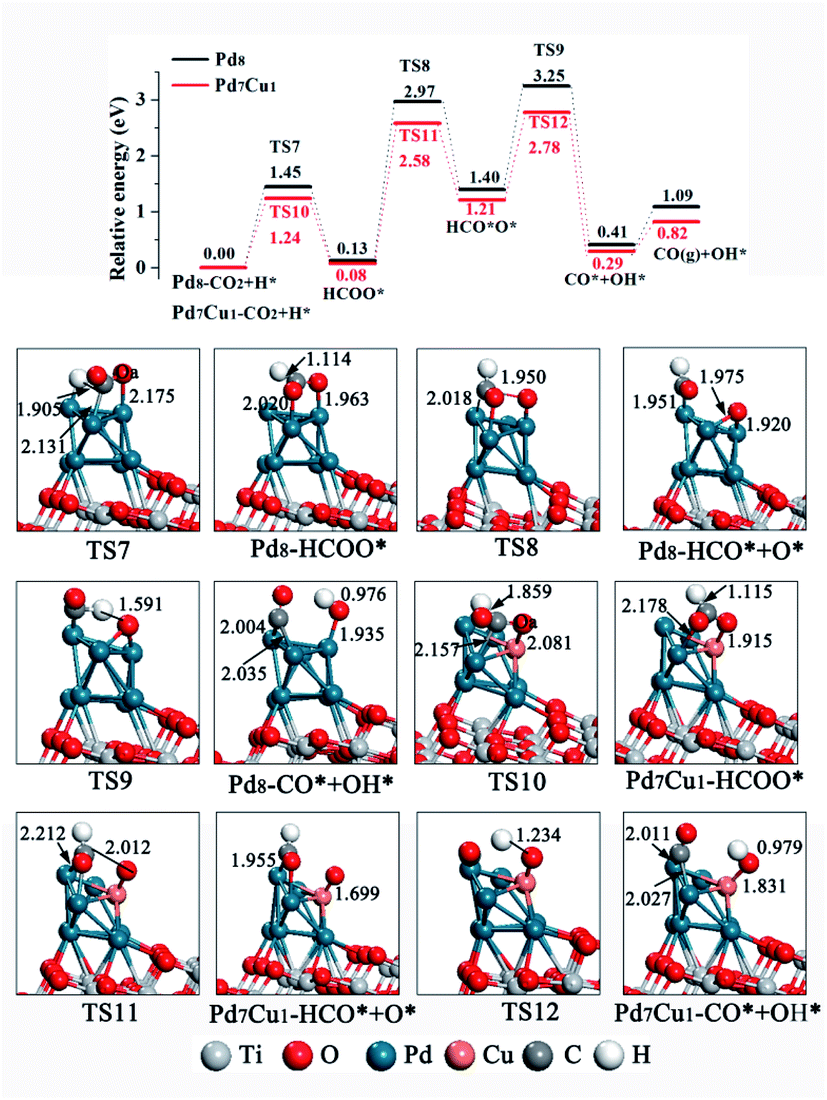 | ||
| Fig. 7 Potential energy profile for the H-assisted dissociation of CO2 via the HCOO* path on the Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces. Distances are in Å. | ||
3.5. Discussion
We have studied adsorption, activation, and dissociation of CO2 over Pd–Cu supported on TiO2(101). In this section, the enhancing effect of the Cu on the catalytic activity of the Pd supported TiO2(101) is discussed. It is a well-known fact that in addition to the surface atomic structure, the surface electronic states are also closely related to the catalytic activity. Therefore, in order to determine the intrinsic factors that promote the catalytic activity of Pd7Cu1/TiO2(101), a detailed comparison of the surface electronic states between the surfaces of Pd8/TiO2(101) and Pd7Cu1/TiO2(101) was conducted.As shown in Fig. 8, the 2D electric potential maps of Pd7Cu1/TiO2(101) shows that the electric potential distribution on the Cu-6 and Pd-8 atoms is no longer uniform due to the large charge polarization between Cu-6 and Pd atoms. The positive polarization potential of the Cu-6 atom is higher than that of the Pd-8 atom. This means that the electric field on the Cu-6 atom is higher than that on the Pd-8 atom. This response can promote the adsorption of CO2 (in which the O atoms are negatively charged) by increasing the electrostatic attraction between the CO2 and the Cu-6 atom. Therefore, the surface activity of the Pd7Cu1/TiO2(101) is improved compared to the Pd8/TiO2(101), and the active center is the introduced Cu atom. It is precisely because of the stable adsorption of CO2 on Cu-6 atoms and the special electric field on Cu-6 atoms that the reactivity of CO2 is enhanced. This result is consistent with previous results. Jia et al.44 investigated the CO oxidation on Ru–Pt supported on TiO2(101) and found that the special electric field on the top-layered Ru atom can enhance the stability and reactivity of CO + O2. Luo et al.45 reported that the optimal performance shows a strong dependence on the interaction between CO2 and the local electric field.
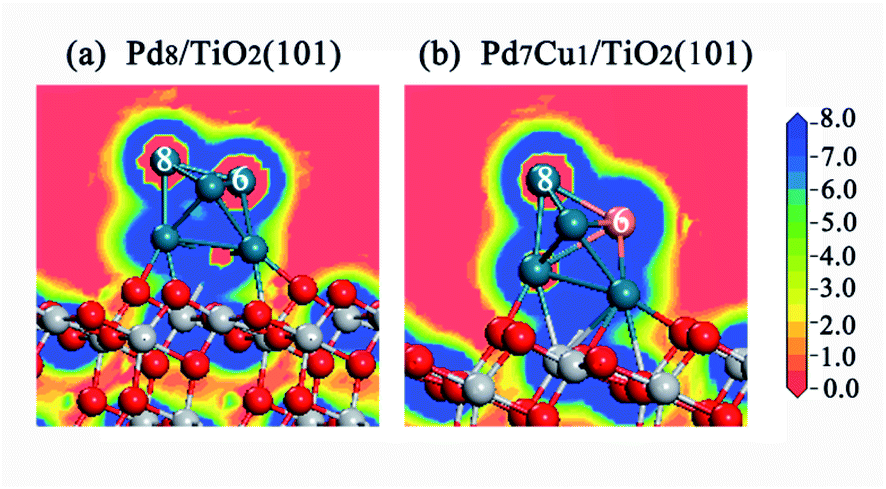 | ||
| Fig. 8 The 2D electric potential maps for the (a) Pd8/TiO2(101) and (b)Pd7Cu1/TiO2(101) surfaces. | ||
Fig. 9 is DOS projected onto the d orbitals of Pd-6 (or Cu-6) and Pd-8 on the Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces. It can be seen from the Fig. 9 that the d-band center (εd) of the Cu-6 atom on Pd7Cu1/TiO2(101) is higher than that of the Pd-6 atom on Pd8/TiO2(101) (−1.31 versus −1.46 eV). It is a well-known fact that, relative to the Fermi level, the higher the energy of the d-band states, the stronger the interaction with the adsorbate.46 This is because when the d-band states is close to the Fermi level, the antibonding states can be pushed above the Fermi level.46 Therefore, Pauli repulsion decreases and the binding strength between the adsorption site and adsorbate increases.46 Thus, the adsorption strength of CO2 on Pd7Cu1/TiO2(101) surface is higher than that of Pd8/TiO2(101) surface. Our results are consistent with previous results that show the adsorption strength of CO2 is controlled by the d-band center of the metal surfaces. For example, Long et al.16 calculated the d-band centers of Cu atom in Pd7Cu1 and Pd1Cu1 lattices, and found that the Cu d-band center of Pd7Cu1 lattice is significantly higher than that of Pd1Cu1 lattice (−1.161 versus −1.452 eV), and the adsorption energy of CO2 on Pd7Cu1 lattice is stronger than that on Pd1Cu1 lattice (−0.463 versus −0.308 eV).
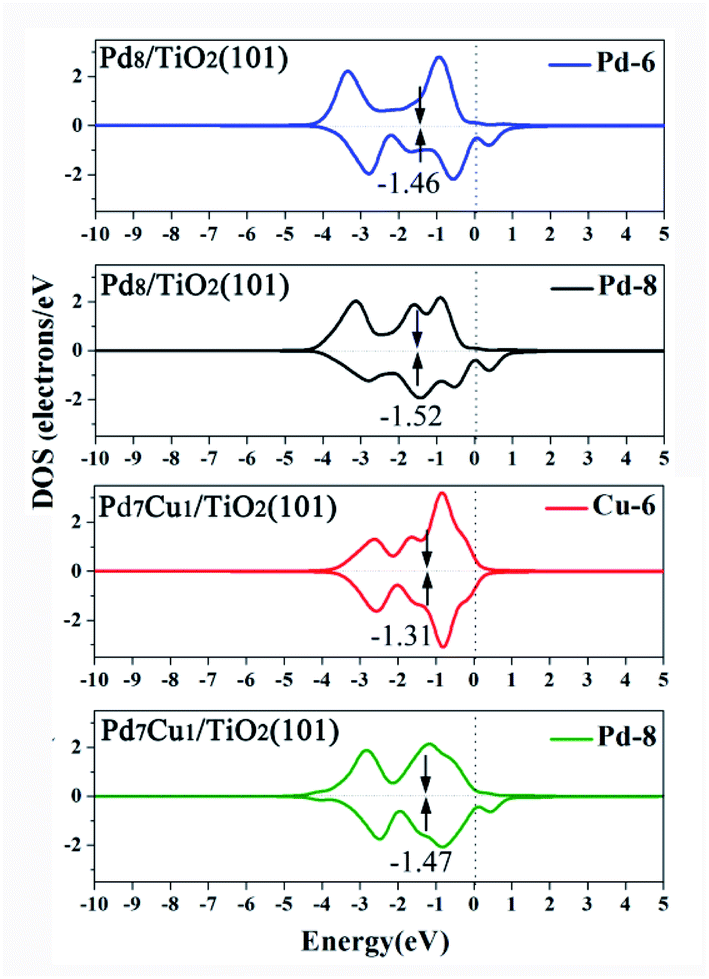 | ||
| Fig. 9 Projected d-band states of Pd and Cu on the Pd8/TiO2(101) and Pd7Cu1/TiO2(101) surfaces. The d-band center is marked by black arrow. The Fermi level is set to 0 eV. | ||
Based on the above discussion, the following conclusions can be drawn: due to the strong positive polarization potential and the elevated Cu d-band center, the Pd7Cu1/TiO2(101) has high activity, which promotes CO2 adsorption and dissociation.
4. Conclusions
In order to understand the origin of the activity of Pd–Cu bimetallic nanoclusters supported on TiO2(101), adsorption, activation, and dissociation of CO2 on TiO2-supported Pd and Pd–Cu clusters were investigated using DFT calculation. The results show that the activity of Pd7Cu1/TiO2(101) can be greatly improved by introducing Cu atom. Because of the significant charge polarization between the Cu atom and the neighboring Pd atoms on the Pd7Cu1/TiO2(101) surface, the positive polarization potential of the Cu atom is much higher than that of the Pd atom. This response can promote the adsorption of CO2 by increasing the electrostatic attraction between CO2 and the Cu site. Meanwhile, the elevated Cu d-band center on the surface of Pd7Cu1/TiO2(101) would increase its surface catalytic activity. Therefore, compared to Pd8/TiO2(101) surface, the activity of Pd7Cu1/TiO2(101) surface is higher, and the active center is the introduced Cu atom. This result is not only immensely helpful for the development of effective CO2 photocatalysts, but also essential for understanding the basic mechanisms of bimetallic catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Foundation (No. ZZ20200133) of State Key Laboratory of Biobased Material and Green Papermaking, Qilu University of Technology (Shandong Academy of Sciences) and the Natural Science Foundation of Shandong Province of China (Grant No. ZR2018LA013).References
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC.
- W. Chu, Q. Zheng, O. V. Prezhdo and J. Zhao, J. Am. Chem. Soc., 2020, 142, 3214–3221 CrossRef CAS PubMed.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro and H. Garcia, ChemSusChem, 2013, 6, 562–577 CrossRef PubMed.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- Ş. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed.
- D. Tan, J. Zhang, J. Shi, S. Li, B. Zhang, X. Tan, F. Zhang, L. Liu, D. Shao and B. Han, ACS Appl. Mater. Interfaces, 2018, 10, 24516–24522 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- V. Vaiano, D. Sannino and P. Ciambelli, Photochem. Photobiol. Sci., 2015, 14, 550–555 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, P. Dong, Z. Huang, X. Nie and X. Zhang, RSC Adv., 2018, 8, 15991–15998 RSC.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS PubMed.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 RSC.
- L. Liu, Z. Liu, H. Sun and X. Zhao, Appl. Surf. Sci., 2017, 399, 469–479 CrossRef CAS.
- X. Zhang, F. Han, B. Shi, S. Farsinezhad, G. P. Dechaine and K. Shankar, Angew. Chem., Int. Ed., 2012, 51, 12732–12735 CrossRef CAS PubMed.
- S. Lee, S. Jeong, W. D. Kim, S. Lee, K. Lee, W. K. Bae, J. H. Moon, S. Lee and D. C. Lee, Nanoscale, 2016, 8, 10043–10048 RSC.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS PubMed.
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Catal., B, 2017, 204, 548–560 CrossRef CAS.
- Q. Chen, X. Chen, M. Fang, J. Chen, Y. Li, Z. Xie, Q. Kuang and L. Zheng, J. Mater. Chem. A, 2019, 7, 1334–1340 RSC.
- J. Jiao, Y. Wei, Y. Zhao, Z. Zhao, A. Duan, J. Liu, Y. Pang, J. Li, G. Jiang and Y. Wang, Appl. Catal., B, 2017, 209, 228–239 CrossRef CAS.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- Y. Yang and D. Cheng, J. Phys. Chem. C, 2014, 118, 250–258 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Perderson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Perderson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4978 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 14999 CrossRef CAS.
- M. Segall, P. Lindan, M. Probert, C. Pickard, P. Hasnip, S. Clark and M. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- C.-T. Yang, B. C. Wood, V. R. Bhethanabotla and B. Joseph, J. Phys. Chem. C, 2014, 118, 26236–26248 CrossRef CAS.
- Y. Han, M. Zhang, W. Li and J. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 8683–8692 RSC.
- C. T. Yang, B. C. Wood, V. R. Bhethanabotla and B. Joseph, Phys. Chem. Chem. Phys., 2015, 17, 25379–25392 RSC.
- D. C. Sorescu, W. A. Al-Saidi and K. D. Jordan, J. Chem. Phys., 2011, 135, 124701 CrossRef PubMed.
- J. X. Liu, Y. Su, I. A. W. Filot and E. J. M. Hensen, J. Am. Chem. Soc., 2018, 140, 4580–4587 CrossRef CAS PubMed.
- Y. Li, C. Liu, Z. Pu, L. Bao, Y. Zhu and J. Ma, J. Phys. Chem. C, 2019, 123, 13739–13747 CrossRef CAS.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- L. Liu and P. Lv, New J. Chem., 2020, 44, 14662–14669 RSC.
- C. Jia, W. Zhong, M. Deng and J. Jiang, J. Chem. Phys., 2018, 148, 124701 CrossRef PubMed.
- H. Jiang, Z. Hou and Y. Luo, Angew. Chem., Int. Ed., 2017, 56, 15617–15621 CrossRef CAS PubMed.
- J. Xiao and T. Frauenheim, J. Phys. Chem. C, 2013, 117, 1804–1808 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01724a |
| This journal is © The Royal Society of Chemistry 2021 |