
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal oxide adsorption on fullerene C60 and its potential for adsorption of pollutant gases; density functional theory studies†
Sanaz Haghgoo and
A.-Reza Nekoei*
and
A.-Reza Nekoei*
Department of Chemistry, Shiraz University of Technology, Shiraz, 71555-313, Iran. E-mail: nekoei@sutech.ac.ir; Tel: +98-07137354501-7
First published on 12th May 2021
Abstract
Combinations of fullerene and metal oxides (MOx) are interesting, not only because they display the individual properties of fullerene and of MOx nanoparticles, but they may also exhibit synergetic properties that are advantageous for gas sensing applications. In the present work, the adsorption of some different MOxs on fullerene C60, and also the NO2 and CO sensing properties of these complexes, have been theoretically studied. All quantum mechanical computations have been carried out using Gaussian G09, employing the DFT method at the B97D/6-311G(d,p) level. NBO theory has been used for analysis of the charge transfers during gas adsorption. The chemical nature of the newly formed bonds in the studied complexes and their relative strength have been analysed using AIM2000 software. The results show that MOx/C60 complexes are much stronger adsorbents for NO2 and CO than C60 is. It is also expected that these complexes have more optical and electrical sensitivity in the selectivity towards gases, including NO2 and CO.
1 Introduction
Metal oxides (MOx) are widely used in environmental reconstruction and destruction of organic pollutants due to their easy accessibility, low cost and non-toxicity and excellent photocatalytic properties.1–4 In the photocatalytic processes, the absorbed photons in MOxs form electron–hole pairs, which can react with molecules on the surface of particles and cause their destruction or degradation. Despite the high ability of MOxs to perform photocatalytic processes, some limitations prevent their widespread use in these processes. Their relatively low quantum efficiency due to the quick recombination of electron–hole pairs is among the limitations of most MOxs, especially titanium dioxide (TiO2) and is a limiting factor in photocatalytic processes.5 The photocatalytic activity of MOxs is improved by some modifications such as coupling with other elements, and surface coating with noble metals and other semiconductors.6,7Conjugation of the MOxs with three-dimensional π systems effectively enhances the electron transfer process between them.8 Fullerenes C60, with spherical closed-shell configuration consisting of 30 bonding molecular orbitals with 60 π-electrons,9 is suitable for conjugation with the MOxs that improves the electron transfer process and leads to the reduction of C60 and minimization of the structural changes.10,11 One of the remarkable properties of the fullerenes (with a high electron-acceptor capacity) in the electron transfer processes is their ability to the quick separation of photoinduced charges, and consequently slows down the electron–hole pair recombination.12 Therefore, conjugation of the MOxs and fullerene C60 provides an ideal system for more efficient electron transfer and charge separation.
MOxs, especially ZnO, have been highly regarded in the field of gas monitoring due to the high mobility of conduction electrons and their chemical and thermal stability. Detecting properties for various gases including toxic and pollutant gases (such as CO, NOx, H2S and SO2) by MOx nanoparticles have been reported in many studies.13–15 However, some factors like photo corrosion processes16,17 and the high HOMO–LUMO energy gap18,19 for the MOxs decrease their activity and stability, and limit their application in the gas monitoring. In addition, the operating temperature of these detectors is high, that leads to the high energy consumption and security problems related to the presence of explosive gases in the environment.20
Interactions between the MOxs and fullerenes can change the bond gaps of the reactants, and thereby change conductivity of the total system. The study on optical conductivity of the complex of fullerene C60 and TiO2 shows that its conductivity has been significantly increased in presence of radiation.21 Interaction between the zinc oxide particles and fullerene C60 enhances stability of the ZnO nanostructure in addition to increase efficient photocatalytic activities.22 Moreover, fullerene has a high surface area that can provide more situations for more effective processes. It has been also shown that interaction between the TiO2 nanoparticles and fullerene C60 reduces toxicity of the C60 in aquatic environments.23
Therefore, studying and understanding the interactions between the MOxs and fullerenes is particularly important. The researches in scientific sources express that such interactions have been only investigated for the TiO2 and ZnO crystalline nanoparticles.21–23 Nonetheless, investigating of the interactions between the MOxs and fullerenes at the molecular levels, understanding behaviour of the orbitals, gaining the energies and other adsorption parameters are only possible through the theoretical calculations and molecular quantum studies, which, based on our literature survey, has not been performed, yet.
In this study, molecular interactions between the ZnO (n-type semiconductor), NiO and Cu2O (p-type semiconductors) MOxs with the fullerene C60 (n-type) have been theoretically studied. Adsorption energies, intermolecular distances in complex structures of the fullerene-MOx (MOx/C60), energy of HOMO–LUMO surfaces and their gap, NBO charges analyses, enthalpy changes, polarizabilities and some reactivity indices in these interactions have been analysed and compared with each other.
Since the adsorbed MOxs on the fullerene C60 almost change the conductivity, dipole moment and the other electronic properties of the C60, it is expected that the MOx/C60 complexes will behave different from the involved molecules (C60 and MOxs) in variety of the reactions including interactions with the gases.
On the other hand, the studies on adsorption and storage of the gas molecules by the fullerene C60 show that the gases such as N2, Ar and CO2 locate in the inter-cavity spaces of this spherical structure. Nevertheless, they are weakly adsorbed on the surface of C60.24 This, besides the weak adsorption of gases like CO, NO, CO2 and N2O on the fullerene surface, indicates that the unmodified surface of the C60 is not suitable for adsorption or storage of these gases.25,26
One of the modifications that improves the gas detection ability of the carbon structure substrates is their combination with single metal atom catalysts.27 The carbon substrates doped with the single atom catalysts based on transition metal atoms can effectively alter the surface's local electrical neutrality and increase effective reaction sites.27–32 For example, Luo et al. showed that fullerenes doped with MnN4 catalyst efficiently adsorb toxic gases. Nevertheless, this substrate (MnN4-C60) is incapable in oxidizing gases.27 Oxidation of a toxic gas ia a significant advantage for its sensing. MOx/C60 adsorbents are predicted to provide oxidation potential of toxic gases due to the presence of oxygen in their structure, in addition to their more efficient gas adsorption. Another advantage is more availability of the substrates and less difficulty of the synthesis. MnN4-C60 substrates (MnN4 doping for C atoms of C60) require more time and knowledge for its synthesis, compared to that of the MOx/C60 exohedral fullerene adsorbents.
Based on our literature survey, there is no experimental or theoretical investigation on gas adsorption on MOx/C60 complexes. Studying adsorption of the NO2 (electrophile) and CO (nucleophile) gases on the MOx/C60, is another purpose of this research and could be useful in gaining the detectors with higher sensitivity and capability. NO2 is one of the pollutants of which million tons is produced every year, and its most important sources are internal combustion engines, thermal power plants and to some extent pulp factories. Inhalation of the NO2 gas is toxic, and long-term exposure to that has adverse effects on health.33,34 CO is another common gas pollutant, which is produced during the combustion processes, and has adverse effects on human health and environment. Although many efforts have been made to reduce these gas pollutants,35,36 more effective methods are still needed to eliminate them.
2 Computational methods
In the weaker interactions, such as interaction of the gases on the fullerene surfaces, dispersion forces or van der Waals forces play a major role.37 One of the DFT based methods that includes dispersion forces in computations is B97D (or Grimm method involving dispersion).38 In this method, the generalized gradient approximation (GGA) is used to calculate the exchange-correlation energy and can comprise electronic long-range correlations (which are responsible for the van der Waals forces) in the calculations.38The geometries of all free structures, including the fullerene C60, MOxs, and NO2 and CO gases, in addition to the complexes of their interactions have been fully optimized at the B97D/6-311G(d,p) theoretical level. Harmonic vibration frequency calculations have been also performed following the geometrical optimizations to confirm the global minimums, at the same method and basis set. Interaction energies have been calculated for all complex systems and these energies have been corrected for the basis set superposition error (BSSE).39,40 All optimizations and energy calculations in this study have been carried out using Gaussian W09 package.41
Analyses of natural bond orbitals (NBOs) have been done using GENNBO 5.0 software42 to investigate the natural atomic charges, charge transfers between donor–acceptor bond orbitals, and the interaction energies between them. Gauss View 5.0 software43 has been also used to visualize and investigate geometries and structural properties.
Chemical nature of new formed bonds in the studied complexes and their relative strength have been analysed using Atoms in Molecules (AIM) theory of Bader.44,45 Electron density (ρ(rc)) at the critical points of the bonds and its Laplacian (∇2ρ(rc)) for the global minimum structures have been estimated using AIM2000 software.44 The magnitude and sign of the electron density's Laplacian by specifying condensation or expulsion zones of charges is the basis for categorizing the nature of interactions.45 Laplacian values of ρ at the rc for atomic interactions in molecules at the equilibrium geometries have a negative sign for the systems with covalent interactions; and have relatively small values with a positive sign for weak closed-shell interactions such as van der Waals interactions.45
3 Results and discussion
3.1 Gases adsorption on fullerene C60
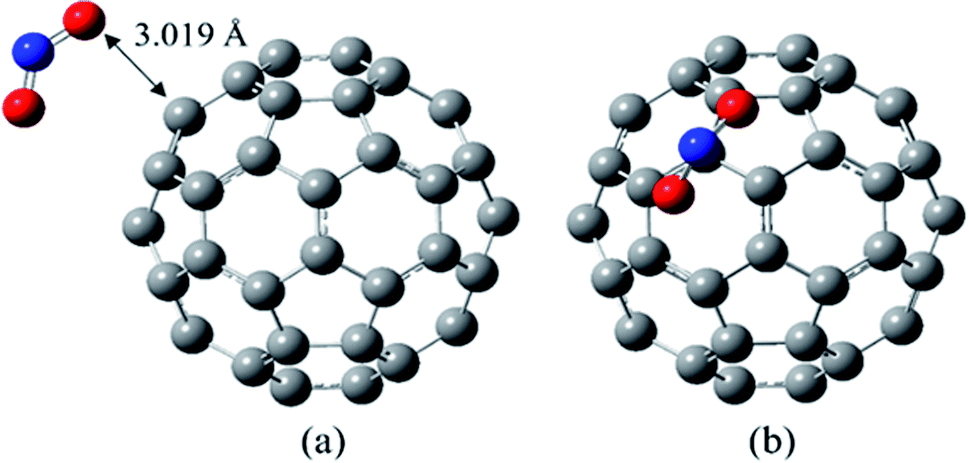 | ||
| Fig. 1 The most stable complex of NO2/C60 in two different views, at B97D/6-311G(d,p). | ||
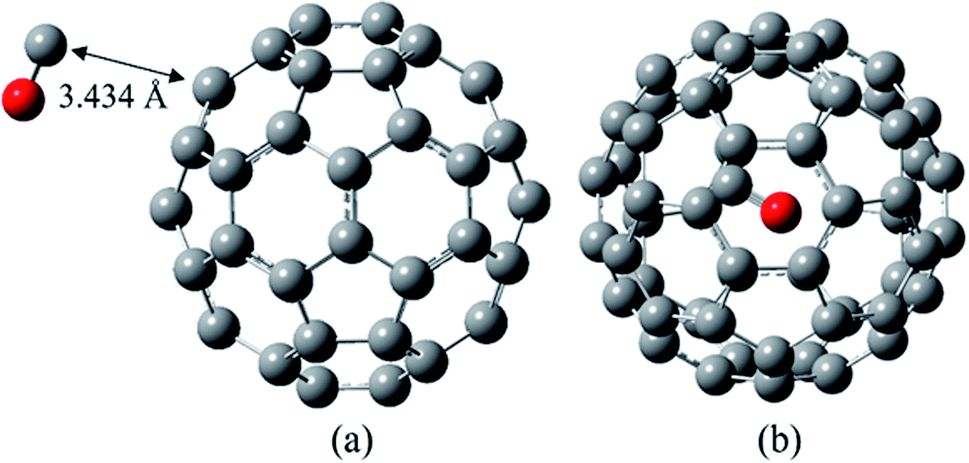 | ||
| Fig. 2 The most stable complex of CO/C60 in two different views, at B97D/6-311G(d,p). | ||
Calculated values of bond lengths and Wiberg bond orders for N–O and C–O bonds of the most stable complexes of NO2/C60 and CO/C60, together with their corresponding values for the NO2 and CO gas molecules have been listed in Table 1. Absolute energies (Eabs.), also their corrected values for zero point energy (ZPE), of NO2, CO and their most stable complexes with C60 have been given in ESI as Table S1.†
| Structure | Bond | Bond length | Bond order |
|---|---|---|---|
| NO2/C60 | N–O | 1.205 | 1.642 |
| CO/C60 | C–O | 1.137 | 2.293 |
| NO2 | N–O | 1.203 | 1.650 |
| CO | C–O | 1.136 | 2.298 |
According to the optimization calculations, those NO2/C60 complexes are the most unstable in which the NO2 group has interacted with the C60 from the nitrogen atom side. In the most stable complex of NO2/C60, the NO2 molecule has interacted with the C60 by its oxygen atoms. As shown in Fig. 1, in this structure, one of the oxygens has placed on a pentagonal ring and the other has located on a hexagonal ring of the C60.
The value of 3.019 Å has been obtained for the shortest distance between the oxygen of the NO2 and carbon atom of the C60, with the Wiberg bond order of 0.010.
In the optimized NO2 molecule, the N–O bond length and the ONO bond angle are 1.203 Å and 133.9°, respectively, which are very close to the experimental values (N–O bond length in the NO2 molecule is 1.188 ± 0.004 Å, and the bond angle is 134.100 ± 0.250°).46 The obtained N–O bond length of NO2 segment in the most stable complex of NO2/C60 is 1.205 Å, and its bond angle is 133.8°. In general, during the NO2 adsorption on the C60, the structural parameters of both molecules have not been significantly changed.
As shown in Fig. 2, in the most stable complex of CO/C60 the CO molecule has almost located on a hexagonal ring of the C60. The calculated distance of the carbon atom of the CO to the nearest carbon atom of the C60 is 3.434 Å, with the Wiberg bond order of 0.010. According to the data of Table 1, the C–O bond length in the optimized CO molecule and also in the most stable complex of CO/C60 is approximately 1.136 Å, at the B97D/6-311G(d,p) computational level. In general, it could be said that during the CO adsorption on the C60, the structural parameters of both molecules have been unchanged.
| Structure | Eads. (kcal mol−1) | Eads. + BSSE (kcal mol−1) | ΔH (kcal mol−1) | Δq (a.u.) | EHOMO (eV) | ELUMO (eV) | Gap (eV) |
|---|---|---|---|---|---|---|---|
| NO2/C60 | −3.93 | −2.03 | −2.78 | −0.011 | −5.72 | −4.08 | 1.64 |
| CO/C60 | −2.15 | −1.54 | −4.73 | 0.000 | −5.75 | −4.09 | 1.66 |
| NO2 | — | — | — | — | −6.37 | −3.45 | 2.91 |
| CO | — | — | — | — | −8.96 | −1.76 | 7.20 |
| C60 | — | — | — | — | −5.74 | −4.08 | 1.66 |
The calculated Eads. values for adsorption of the NO2 and CO molecules on the C60 are small, and even smaller with the BSSE corrections. These, beside the small values of the enthalpy changes, confirm the weakness of these interactions, in agreement with the previously discussed results of structural and AIM studies.
According to the data in Table 2, the HOMO–LUMO levels and their gap in the fullerene C60 have been not or negligibly changed after adsorption of the studied gas molecules.
All structural, AIM, energy and NBO studies indicate that the adsorptions of the NO2 and CO gas molecules on the C60 are very weak. Therefore, it is confirmed that fullerene alone cannot be a suitable adsorber or detector for the target gases.
3.2 Gases adsorption on the MOxs
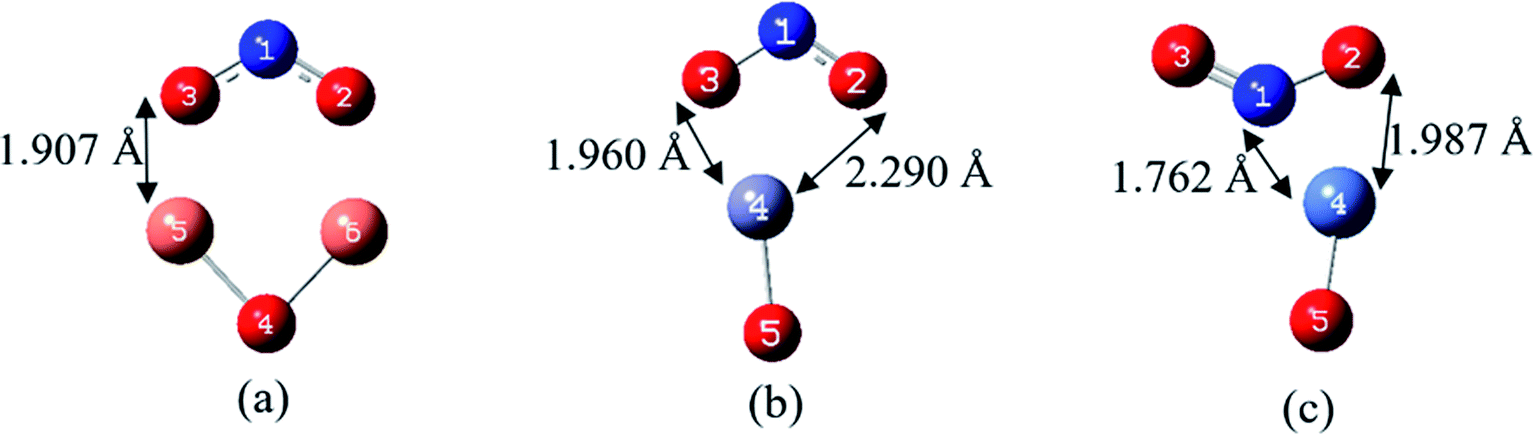 | ||
| Fig. 3 The most stable complexes and the atom numbering of (a) NO2/Cu2O, (b) NO2/ZnO and (c) NO2/NiO, at B97D/6-311G(d,p). | ||
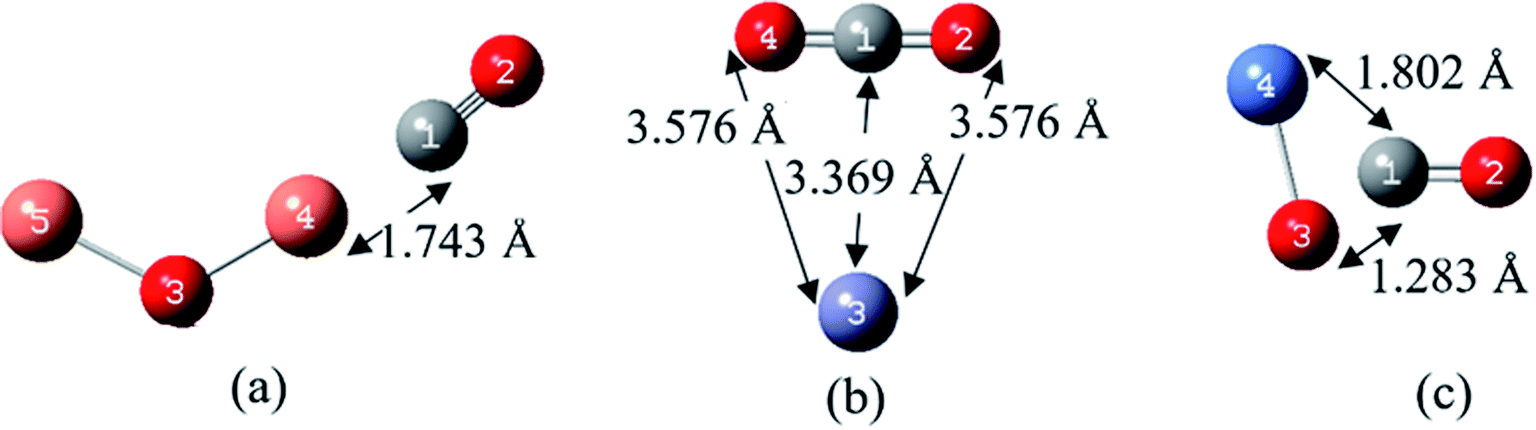 | ||
| Fig. 4 The most stable complexes and the atom numbering of (a) CO/Cu2O, (b) CO/ZnO and (c) CO/NiO, at B97D/6-311G(d,p). | ||
Also, the calculated values of the selected bond lengths and their Wiberg bond orders for the mentioned most stable complexes, together with those corresponding values of the MOx have been given in Table 3. The obtained Eabs. values with the ZPE corrections for these complexes have been given in Table S1.†
| Structure | Bond | Bond length | Bond order |
|---|---|---|---|
| NO2/Cu2O | Cu5–O4, Cu6–O4 | 1.792 | 0.515 |
| Cu5–O3 | 1.907 | 0.234 | |
| N1–O2, N1–O3 | 1.276 | 1.436 | |
| NO2/ZnO | Zn4–O5 | 1.814 | 0.485 |
| Zn4–O2 | 2.290 | 0.133 | |
| Zn4–O3 | 1.960 | 0.257 | |
| N1–O2 | 1.238 | 1.630 | |
| N1–O3 | 1.294 | 1.387 | |
| NO2/NiO | Ni4–O5 | 1.629 | 1.094 |
| Ni4–O2 | 1.987 | 0.297 | |
| Ni4–N1 | 1.762 | 0.503 | |
| N1–O2 | 1.284 | 1.479 | |
| N1–O3 | 1.193 | 1.471 | |
| CO/Cu2O | Cu4–O3 | 1.751 | 0.434 |
| Cu5–O3 | 1.758 | 0.745 | |
| Cu4–C1 | 1.743 | 0.834 | |
| C1–O2 | 1.154 | 2.118 | |
| CO/ZnO | Zn3–O2, Zn3–O4 | 3.576 | 0.002 |
| Zn3–C1 | 3.369 | 0.015 | |
| C1–O2, C1–O4 | 1.169 | 1.909 | |
| CO/NiO | Ni4–O3 | 1.820 | 0.527 |
| Ni4–C1 | 1.802 | 0.707 | |
| C1–O3 | 1.283 | 1.384 | |
| C1–O2 | 1.197 | 1.786 | |
| Cu2O | Cu–O | 1.766 | 0.795 |
| Cu–Cu | 2.957 | 0.158 | |
| ZnO | Zn–O | 1.776 | 0.992 |
| NiO | Ni–O | 1.673 | 1.030 |
According to Fig. 3(a), for the most stable structure of NO2/Cu2O complex, each of the copper atoms has located in adjacent to one of the oxygen atoms of the NO2 gas, with a distance of 1.907 Å and a bond order of 0.223. In the most stable structure of NO2/ZnO complex, Fig. 3(b), ZnO has been approached to the oxygen atoms of NO2 from the Zn side. This is in agreement with the findings of Spencer et al. in their theoretical study on adsorption of the NO2 on the ZnO nanowires.48 As shown in the Fig. 3 and Table 3, the distances of the Zn atom to the oxygen atoms are 1.960 and 2.290 Å. In the case of the most stable structure of NO2/NiO complex, as shown in Fig. 3(c), NO2 has interacted with the Ni atom more closely from its nitrogen side. Previously, Wang et al. studied adsorption of the NO2 molecule on a surface of NiO using molecular simulation and DFT methods, and understood that the strongest interaction occurs when NO2 approaches to the NiO surface from its N atom side.49
According to the data in Table 3, the bond lengths of N–O in the structure of all NO2/MOx complexes have been slightly increased in comparison to that in the NO2 free molecule. The lengths of metal–oxygen bond of MOx in both complexes of NO2/Cu2O and NO2/ZnO are slightly higher than their corresponding values in the Cu2O and ZnO molecules. These could be due to weak donor–acceptor interactions in the NO2/Cu2O and NO2/ZnO complexes. Whereas for the most stable complex of NO2/NiO, with the relatively good donor–acceptor interactions, the Ni–O bond length is shorter than that in the NiO molecule.
According to Fig. 4(a) at the most stable structure of CO/Cu2O complex, the CO has been approached to the Cu atom from its carbon atom side, with a distance of 1.743 Å and a bond order of 0.834.
In the most stable form of CO/ZnO complex (Fig. 4(b)), interestingly, the adsorption is more a chemical interaction, which the CO molecule has adsorbed the oxygen atom of the ZnO, and thereby, a CO2 molecule has been produced and Zn (with a partial charge of 0.009) released. The ZnO nanoparticles oxidize their adjacent species by creation of oxygen active species and have been always studied as a toxic MOx in biological and health issues.50,51 The rapid and strong interaction of carbon monoxide gas on the MOx substrate of ZnO and the releasing of CO2 gas has been demonstrated in various experiments.52
In the case of CO/NiO complex, the CO molecule has been approached to the NiO from its carbon side. The values of bond lengths and bond orders in this complex have been given in the Fig. 4(c) and Table 3. In this structure, the O3 of NiO is very close to the CO molecule and bonds with it (with a bond length of 1.283 Å and bond order of 1.384), but (unlike to the CO complex with ZnO) its affinity for bonding with the Ni prevents releasing of CO2.
According to the data in Table 3, the length of the CO bond in complex with the target MOxs has become slightly longer than that in the free CO molecule (which is 1.136 Å). The metal and oxygen atoms distances of MOxs in both complexes of CO/ZnO and CO/NiO are higher than their corresponding values in the ZnO and NiO molecules. However, for the complex of CO/Cu2O, the Cu–O bond length is shorter than that in the Cu2O molecule, which may be due to the notable donor–acceptor interaction from lone pair (LP) of O3 to σ* of C1–Cu4 bond (78.98 kcal mol−1).
According to the AIM calculations, in the most stable structure of CO/Cu2O complex, the value of ∇2ρ at the critical point between Cu4 and carbon atom is 0.6340 (ρ = 0.1583 a.u.), which indicates a significant van der Waals type interaction between the two molecules. In the case of CO/ZnO, the ∇2ρ value at the critical point of the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O4 bonds (about −0.0174 with ρ = 0.4461 a.u.) can be considered as a confirmation of the covalent type interaction here. For the CO/NiO complex, the calculated ∇2ρ values at the critical points between the Ni–C and O3–C bonds are respectively 0.2945 (ρ = 0.1560 a.u.) and −0.3894 (ρ = 0.3466 a.u.). The negative value of ∇2ρ (with the acceptable value of ρ) indicates a covalent interaction between the CO molecule and the oxygen of NiO.
O4 bonds (about −0.0174 with ρ = 0.4461 a.u.) can be considered as a confirmation of the covalent type interaction here. For the CO/NiO complex, the calculated ∇2ρ values at the critical points between the Ni–C and O3–C bonds are respectively 0.2945 (ρ = 0.1560 a.u.) and −0.3894 (ρ = 0.3466 a.u.). The negative value of ∇2ρ (with the acceptable value of ρ) indicates a covalent interaction between the CO molecule and the oxygen of NiO.
| Structure | Eads. (kcal mol−1) | Eads. + BSSE (kcal mol−1) | ΔH (kcal mol−1) | Δq (a.u.) | EHOMO (eV) | ELUMO (eV) | Gap (eV) |
|---|---|---|---|---|---|---|---|
| NO2/Cu2O | −67.14 | −41.50 | −66.39 | −0.717 | −4.72 | −3.07 | 1.64 |
| NO2/ZnO | −69.13 | −59.37 | −68.37 | −0.655 | −6.71 | −3.43 | 3.28 |
| NO2/NiO | −134.70 | −69.98 | −133.42 | −0.354 | −6.25 | −4.83 | 1.42 |
| CO/Cu2O | −55.60 | −39.02 | −54.10 | −0.256 | −3.64 | −2.20 | 1.43 |
| CO/ZnO | −130.43 | −125.03 | −127.73 | 0.468 | −5.59 | −0.21 | 5.39 |
| CO/NiO | −134.44 | −128.03 | −132.60 | 0.089 | −4.19 | −3.00 | 1.20 |
| Cu2O | — | — | — | — | −3.23 | −2.28 | 0.95 |
| ZnO | — | — | — | — | −3.69 | −3.60 | 0.09 |
| NiO | — | — | — | — | −1.25 | −0.59 | 0.66 |
In general, the calculated energies for adsorption of the NO2 and CO on the studied MOxs, in comparison with their adsorption on the C60 (Table 2), show much larger values. In addition, the high values of enthalpy changes for adsorption of these gas molecules on the MOxs, compared to those values in the C60, are another confirmation of strength of these interactions. Enthalpy changes with a negative sign also indicate that these adsorption processes are exothermic. Eads. data in the Table 4 show that NiO is a much stronger adsorbent for the NO2 and CO than the two other MOxs.
According to the data in Table 4, the energy values of HOMO and LUMO levels and the gaps between them in the complexes of NO2/MOx and CO/MOx have been significantly changed in comparison with those in the corresponding MOxs.
According to the NBO calculation, the most notable donor–acceptor interactions in the complex of NO2/NiO are from LP of Ni4 to σ* of N1–O3 (7.17 kcal mol−1), and from LP of O5 to σ* of N1–Ni4 (12.22 kcal mol−1). In the case of the other two complexes (NO2/Cu2O and NO2/ZnO) no significant interaction is observed in the NBO calculations.
In the complex of CO/Cu2O, 0.256 a.u. of charge has been transferred from Cu2O to the CO unit (according to the NBO charges calculations), and the total charge of CO molecule has become negative after adsorption on the Cu2O. The ZnO molecule has donated its oxygen atom to the CO during the interaction with it, and a neutral CO2 molecule has been produced. The calculated initial charge of CO unit in the produced CO2 molecule is 0.468 a.u., which indicates oxidation of CO against ZnO. In the complex of CO/NiO, the CO molecule has become similar to a CO2 molecule by forming a new bond with the oxygen of NiO, but the tendency of this oxygen to keep a bond with the Ni has prevented releasing of CO2. Therefore, the oxidation process here is weaker than that in the CO/ZnO and is associated with lower charge transfer. In general, the charge transfer between the MOxs and CO is much more significant compared to the amount of charge (near to zero) that C60 transfers to this gas molecule during their interaction.
As the NBO calculations show the most significant donor–acceptor interactions in the CO/Cu2O complex is from LP of O3 to σ* of C1–Cu4 (78.98 kcal mol−1). The strongest donor–acceptor interactions in the most stable complex of CO/ZnO are from LP of O2 to π* of C1–O4 (102.87 kcal mol−1) and to σ* of C1–O4 (103.27 kcal mol−1). In the case of CO/NiO complex, the strongest donor–acceptor interactions are from LP of O2 to σ* of C–Ni (49.77 kcal mol−1) and to σ* of C–O3 (12.81 kcal mol−1), and from LP of O3 to σ* of C–O2 (62.68 kcal mol−1).
The changes in the HOMO–LUMO levels energies of metal oxides before and after adsorption of gas molecules, along with the high values of Eads., notable charge transfers during interactions, and significant changes in the enthalpy values indicate the strong interactions of the NO2 and CO molecules with the studied MOxs. All calculations have shown that both studied gas molecules have stronger adsorption on NiO, compared to the other two MOxs.
3.3 Gases adsorption on MOx/C60
Structural study. The target Cu2O, ZnO and NiO metal oxides have been placed in different sites of the fullerene C60 (on pentagonal and hexagonal rings, on single bonds (between pentagonal and hexagonal rings) or double bonds (between two hexagonal rings)). The most stable structure for each complex of the MOx/C60 (shown in Fig. 5) have been identified by calculating the Eads. Harmonic vibrational frequency calculations following all the optimization calculations show that all frequencies have positive values. The calculated values of the bond lengths and Wiberg bond orders for the new formed bonds of the most stable MOx/C60 complexes, along with those values for the initial C60 have been listed in Table 5. The obtained Eabs. values for these complexes have been also given in Table S1.†
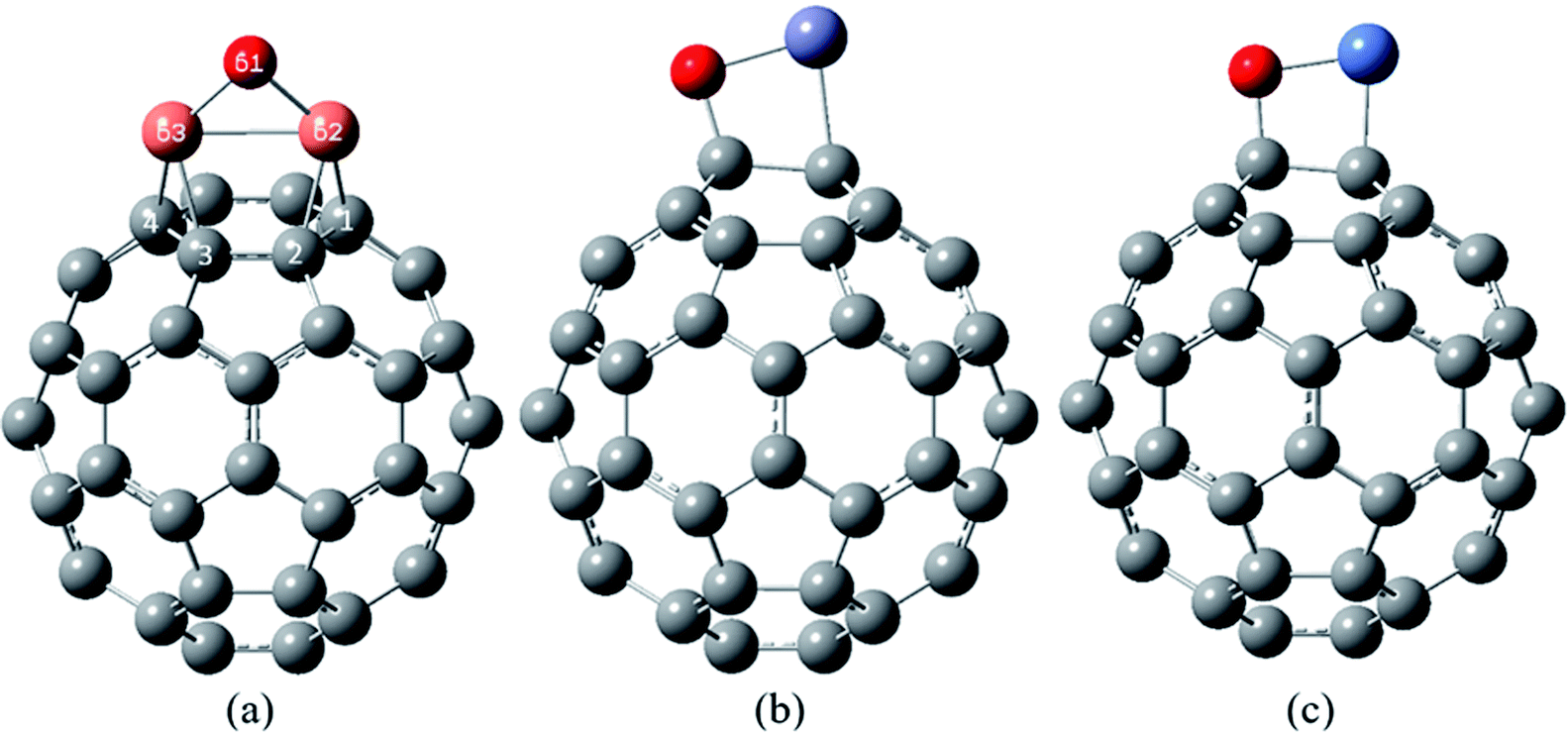 | ||
| Fig. 5 The most stable complexes of (a) Cu2O/C60, (b) ZnO/C60 and (c) NiO/C60, at B97D/6-311G(d,p). | ||
| Structure | Bond | Bond length | Bond order |
|---|---|---|---|
| Cu2O/C60 | Cu–O | 1.817 | 0.493 |
| Cu–Cu | 2.443 | 0.037 | |
| Cu–C | 1.970 | 0.295 | |
| C–C | 1.489 | 1.082 | |
| CC |
1.456 | 1.194 | |
| ZnO/C60 | Zn–O | 1.944 | 0.282 |
| Zn–C | 2.122 | 0.424 | |
| O–C | 1.408 | 0.994 | |
| C–C | 1.612 | 0.947 | |
| NiO/C60 | Ni–O | 1.762 | 0.664 |
| Ni–C | 1.847 | 0.621 | |
| O–C | 1.435 | 0.925 | |
| C–C | 1.563 | 0.939 | |
| C60 | C–C | 1.454 | 1.162 |
| CC |
1.401 | 1.368 |
The most stable structure of the Cu2O/C60 complex has been shown in Fig. 5(a). In this complex the Cu2O molecule has located on a hexagonal ring of the C60, and each of the Cu atoms has formed a quasi-trigonal ring with the atoms of the double bonds of the hexagonal ring (the bonds between hexagonal rings). This is consistent with the reports on exohedral cuprofullerenes.53–55 The adsorption of Cu3 units on the C60 fullerene demonstrates that each of the Cu atoms bridges on a double bond of the C60 and forms a trigonal. This trend continues until the increase of eight Cu3 units and synthesis of C60@Cu24.53,54 Also, according to the calculations, in the complex of Cu2O/C60, the Cu–O bond length is 1.817 Å, which is higher than that in a Cu2O free molecule (1.766 Å).
In the most stable complex of ZnO/C60, Fig. 5(b), the ZnO molecule has been placed exactly parallel to a bond between two hexagonal rings of the fullerene C60, and has formed a quasi-tetragonal ring with the atoms of this CC bond. According to the data in Table 5, the Zn–O bond in this complex has a calculated length of 1.944 Å, which is much longer than that in the ZnO free molecule (1.776 Å). The calculated length of new formed bonds of Zn–C and O–C in this complex are 2.122 and 1.408 Å, respectively.
Fig. 5(c) shows the most stable structure of NiO/C60 complex, in which NiO (like ZnO) has posited in parallel with a CC bond between two hexagonal rings of the C60. According to the data in Tables 3 and 5, the Ni–O bond length in this complex is longer than this corresponding value in the NiO molecule. The bond lengths of Ni–C and O–C in this complex are respectively 1.847 and 1.435 Å.
AIM study. The values of ρ and ∇2ρ at all bond critical points between the atoms involved in the interactions of the MOxs and the C60 have been obtained and analysed. In the most stable structure of Cu2O/C60 complex, the values of ∇2ρ and ρ at the bond critical points between the Cu atoms and the carbon atoms involved in the interaction (Fig. 5(a)) are 0.2313 and 0.0974 a.u., respectively. In the ZnO/C60 complex structure, the values of ∇2ρ and ρ at the bond critical point between the oxygen and the nearest carbon atom of the C60 are respectively −0.6537 and 0.2748 a.u. The values of ∇2ρ at the bond critical points between Ni and oxygen of the MOx and the nearest carbon atoms in the NiO/C60 complex are 0.1764 (ρ is 0.1378 a.u.) and −0.5692 (ρ is 0.2568 a.u.), respectively.
Accordingly, it can be said that the positive values of ∇2ρ at the bond critical points between the metal-carbon show van der Waals type interactions; and relatively large and negative values of ∇2ρ (with larger values of ρ) at the critical points between oxygen–carbon bonds display significant covalent type interactions in the studied complexes.
Energy study. The binding energies (Eb) and their corrected values for the BSSE, interaction enthalpy changes (ΔH), ionization potential (IP), energies of HOMO–LUMO and their gaps for the MOxs and the most stable MOx/C60 complexes have been given in Table 6.
| Structure | Eb. (kcal mol−1) | Eb. + BSSE (kcal mol−1) | ΔH (kcal mol−1) | Δq (a.u.) | IP (eV) | EHOMO (eV) | ELUMO (eV) | Gap (eV) |
|---|---|---|---|---|---|---|---|---|
| Cu2O/C60 | −89.26 | −56.07 | −89.02 | 0.903 | 6.50 | −4.84 | −4.14 | 0.70 |
| ZnO/C60 | −66.93 | −45.31 | −66.44 | 0.191 | 6.72 | −5.27 | −4.18 | 1.09 |
| NiO/C60 | −129.42 | −65.75 | −128.61 | 0.129 | 6.36 | −4.68 | −3.99 | 0.69 |
| Cu2O | — | — | — | — | 6.25 | −3.23 | −2.28 | 0.95 |
| ZnO | — | — | — | — | 7.84 | −3.69 | −3.60 | 0.09 |
| NiO | — | — | — | — | 5.43 | −1.25 | −0.59 | 0.66 |
Comparison of the ionization potential values of the MOxs before adsorption on the C60 in Table 6 shows that the first ionization energy for the NiO is less than that for the other studied MOxs, therefore the NiO more easily donates electron to the C60 and makes stronger interaction with it.
As data in the tables show, energy of the HOMO–LUMO levels in the complexes of MOx/C60 are more similar to those in the C60 (Table 2) in comparison with those in the MOxs. Since the fullerene C60 receives the electrons from the MOxs surfaces during interaction with them, its LUMO orbital surface is expected to be involved in the interaction with the MOxs. However, since the C60 molecule is several times larger than the studied MOxs, its LUMO surface have not been significantly changed in the interaction with the HOMO surface of the MOxs.
Comparison between the energies of the HOMO surfaces in the MOx/C60 complexes shows that the HOMO surface energy in the NiO/C60 complex (as the most stable complex of the studied MOx/C60 complexes) relative to the HOMO in NiO has more changed in comparison with the other two cases. On the other hand, according to the data in Table 6, comparison of the calculated values of the HOMO–LUMO gaps in the fullerene C60 (1.66 eV) and those in the MOx/C60 complexes displays that the ZnO/C60 complex (which is the most unstable MOx/C60 complex) has less difference than the other two cases.
NBO study. Analysis of the NBO charges shows that the MOxs of the Cu2O, ZnO and NiO have transferred a little charge to the fullerene C60 during interaction with it, and their total charges have become positive compared to the neutral forms before the interactions. Because NiO has a stronger interaction with the C60 than the other MOxs, more charge transfer during this interaction is expected. However, according to the data of Table 6, the charge transfer from the NiO to the C60 surface is less than that from the other two MOxs. Electron transfer process in an interaction depends extensively on the electronegativity, charge and valence electrons of the involved atoms in the interaction. The NiO and ZnO have interacted with the C60 by a metal atom and a non-metal electronegative oxygen atom, whereas Cu2O has participated in the interaction through two metal atoms.
The most significant donor–acceptor interactions in the Cu2O/C60 complex, are from LP of O61 to LP* of Cu62 and Cu63 atoms (that energies of both interactions are 132.51 kcal mol−1), also from LP of Cu62 and Cu63 atoms to π* of C1–C2 and C3–C4 bonds, respectively (both 20.12 kcal mol−1). The strongest donor–acceptor interactions in the most stable complex of ZnO/C60 are from LP of O to σ* of Zn–C and to σ* of C–C bonds, also from σ* of Zn–C bond to π* of C–C bonds (27.37, 30.93 and 16.24 kcal mol−1, respectively). In the case of NiO/C60 complex, the strongest donor–acceptor interactions are from σ* and σ of Ni–O bond to σ* of Ni–C bond, from σ of Ni–C bond to π* of C–C bonds, and from LP of O to σ* of C–C bonds (20.66, 12.36, 19.98 and 13.16 kcal mol−1, respectively).
Consequently, the high Ebs, notable charge transfers, and significant changes in the enthalpy, and the HOMO–LUMO values of the fullerene C60 after adsorbing the studied MOxs confirm their strong interactions. According to the mentioned calculations, the NiO/C60 complex is more stable than the Cu2O/C60 and ZnO/C60 complexes.
Structural study. In order to obtain the most stable structures of NO2/MOx/C60 and CO/MOx/C60 complexes, all possible structures for placing the NO2 and CO molecules relative to the MOx/C60 complexes have been considered. After optimization calculations and analysing the calculated adsorption energies for all possible structures, the given structures in Fig. 6 and 7 have been obtained as the most stable complexes.
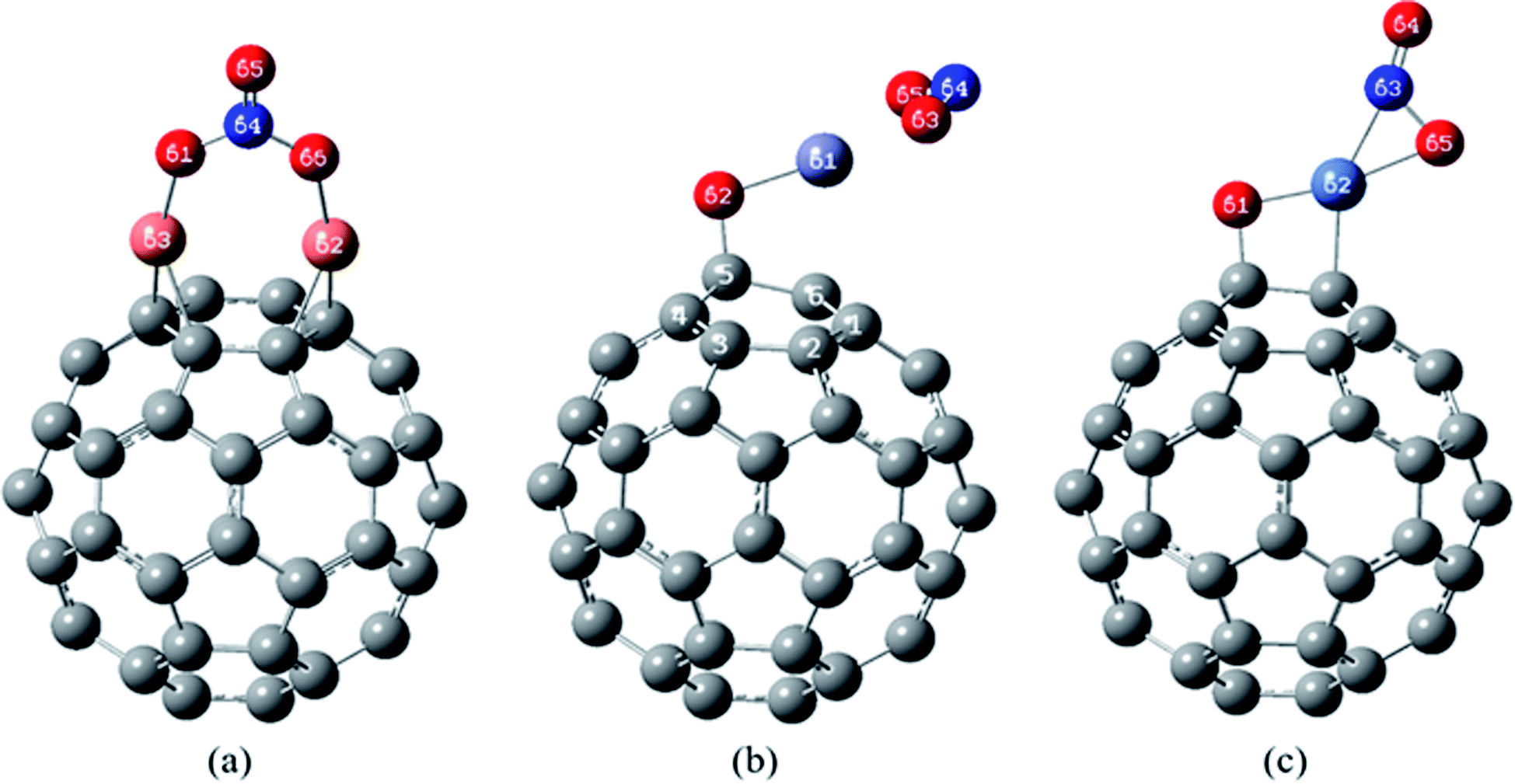 | ||
| Fig. 6 The most stable complexes of (a) NO2/Cu2O/C60, (b) NO2/ZnO/C60 and (c) NO2/NiO/C60, at B97D/6-311G(d,p). | ||
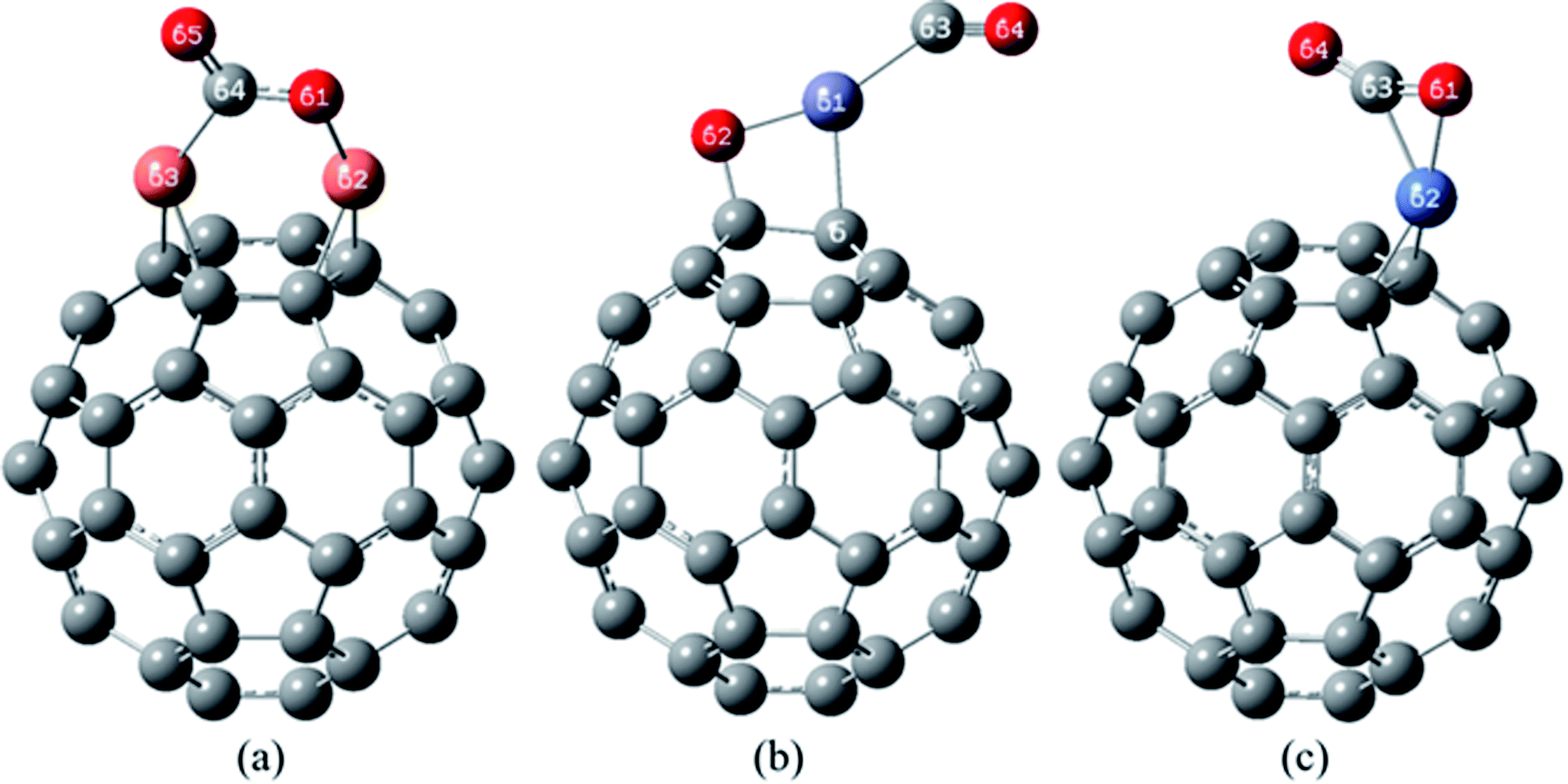 | ||
| Fig. 7 The most stable complexes of (a) CO/Cu2O/C60, (b) CO/ZnO/C60 and (c) CO/NiO/C60, at B97D/6-311G(d,p). | ||
The selected bond lengths values and their Wiberg bond orders have been given in Table 7 (and the calculated Eabs. values have been summarized in Table S1†).
| Structure | Bond | Bond length | Bond order |
|---|---|---|---|
| NO2/Cu2O/C60 | Cu63–O61 | 1.888 | 0.225 |
| N64–O61, N64–O66 | 1.302 | 1.194 | |
| N64–O65 | 1.205 | 1.586 | |
| Cu62–C, Cu63–C | 2.036 | 0.215 | |
| Cu62–C, Cu63–C | 1.973 | 0.253 | |
| NO2/ZnO/C60 | Zn61–O62 | 1.856 | 0.278 |
| Zn61–O63, Zn61–O65 | 2.096 | 0.205 | |
| N64–O63, N64–O65 | 1.264 | 1.507 | |
| O62–C | 1.421 | 0.943 | |
| Zn61–C | 2.370 | 0.080 | |
| NO2/NiO/C60 | Ni62–O61 | 1.769 | 0.537 |
| Ni62–N63 | 1.930 | 0.262 | |
| Ni62–O65 | 1.890 | 0.315 | |
| N63–O65 | 1.324 | 1.278 | |
| N63–O64 | 1.193 | 1.741 | |
| Ni62–C | 1.948 | 0.411 | |
| O61–C | 1.437 | 0.915 | |
| CO/Cu2O/C60 | Cu62–O61 | 1.864 | 0.278 |
| Cu63–C64 | 1.994 | 0.554 | |
| C64–O61 | 1.321 | 1.224 | |
| C6–O65 | 1.217 | 1.712 | |
| Cu62–C, Cu63–C | 1.976 | 0.342 | |
| Cu62–C, Cu63–C | 2.200 | 0.140 | |
| CO/ZnO/C60 | Zn61–O62 | 1.860 | 0.290 |
| Zn61–C63 | 2.009 | 0.347 | |
| C63–O64 | 1.146 | 2.246 | |
| Zn61–C | 2.087 | 0.238 | |
| O62–C | 1.430 | 0.948 | |
| CO/NiO/C60 | Ni62–O61 | 1.898 | 0.304 |
| Ni62–C63 | 1.891 | 0.521 | |
| O61–C63 | 1.253 | 1.496 | |
| C63–O64 | 1.187 | 1.846 | |
| Ni62–C | 1.929 | 0.393 |
In the most stable structure of NO2/Cu2O/C60 complex, Fig. 6(a), adsorption of the NO2 molecule causes a significant change in the initial structure of Cu2O/C60. The two Cu atoms have formed quasi-trigonal rings with the atoms of the bonds between hexagonal rings, and oxygen atom of the Cu2O (O61 atom) has interacted with the nitrogen atom of the NO2 in a way that the NO2 molecule is oxidized to NO3. This process can significantly reduce the volume of the NO2 toxic gas, which is not observed in the interaction of the NO2 on the Cu2O.
Structure (b) in Fig. 6 shows the most stable complex of NO2/ZnO/C60, in which (similar to the most stable complex of NO2/ZnO) NO2 has been approached to the Zn atom from the oxygen atoms side. The distance between the Zn atom and the nearest carbon atom of the C60 is 2.370 Å, which has become higher than the length of Zn–C bond in the most stable complex of ZnO/C60 (2.122 Å).
The most stable structure of NO2/NiO/C60 complex has been shown in Fig. 6(c). Based on data comparison in Tables 5 and 7, in this complex, NO2 adsorption has not significantly changed the initial structure of NiO/C60 and only the length of C–Ni bond has been slightly increased. In this complex NO2 interacts with the Ni atom through the nitrogen and one of the oxygen atoms. The values of the bond lengths and bond orders for the new formed bonds have been given in Table 7.
In the most stable structure of CO/Cu2O/C60 complex (Fig. 7(a)), adsorption of a CO molecule, like NO2, has significantly changed the initial structure of Cu2O/C60. In the mentioned structure, the CO molecule has approached the Cu2O from the carbon side. The two Cu atoms have formed quasi-trigonal rings with the atoms of CC bonds between hexagonal rings. One of Cu–O bond of Cu2O/C60 has been broken, and the carbon atom of the CO has placed between these Cu and O atoms, making new bonds with both of them (the characteristics of these bonds have been given in Table 7). During this desirable process, the CO gas has relatively changed into the oxidized form of CO2.
The most stable complex of CO/ZnO/C60 has been shown in Fig. 7(b). The figure, along with the data from Tables 5 and 7 demonstrate that the initial structure of the ZnO/C60 has been maintained after CO adsorption. The distances of Zn and O atoms to carbon atoms of the C60 in CO/ZnO/C60 are greatly comparable to those in the most stable complex of the ZnO/C60. In this structure, the CO molecule has interacted with the Zn atom from the carbon atom side, and the distance between Zn and carbon of the CO is 2.009 Å (with a bond order of 0.347).
Fig. 7(c) shows the most stable structure of CO/NiO/C60 complex. According to the data of Tables 5 and 7, in this structure, adsorption of the CO causes significant changes in the initial structure of NiO/C60 (Fig. 5(c)). In the mentioned structure, the NiO has interacted with the C60 only by its Ni atom in such way that has formed a quasi-trigonal ring with the carbon atoms of a CC bond between two hexagonal rings of the C60. The CO molecule has approached the NiO molecule from its carbon side and has interacted extensively with the both O and Ni atoms. In this complex, the distance between the carbon atom of CO to the oxygen atom of the NiO/C60 unit is 1.253 Å (with bond order of 1.496), which indicates the strong interaction of the CO molecule with the complex of NiO/C60 and the relatively oxidation of the CO to the CO2.
AIM study. The values of ρ and ∇2ρ at all bond critical points between the atoms involved in the interactions of the target gas molecules and the MOx/C60 have been analysed. In the NO2/Cu2O/C60 complex, according to the obtained data from the AIM calculations, the ∇2ρ value at the critical point of the bond between the N and the O61 (of the metal oxide) is exactly equal to that between the N and the O66 (of the initial NO2). The magnitude and sign of the mentioned Laplacian (−0.5760, with ρ = 0.4093 a.u.) confirm two strong covalent interactions.
In the most stable complex of NO2/ZnO/C60, the ∇2ρ value for the bond between the Zn and the oxygen of NO2, which is 0.2529 (with ρ = 0.0637 a.u.), shows a van der Waals type interaction between the NO2 and ZnO/C60 in this complex.
In the case of NO2/NiO/C60 complex, the ∇2ρ value at the bond critical point between the Ni and the N atoms is 0.4779 (ρ = 0.0966 a.u.).
In the most stable structure of CO/Cu2O/C60 complex, the value of ∇2ρ at the critical point of C64–O61 bond is −0.4466 (ρ = 0.3139 a.u.). These magnitude and sign indicate a strong covalent interaction in this gas adsorption.
In the CO/ZnO/C60 complex, the ∇2ρ and ρ values (0.2175 and 0.0884 a.u., respectively) at the bond critical point between carbon atom of the gas molecule and the Zn of the metal oxide show a van der Waals type interaction between CO and ZnO/C60.
For the CO/NiO/C60 complex, the calculated values of ∇2ρ at the critical points of Ni–C63 and O61–C63 bonds (in Fig. 7(c)) are respectively 0.2429 (ρ = 0.1189 a.u.) and −0.3324 (ρ = 0.3701 a.u.), that the negative value of ∇2ρ (with relatively large value of ρ) also confirms the strong covalent interaction between the carbon of the CO gas and the oxygen of the NiO metal oxide.
Energy study. The values of Eads. with the BSSE corrections, the enthalpy changes, energies of the HOMO–LUMO levels and their related gaps for the most stable complexes of NO2/MOx/C60 and CO/MOx/C60 have been calculated and the results have been summarized in Table 8.
| Structure | Eads. (kcal mol−1) | Eads. + BSSE (kcal mol−1) | ΔH (kcal mol−1) | Δq (a.u.) | EHOMO (eV) | ELUMO (eV) | Gap (eV) |
|---|---|---|---|---|---|---|---|
| NO2/Cu2O/C60 | −32.98 | −19.36 | −30.70 | −0.553 | −5.28 | −4.31 | 0.97 |
| NO2/ZnO/C60 | −53.36 | −45.99 | −52.08 | −0.651 | −5.34 | −4.18 | 1.16 |
| NO2/NiO/C60 | −53.68 | −62.30 | −52.58 | −0.492 | −5.59 | −4.34 | 1.25 |
| CO/Cu2O/C60 | −36.32 | −28.17 | −33.83 | −0.206 | −4.81 | −4.00 | 0.80 |
| CO/ZnO/C60 | −18.42 | −10.07 | −17.08 | −0.170 | −5.17 | −4.20 | 0.97 |
| CO/NiO/C60 | −82.27 | −71.12 | −80.01 | 0.234 | −5.57 | −4.12 | 1.45 |
The calculated energies for adsorption of the NO2 and CO gases on the studied complexes of MOx/C60 show the much bigger values, compared to the values of the adsorption energies of these gas molecules on the C60 (Table 2). The high values of enthalpy changes in these interactions are also another confirmation of the strength of these adsorption processes compared to those that occur on the C60. Moreover, these enthalpy changes with negative sign indicate that these adsorption processes are exothermic.
According to the calculated Δq in Table 8, during gas adsorptions on the MOx/C60, some charge transfer is occurred from the MOx/C60 to the gases. For this, the LUMO of the NO2 is involved in the interaction with the HOMOs of the MOx/C60 complexes. According to the data in Tables 6 and 8, after NO2 adsorption, the energy changes in the HOMO levels of the MOx/C60 complexes are more than that in their LUMO levels, which is in agreement with the charge transfer from the HOMO of the MOx/C60 complexes. According to the calculated data, in comparison to the other two MOx/C60 complexes, the HOMO level in the NiO/C60 complex is closer to the LUMO level of the NO2 molecule, which results in a stronger interaction between them. The changes in energy of the HOMO surfaces and the HOMO–LUMO gaps in MOx/C60 complexes, after NO2 adsorption, are much more significant in comparison with the corresponding changes in the C60 (Table 2).
According to data in Tables 6 and 8, in comparison to the other CO/MOx/C60 complexes, both HOMO and LUMO levels in the CO/NiO/C60 complex have become more stable during gas adsorption. The change in the HOMO–LUMO gap value before and after CO adsorption on the NiO/C60 complex is also greater than those on the other MOx/C60 complexes, which confirm the stronger adsorption of this gas molecule on the NiO/C60 complex.
NBO study. According to the NBO charge changes, given in Table 8, the total charge of the NO2 molecule has become negative after adsorption on the MOx/C60 complexes, which indicates the electron transfer from these complexes to the NO2. Because the NiO/C60 has a stronger interaction with the NO2 than the other MOx/C60 complexes, more charge transfer is expected during this adsorption process. However, according to the Table 8, the charge transfer for the interaction of NO2 with ZnO/C60 is higher than the other two cases. As mentioned, NO2 has interacted with the ZnO/C60 from the oxygen atoms side. Since oxygen has a high electronegativity, NO2 receives more electrons from the ZnO/C60 surface during adsorption on it. Comparison between the ionization potential of MOx/C60 complexes before NO2 adsorption in Table 6 shows that the IP for NiO/C60 is lower than those for the other MOx/C60 complexes, and therefore NiO/C60 more easily donates its first electron to the NO2 and interacts more strongly with it.
Based on the NBO results, donor–acceptor interactions are in agreement to the charge transfers during these processes, and confirm the changes of atomic charges. The most notable donor–acceptor interactions in the complex of NO2/Cu2O/C60 are from LP of O61 and O66 atoms to σ* of N64–O65 bond (both 28.76 kcal mol−1). In the case of NO2/ZnO/C60 complex the most significant interactions is from LP of O62 atom to LP* of Zn61 atom (34.00 kcal mol−1). These interactions in the NO2/NiO/C60 are from LP of O61 atom to LP* of Ni62, from σ* of C–Ni62 to LP* of Ni62 atom, and from LP of O65 to σ* of N63–O64 (36.66, 22.03 and 26.27 kcal mol−1, respectively).
In the complexes of CO/Cu2O/C60 and CO/ZnO/C60, the electrons has been transferred to the CO gas throughout the bond formed between its C atom and the metal atom of the MOx, and (according to the NBO charge calculations) the total charge of the CO molecule has become negative after adsorption on the Cu2O/C60 and ZnO/C60 complexes. In the complex of CO/NiO/C60, the CO molecule has reasonably changed into a CO2 molecule by creating a strong covalent bond with the oxygen (non-metal) atom of the NiO, but tendency of the mentioned oxygen to keep a bond with the Ni prevents releasing of that as the free CO2.
According to the NBO calculations, donor–acceptor interactions also confirm the charge transfer processes. Interactions from LP of O61 atom to π* and to σ* of C64–O65 bond (14.11 and 26.01 kcal mol−1, respectively) in the CO/Cu2O/C60, and from σ* of C6–Zn61 to LP* of C63 atom (47.64 kcal mol−1) in the CO/ZnO/C60 are some examples. In the complex of CO/NiO/C60, the most notable interactions are from LP of O64 atom to σ* of Ni62–C63 bond, from σ* of Ni62–C63 to LP* of Ni62 atom (97.71 and 60.38 kcal mol−1, respectively), and from LP of O61 atom to σ* of C63–O64 and Ni62–C63 bonds (71.57 and 67.52 kcal mol−1, respectively).
Comparing the data in Tables 2 and 8 clearly demonstrate that amount of charge transfer during gas adsorption for the MOx/C60 complexes is higher than that for the C60.
Consequently, the high Eads., significant charge transfers during interactions, and notable changes in the enthalpy values, energies of HOMO–LUMO levels and their gaps for the MOx/C60 complexes after adsorption of the NO2 and CO gases indicate strong interactions between these gas molecules and the MOx/C60 complexes compared to the weak interactions between them and the fullerene C60.
3.4 Investigation of some reactivity indices in the studied adsorbents
The values of the HOMO–LUMO gap for the fullerene C60 (ref. 56) and the Cu2O,57 ZnO58 and NiO59,60 have been measured during various experiments. These experimental values along with their calculated values in the present study have been given in Table 9.| Structure | αcalc. (3Å) | αexp. (3Å)a | Gapcalc. (eV) | Gapexp. (eV) |
|---|---|---|---|---|
| a By empirical findings and Lorentz–Lorenz relation. | ||||
| C60 | 75.79 | 76.50 ± 0.80 (ref. 63) | 1.66 | 1.80–1.60 (ref. 56) |
| Cu2O | 6.68 | 6.54 (ref. 65) | 0.95 | 2.28 (ref. 57) |
| ZnO | 2.31 | 2.61 (ref. 64) | 0.09 | 3.37 (ref. 58) |
| NiO | 1.35 | 2.20 (ref. 64) | 0.66 | 4.00 (ref. 60) |
| Cu2O/C60 | 87.92 | — | 0.70 | — |
| ZnO/C60 | 87.35 | — | 1.09 | — |
| NiO/C60 | 83.59 | — | 0.69 | — |
According to the data in this table, the calculated value of HOMO–LUMO gap for the C60 at the theoretical level of B97D/6-311G (d,p) is in good agreement with the experimental value.56 On the other hand, in the case of the studied MOxs, their calculated gap values are much lower than their experimental values. Despite the remarkable successes of DFT theory in the framework of LDA and GGA approximations, these methods are incapable for prediction some parameters related to the strongly correlated systems.61,62 The reason is misbehavior of the exchange interactions in these approximations, which do not sufficiently eliminate the electron self-interaction errors, and therefore estimate the energy values of the unoccupied surfaces (LUMO) less than the actual values.61 Because the intermediate MOxs are the most prominent examples for the strongly correlated systems, the calculated values of their HOMO–LUMO gap using DFT methods are much smaller than the actual values and are not reliable.61,62
Since the calculated values of the HOMO–LUMO gap for the MOx molecules are not enough accurate, the study of parameters such as chemical hardness or chemical potential, that are directly related to the HOMO–LUMO gap, are not suitable for checking the stability or reactivity of the studied adsorbent species. The polarizability parameter is one of the reliable indices in computational methods and also determines the chemical hardness of considered systems. The more realistic values of polarizability using empirical findings and Lorentz–Lorenz equation for the C60 (ref. 63) and all three MOx molecules (NiO,64 ZnO64 and Cu2O65) have been given in Table 9. These values are reasonably consistent with the calculated values here at the studied theoretical level.
According to the obtained data for the polarizability, it can be clearly seen that the adsorbents containing the C60 are much softer than the MOx adsorbents, and it is predicted that they exhibit higher conductivity and reactivity. In addition, organic compounds (such as fullerenes) are more readily modified than inorganic materials (including MOxs) and it is expected the MOx/C60 complexes would be better detectors or filters for the gas species including CO and NO2 in comparison with the MOx nanoparticles which have very low selectivity.66,67
Interaction between organic–inorganic interfaces alternates electronic states, bond gap and magnetic moment of the substrates. For example, the presence of fullerene in CdSe-C60 nanocomposites allows energy levels and band gap of the system to be adjusted by resizing CdSe substrate.68 In the reaction between the surfaces of C60 and tungsten (W), the bond gap can be controlled by selecting the annealing temperature.69 These are just some example of the bond gap engineering. A selective substrate for adsorption of gases or vapors can be achieved by bond gap engineering in gas adsorbents combined with modified fullerene.70–74 Other example is modifying polyphenylene oxide membranes (PPO) with fullerene that exhibits a lower gas permeability and enhanced selectivity for gas separation in comparison with the PPO.70 Combining polystyrene with the fullerene C60, as a hybrid gas separation membrane, leads to a change in the crystallinity, density, and free volume in this membrane. This improve some transport properties which in turn leads to an improvement of the selectivity in the gas separation.71 Therefore, the modified MOx/C60 complexes are expected to show more potent in selectivity of gas adsorptions.
4 Conclusions
Combination of fullerene and MOx, to make composite nanostructures, are interesting not only because they display the individual properties of fullerene and MOx nanoparticles but they may also exhibit synergetic properties that are advantageous for gas sensing applications. The studied MOx molecules, during adsorption on the fullerene C60 surface, transfer some electrons to the C60. High adsorption energies, significant changes in the both enthalpy and the HOMO–LUMO values of the fullerene C60, after adsorption of the MOxs, indicate strong interactions in the resulting MOx/C60 complexes. According to calculations, the NiO is more stably absorbed on the surface of the C60 than the Cu2O and ZnO.According to the results, the MOx/C60 complexes are much stronger adsorbents than the C60 in the gas adsorption. The charge transfers to the NO2 and CO gas molecules from the MOx/C60 complexes are much more significant compared to those from C60. The calculated adsorption energies and the enthalpy changes for adsorption of the studied gas molecules on the MOx/C60 complexes are several times larger than those on the C60. These adsorptions on the NiO/C60 are stronger than on the other MOx/C60 complexes.
According to the obtained data for the polarizability, it can be clearly seen that the adsorbents containing the C60 are much softer than the MOx adsorbents, and it is predicted that they exhibit higher conductivity and reactivity. Although the MOx/C60 complexes are weaker adsorbents for the NO2 and CO gases in comparison with the MOxs, but because the organic compounds (such as fullerenes) are more readily modified than inorganic materials (including MOxs), the MOx/C60 complexes are more potent in selectivity of adsorption of different gases, such as CO and NO2, and they may require lower operating temperatures than the MOxs.
It is expected that combination of the MOxs and the C60 three-dimensional system, with the rapid recombination of electron–hole pairs, reduction photo-corrosion processes, also with increase the interaction area surface, would provide ideal gaseous adsorbents with more stability than the MOx adsorbents.
Author contributions
A-Reza Nekoei: supervision, conceptualization, methodology, resources, writing-review & editing. Sanaz Haghgoo: conceptualization, formal analysis, investigation, writing-original draft.Conflicts of interest
There are no conflicts to declare.References
- R. Aitken, M. Q. Chaudhry, A. B. A. Boxall and M. Hull, Occup. Med., 2006, 56, 300–306 CrossRef CAS PubMed.
- K. I. Hadjiivanov and D. K. Klissurski, Chem. Soc. Rev., 1996, 25, 61–69 RSC.
- W. T. Qiao, G. W. Zhou, X. T. Zhang and T. D. Li, Mater. Sci. Eng. Carbon, 2009, 29, 1498–1502 CrossRef CAS.
- D. L. Jiang, S. Q. Zhang and H. J. Zhao, Environ. Sci. Technol., 2007, 41, 303–308 CrossRef CAS PubMed.
- H. S. Yun, K. Miyazawa, I. Honma, H. S. Zhou and M. Kuwabara, Mater. Sci. Eng. Carbon, 2003, 23, 487–494 CrossRef.
- N. Todorova, T. Giannakopoulou and T. Vaimakis, Mater. Sci. Eng., B, 2008, 152, 50–54 CrossRef CAS.
- G. P Lepore, C. H. Langford, J. Vichova and A. Vlcek, J. Photochem. Photobiol., A, 1993, 75, 67–75 CrossRef.
- S. Fukuzumi, K. M. Kadish, K. Smith and R. Guilard, in The Porphyrin Handbook: Inorganic, Organometallic and Coordination Chemistry, Academic Press, San Diego, 2000 Search PubMed.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593–601 CrossRef CAS.
- S. Fukuzumi and D. M. Guldi, in Electron Transfer in Chemistry: Electron-Transfer Chemistry of Fullerenes, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, p. 270 Search PubMed.
- S. Fukuzumi and H. Imahori, in Electron Transfer in Chemistry: Electron-Transfer Chemistry of Fullerenes, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, p. 927 Search PubMed.
- M. Melle-Franco, M. Marcaccio, D. Paolucci, F. Paolucci, V. Georgakilas and D. Guldi, J. Am. Chem. Soc., 2004, 126, 1646–1647 CrossRef CAS PubMed.
- T. G. Nenov and S. P. Yordanov, Ceramic Sensors-Technology and Applications, Technomic Publishing, Lancaster, PA, 1996 Search PubMed.
- H. Meixner and U. Lampe, Sens. Actuators, B, 1996, 33, 198–202 CrossRef CAS.
- P. T. Moseley, Meas. Sci. Technol., 1997, 8, 223–237 CrossRef CAS.
- B. Cao and W. J. Cai, J. Phys. Chem. C, 2008, 112, 680–685 CrossRef CAS.
- Q. Li, V. Kumar, Y. Li, H. Zhang, T. J. Marks and R. P. H. Chang, Chem. Mater., 2005, 17, 1001–1006 CrossRef CAS.
- S. Zhang, H. S. Chen, K. Matras-Postolek and P. Yang, Phys. Chem. Chem. Phys., 2015, 17, 30300–30306 RSC.
- M. H. Huang, S. Mao, H. Feick, H. Yan, Y. Wu, H. Kind, E. Weber, R. Russo and P. Yang, science, 2001, 292, 1897–1899 CrossRef CAS PubMed.
- S. G. Chatterjee, S. Chatterjee, A. K. Ray and A. K. Chakraborty, Sens. Actuators, B, 2015, 221, 1170–1181 CrossRef.
- Z. D. Meng, L. Zhu, J. G. Choi, M. L. Chen and W. C. Oh, J. Mater. Chem., 2011, 21, 7596–7603 RSC.
- H. Fu, T. Xu, S. Zhu and Y. Zhu, Environ. Sci. Technol., 2008, 42, 8064–8069 CrossRef CAS PubMed.
- S. B. Lovern and R. Klaper, Environ. Toxicol. Chem., 2006, 25, 1132–1137 CrossRef CAS PubMed.
- A. Martínez-Alonso, J. M. D. Tascón and E. J. Bottani, J. Phys. Chem. B, 2001, 105, 135–139 CrossRef.
- A. M. El Mahdy, Appl. Surf. Sci., 2016, 383, 353–366 CrossRef CAS.
- B. Gao, J. X. Zhao, Q. H. Cai, X. G. Wang and X. Z. Wang, J. Phys. Chem. A, 2011, 115, 9969–9976 CrossRef CAS PubMed.
- M. Luo, Z. Liang, S. G. Peera, M. Chen, C. Liu, H. Yang, J. Liu, U. P. Kumar and T. Liang, Appl. Surf. Sci., 2020, 525, 146480 CrossRef CAS.
- M. Luo, Z. Liang, M. Chen, C. Liu, X. Qi, S. G. Peera, J. Liu and T. Liang, New J. Chem., 2020, 44, 15724–15732 RSC.
- M. Luo, Z. Liang, C. Liu, X. Qi, M. Chen, H. Yang and T. Liang, RSC Adv., 2020, 10, 27856–27863 RSC.
- M. Luo, Z. Liang, C. Liu, M. Liu, X. Qi, M. Chen, H. Yang and T. Liang, ACS Omega, 2020, 5, 21203–21210 CrossRef CAS PubMed.
- M. Luo, Z. Liang, M. Chen, S. G. Peera, C. Liu, H. Yang, X. Qi, J. Liu and T. Liang, New J. Chem., 2020, 44, 9402–9410 RSC.
- M. Luo, Z. Liang, C. Liu, X. Qi, M. Chen, R. U. R. Sagar, H. Yang and T. Liang, Appl. Surf. Sci., 2021, 536, 147809 CrossRef CAS.
- B. Son, W. Yang, P. Breysse, T. Chung and Y. Lee, Environ. Res., 2004, 94, 291–296 CrossRef CAS PubMed.
- Report on a WHO Working Group, Health Aspects of Air Pollution with Particulate Matter, Ozone and Nitrogen Dioxide, Bonn, Germany, 2003, vol. 13–15 Search PubMed.
- Y. Xie, Y. P. Huo and J. M. Zhang, Appl. Surf. Sci., 2012, 258, 6391–6397 CrossRef CAS.
- M. Shelef, Chem. Rev., 1995, 95, 209–225 CrossRef CAS.
- S. L. Schroeder and M. Gottfried, Adv. Phys. Chem. Lab., 2002, 1–22 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- F. B. Van Duijneveldt, J. G. van Duijneveldt-van de Rijdt and J. H. van Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS.
- D. W. Schwenke and D. G. Truhlar, J. Chem. Phys., 1985, 82, 2418–2426 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers,K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts,R. E. Stratmann,O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Wallingford CT, 2013 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison WI, 2001 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView Version 5, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- F. W. Biegler Konig, J. Schonbohm and D. Bayles, AIM2000, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- R. F. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- L. E. Sutton, J. Chem. Soc., 1965, 1956–1959 Search PubMed.
- C. B. Leffert, W. M. Jackson and E. W. Rothe, J. Chem. Phys., 1973, 58, 5801–5806 CrossRef CAS.
- M. Spencer, I. Yarovsky, W. Wlodarski and K. Kalantar-Zadeh, in Solid-State Sensors, Actuators and Microsystems Conference, 2007, pp. 987–990 Search PubMed.
- B. Wang, J. Nisar and R. Ahuja, ACS Appl. Mater. Interfaces, 2012, 4, 5691–5697 CrossRef CAS PubMed.
- X. Zhao, X. Ren, R. Zhu, Z. Luo and B. Ren, Aquat. Toxicol., 2016, 180, 56–70 CrossRef CAS PubMed.
- C. T. Ng, L. Q. Yong, M. P. Hande, C. N. Ong, L. E. Yu, B. H. Bay and G. H. Baeg, Int. J. Nanomed., 2017, 12, 1621–1637 CrossRef CAS PubMed.
- J. C. Lavalley, J. Saussey and T. Rais, J. Mol. Catal., 1982, 17, 289–298 CrossRef CAS.
- S. Z. Zhan, G. H. Zhang, J. H. Li, J. L. Liu, S. H. Zhu, W. Lu, J. Zheng, S. W. Ng and D. Li, J. Am. Chem. Soc., 2020, 142, 5943–5947 CrossRef CAS PubMed.
- D. Chu, Y. Liu, Y. Li, Y. Liu and Y. Cui, Inorg. Chem. Front., 2020, 7, 2556–2559 RSC.
- S. Z. Zhan, J. H. Li, Y. Li, G. Xu, D. F. Luo, L. Dang and D. Li, Sci. China Mater., 2021, 1–7, DOI:10.1007/s40843-020-1589-7.
- C. H. Andersson, Chemistry of Carbon Nanostructures: Functionalization of Carbon Nanotubes and Synthesis of Organometallic Fullerene Derivatives, Doctoral dissertation, Uppsala University, 2011 Search PubMed.
- I. Grozdanov, Mater. Lett., 1994, 19, 281–285 CrossRef CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829–R858 CrossRef CAS.
- S. Hüfner, J. Osterwalder, T. Riesterer and F. Hulliger, Solid State Commun., 1984, 52, 793–796 CrossRef.
- G. A. Sawatzky and J. W. Allen, Phys. Rev. Lett., 1984, 53, 2339–2342 CrossRef CAS.
- R. Gillen and J. Robertson, J. Phys.: Condens. Matter, 2013, 25, 165502 CrossRef PubMed.
- J. P. Perdew and A. Zunger, Phys. Rev. B, 1981, 23, 5048–5079 CrossRef CAS.
- R. Antoine, P. Dugourd, D. Rayane, E. Benichou, M. Broyer, F. Chandezon and C. Guet, J. Chem. Phys., 1999, 110, 9771–9772 CrossRef CAS.
- V. Dimitrov and S. Sakka, J. Appl. Phys., 1996, 79, 1736–1740 CrossRef CAS.
- A. C. Lasaga and R. T. Cygan, Am. Mineral., 1982, 67, 328–334 CAS.
- S. Zampolli, I. Elmi, J. Stürmann, S. Nicoletti, L. Dori and G. C. Cardinali, Sens. Actuators, B, 2005, 105, 400–406 CrossRef CAS.
- B. Bahrami, A. Khodadadi, M. Kazemeini and Y. Mortazavi, Sens. Actuators, B, 2008, 133, 352–356 CrossRef CAS.
- S. Sarkar, B. Rajbanshi and P. Sarkar, J. Appl. Phys., 2014, 116, 114303 CrossRef.
- E. Monazami, J. B. McClimon, J. Rondinelli and P. Reinke, ACS Appl. Mater. Interfaces, 2016, 8, 34854–34862 CrossRef CAS PubMed.
- G. A. Polotskaya, D. V. Andreeva and G. K. El’yashevich, Tech. Phys. Lett., 1999, 25, 555–557 CrossRef CAS.
- A. Y. Pulyalina, V. A. Rostovtseva, Z. Pientka, L. V. Vinogradova and G. A. Polotskaya, Pet. Chem., 2018, 58, 296–303 CrossRef CAS.
- V. A. Karachevtsev, A. M. Plokhotnichenko, V. A. Pashynska, A. Y. Glamazda, O. M. Vovk and A. M. Rao, Appl. Surf. Sci., 2007, 253, 3062–3065 CrossRef CAS.
- H. B. Lin and J. S. Shih, Sens. Actuators, B, 2003, 92, 243–254 CrossRef CAS.
- S. Keshtkar, A. Rashidi, M. Kooti, M. Askarieh, S. Pourhashem, E. Ghasemy and N. Izadi, Talanta, 2018, 188, 531–539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI:10.1039/d1ra02251b |
| This journal is © The Royal Society of Chemistry 2021 |