DOI:
10.1039/D1RA01738A
(Paper)
RSC Adv., 2021,
11, 13970-13979
Direct use of the solid waste from oxytetracycline fermentation broth to construct Hf-containing catalysts for Meerwein–Ponndorf–Verley reactions†
Received
4th March 2021
, Accepted 7th April 2021
First published on 13th April 2021
Abstract
The oxytetracycline fermentation broth residue (OFR) is an abundant solid waste in the fermentation industry, which is hazardous but tricky to treat. The resource utilization of the waste OFR is still challenging. In this study, a novel route of using OFR was proposed that OFR was used as the organic ligands to construct a new hafnium based catalyst (Hf-OFR) for Meerwein–Ponndorf–Verley (MPV) reactions of biomass-derived platforms. The acidic groups in OFR were used to coordinate with Hf4+, and the carbon skeleton structures in OFR were used to form the spatial network structures of the Hf-OFR catalyst. The results showed that the synthesized Hf-OFR catalyst could catalyze the MPV reduction of various carbonyl compounds under relatively mild reaction conditions, with high conversions and yields. Besides, the Hf-OFR catalyst could be recycled at least 5 times with excellent stability in activity and structures. The prepared Hf-OFR catalyst possesses the advantages of high efficiency, a simple preparation process, and low cost in ligands. The proposed strategy of constructing catalysts using OFR may provide new routes for both valuable utilization of the OFR solid waste in the fermentation industry and the construction of efficient catalysts for biomass conversion.
Introduction
With the development of the social economy, the people's demands for energy and carbon resources are also increasing year by year, but fossil resources are over-exploited, which directly or indirectly leads to the excessive depletion of fossil resources and the persistence of environmental pollution.1 To alleviate the above problems, on one hand, increasing the utilization efficiency of carbon-containing organic waste from industrial production processes, improving the valuable use of the organic waste, and finally realizing “change waste materials into things of value” is an important approach to improve the utilizing efficiency of the carbon sources.2 On the other hand, how to promote the high-value conversion of renewable biomass resources, such as converting lignin and cellulose into chemicals, fuels, materials, etc., becomes more and more important.
Antibiotics are one of the most successful drug families ever developed in the history of pharmaceutical development. They are widely used to treat human health and promote the development of agriculture and animal husbandry.3 The antibiotic fermentation broth residue (AFR) is the main by-product after the production of antibiotics, mainly includes residual media, solvent, mycelium, metabolites and leftover antibiotics due to the incomplete extraction.4 It has high content of water, volatiles, organic matters (more than 90% for the dry AFR) and so on.4 China is the world's largest producer of antibiotics, accounting for 70% of global production.5 Producing 1 ton of antibiotics will produce about 8–10 tons of AFR. Due to its vast amount of nutrients, AFR has been widely used in fertilizers and animal feed additives to promote the growth of plants in recent years, and has visible promotion effects on the growth of animals and plants.4 However, because AFR contains some antibiotic residues, this type of utilization could pollute the land, groundwater, and may cause the accumulation of antibiotics in the animal, leading to drug resistance, which will eventually harm humans.4 Therefore, China listed AFR as a national-level hazardous waste in 2008, which was banned from use in animal feed. How to deal with AFR with the awareness of environmental protection and value-added use has become a hot issue in recent years.6 Direct combustion or landfill is a routine method for pharmaceutical plants to handle such organic waste. However, the drawbacks of these methods are obvious: due to the high content of water in AFR, the burning process will consume a lot of heat, and the processing cost (about 2000 RMB per ton) will undoubtedly bring high economic pressure for the companies. Direct landfilling may cause residual antibiotics in the AFR to penetrate the groundwater, contaminate the water source, and waste the abundant organic matter in the AFR.7 In recent years, many new AFR treatment methods have been reported, such as pyrolysis of AFR to make it a bio-carbon material,6 hydrothermal treatment to convert it into the clean solid biomass fuel,8 anaerobic digestion technology to produce flammable gas9 and composting technology.4 As can be seen from the above treatment methods, AFR is a potential bio-carbonaceous resource due to the high abundance of organic matter. Although these treatment methods can effectively utilize AFR to a certain extent, they still have their shortcomings, such as requires significant energy input, organic structures/functional groups are not targeted for use, etc. Therefore, the exploration of more green and value-added approaches to utilize AFR, in particular, the use of inherent organic structures and functional groups, is still a meaningful subject during the utilization of such solid organic waste.10
In addition to exploring how to use the solid organic waste in industrial production, finding renewable carbon resources is also an essential way for humans to access clean energy and chemicals.11 Biomass resources are a renewable natural carbon resource. Through the appropriate processes, useful molecules can be obtained from biomass raw materials, such as triglycerides,12 glycerol,13 5-hydroxymethylfurfural,14 cellulose,15 hemicelluloses and pentoses,16 lignin17 and lignocellulose18 and then these intermediates can be further converted into the liquid fuels and commercial chemicals.11 The Meerwein–Ponndorf–Verley (MPV) reaction was commonly used to convert biomass derived carbonyl compounds into alcohols or their derivatives, an essential step in biomass catalytic conversion.19 The reaction process has the advantages of high chemoselectivity, mild reaction conditions, operational simplicity, low cost, and scalability. The carbon–oxygen double bonds could be selectively reduced with secondary alcohols as hydrogen sources.19 It has been reported that MPV reaction can be efficiently catalyzed by diverse catalysts, such as using transition metals RANEY®Ni,20 Cu-catalyst derived from hydrotalcite,21 aluminum based catalysts,22 nano MgO,23 and a range of Zr-containing catalysts including zirconium hydroxide,24 zirconia,25 zirconium-containing beta zeolite,26 etc. These catalysts generally have a cumbersome synthesis process, high cost of the organic ligands, and the reaction conditions are not mild enough. In recent years, it has been found that the coordination of natural organic molecules with metal ions can be used to construct catalysts with excellent stability, such as zirconium–phytic acid,27 hafnium-furandicarboxylic acid,28 hafnium phosphonate.29 This idea of using natural organics to construct catalysts offers new route to construct efficient catalysts with low-cost and simple preparation processes. In our recent work, we have successfully utilized humic acid (HAs) extracted from lignite to prepare Zr-HAs catalyst,30 which once again verified the feasibility of the above idea. These reported studies show that using acidic oxygen-containing functional groups in natural organic molecules to construct catalysts has the enormous advantages in viewpoint of cost and efficiency, and exploring cheaper organic ligands remains highly required for the construction of MPV catalysts in the field of biomass conversion.
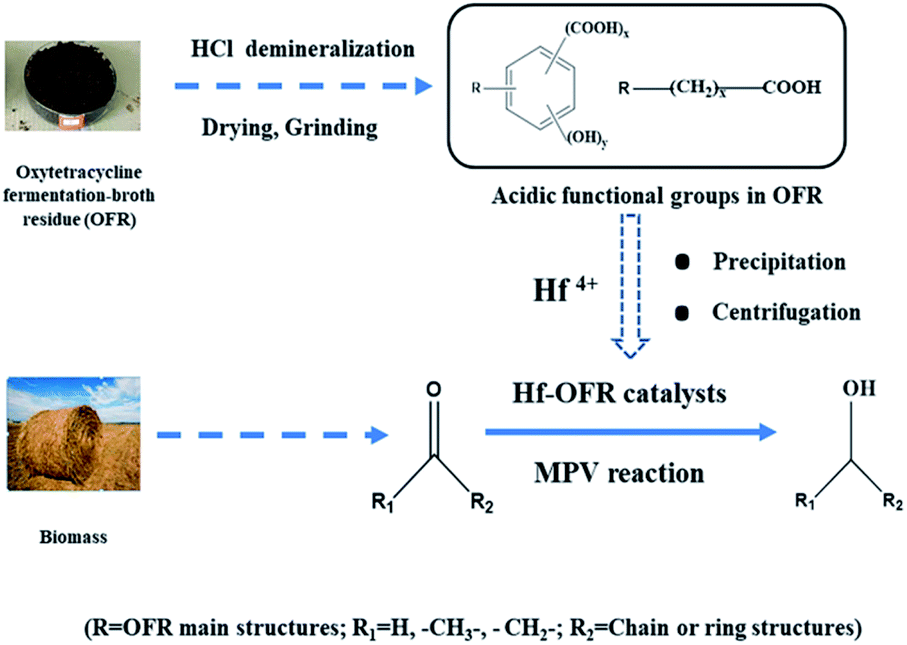
In this study, we used the oxytetracycline fermentation broth residue (OFR) to construct a novel and efficient Hf-based catalyst (termed as Hf-OFR, Scheme 1). The catalyst was applied into the hydrogenation reaction of ethyl levulinate (EL) into γ-valerolactone (GVL). The preparation and reaction conditions of the catalyst were investigated in detail. The structures of the catalysts before and after use were also characterized. The results showed that the prepared novel Hf-OFR catalyst exhibited high activity and stability for the MPV reaction of different carbonyl compounds. The proposed route of preparing Hf-based catalyst in this work may be beneficial to promote the green and high-value utilization of both pharmaceutical solid wastes and biomass resources.
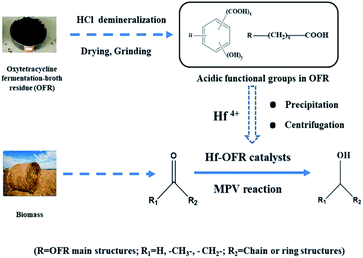 |
| | Scheme 1 Schematic of the direct use of OFR to prepare the Hf-OFR catalysts and its application in the conversion of biomass-derived carboxyl compounds via the MPV reaction. | |
Experiment section
Catalyst preparation
Firstly, in order to eliminate the possible influence of metal elements in the raw oxytetracycline fermentation broth residue (ROFR) in the process of building the catalysts, we used the 18 wt% HCl to demineralize the ROFR sample (about demineralization methods, see Page S3†). The 0.5 g HfCl4 was dissolved in 75 mL DMF, and then 0.5 g of the demineralized OFR (DOFR) was added into the above solution under constant stirring. Then 1.578 g of triethylamine as the deprotonic agent was added into the above mixtures dropwise at a constant rate within 10 min. The mixed slurry was stirred at room temperature for 5 h, and then aged under 70 °C for 3 h. After the reaction, the obtained precipitate was separated by centrifugation, and washed with DMF for five times and then with ethanol for five times. The supernatant was detected using AgNO3 until no Cl− was checked out. So far, the obtained precipitate was dried under vacuum at 80 °C for 12 h, ground into powders, and denoted as Hf-OFR for use. The catalyst constructed with the raw OFR directly coordinated with Hf was marked as Hf-ROFR. The preparation parameters were investigated to establish the optimal preparation conditions, including the ratios of Hf precursor to the DOFR, the dosage of trimethylamine, the aging temperature. The basic processes were similar to the above description.
Reaction
The selective hydrogenation reaction of carbonyl compounds to alcohols or their derivatives was carried out in a 10 mL Teflon-lined stainless steel autoclave equipped with a magnetic stirrer and using isopropanol as the solvent and hydrogen-donor. (Isopropanol is one of the hydrogen sources with the most excellent transfer hydrogenation performance in the MPV reaction system.31) Typically, EL (1 mmol), isopropanol (5 mL), and the catalyst (200 mg) were added into the reactor. After that, the reactor was transferred to an oil bath under constant temperature with magnetic stirring and allowed to react at a preset temperature and time. After the reaction, the reactor was cooled down in the cold water to quench the reaction, and the organic phase was diluted by isopropanol. The liquid samples were analyzed quantitatively by gas chromatography (TECHCOMP GC7900) with a flame ionization detector using decane as the internal standard. The identification of the products was made by GC-MS (Agilent 7890B-7000D). To check the reusability of the Hf-OFR catalyst, the solid catalyst were removed from the reaction system by centrifugation within a short time after quenching the reaction, washed three times with fresh isopropanol, and then reused for the next run without other treatment. In the heterogeneity experiments, the solid catalyst was separated out from the reaction system after one hour of reaction, and the supernatant continued to maintain the reaction without the solid catalyst. It was checked whether the target reaction continued after the solid catalyst was removed. All the experimental devices used were checked by repeating experiments for at least two times to obtain good operation stability during the experiment. The quantitative analysis of the reaction mixtures was repeated twice or three times to assure the accuracy of the data, and average values were used.
Results and discussion
Characterization and analysis of the OFR
Firstly, the basic composition of ROFR and the DOFR were analyzed, including industry analysis and basic element analysis (Tables S1 and S2, about industrial analysis methods, see Page S3†). From Table S1,† industrial analysis results showed that the volatile substance in ROFR accounted for 61.41 wt%, indicating that ROFR contained a large amount of organic matter. By comparing the carbon and oxygen content in the elemental analysis (C: 40.48 wt%, O: 22.80 wt%), it could be speculated that organic matter contained a large amount of C and O element. Besides C, H and O, ROFR also contained N, P, and S element. After demineralization, the mass ratio of inorganic elements in the ROFR was significantly increased, indicating that most of the metal elements contained in the ROFR have been removed by demineralization. The ash content was 12.50 wt% and the significant metal elements in ROFR was also analyzed by ICP-AES (Table S3†), and it was shown that ROFR contains a certain amount of Ca, Zn, Fe, Na, K, Al, Si, etc., the ash content of the DOFR decreased significantly, indicating that most of the mineral salts in the bacteria residue were removed by the hydrochloric acid, only a small amount of iron, zinc and silicon that were hard to dissolve in hydrochloric acid remained.
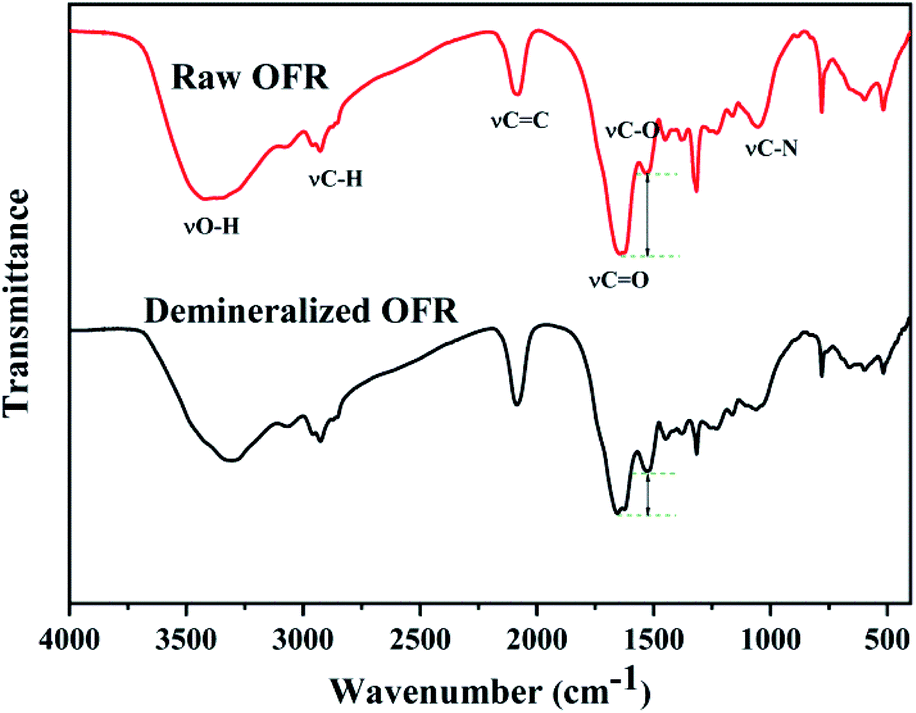
From infrared spectrum curve of the OFR in Fig. 1, a strong and broad absorption peak around 3400 cm−1 generated by the stretching vibration of alcohols or phenols O–H appears. Near 2900 cm−1 there were two absorption peaks, the larger wavenumber was the methyl C–H stretching vibration absorption peak, the smaller one was the methylene C–H stretching vibration absorption peak. The peak at the wavenumber 2078 cm−1 was assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. The peaks around 1659 cm−1 and 1523 cm−1 could be assigned to the strong asymmetrical stretching and relatively weaker symmetrical stretching of –COO– groups, which revealed the presence of acidic carboxyl groups.32 There was a weaker peak at 1030–1220 cm−1, which could be attributed to C–N stretching vibration of aliphatic amines in ROFR.33 Compared with the ROFR, the intensity of the weak symmetrical stretching of –COO– groups at 1523 cm−1 of DOFR became stronger compared to the peak of the asymmetrical stretching of –COO– groups at 1659 cm−1 (the intensity of the transmittance differs by 16.12%), which may be due to the elution of minerals by hydrochloric acid, resulting in the release of occupied acidic functional groups.34 The results of elemental analysis also showed that the demineralization could effectively remove the inherent minerals, which occupied the acidic groups in ROFR, and make it possible for Hf4+ to coordinate with the acidic groups in the preparation process.34 Fig. S1† is the possible structural formula for the oxygen-containing acidic functional groups in ROFR and DOFR.
C. The peaks around 1659 cm−1 and 1523 cm−1 could be assigned to the strong asymmetrical stretching and relatively weaker symmetrical stretching of –COO– groups, which revealed the presence of acidic carboxyl groups.32 There was a weaker peak at 1030–1220 cm−1, which could be attributed to C–N stretching vibration of aliphatic amines in ROFR.33 Compared with the ROFR, the intensity of the weak symmetrical stretching of –COO– groups at 1523 cm−1 of DOFR became stronger compared to the peak of the asymmetrical stretching of –COO– groups at 1659 cm−1 (the intensity of the transmittance differs by 16.12%), which may be due to the elution of minerals by hydrochloric acid, resulting in the release of occupied acidic functional groups.34 The results of elemental analysis also showed that the demineralization could effectively remove the inherent minerals, which occupied the acidic groups in ROFR, and make it possible for Hf4+ to coordinate with the acidic groups in the preparation process.34 Fig. S1† is the possible structural formula for the oxygen-containing acidic functional groups in ROFR and DOFR.
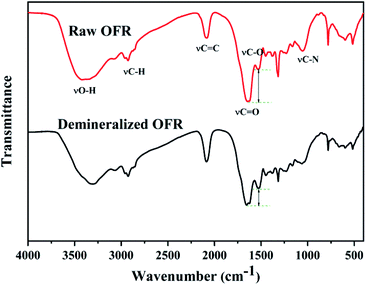 |
| | Fig. 1 FTIR spectra of raw OFR and demineralized OFR. | |
Optimization and characterization of the Hf-OFR catalysts
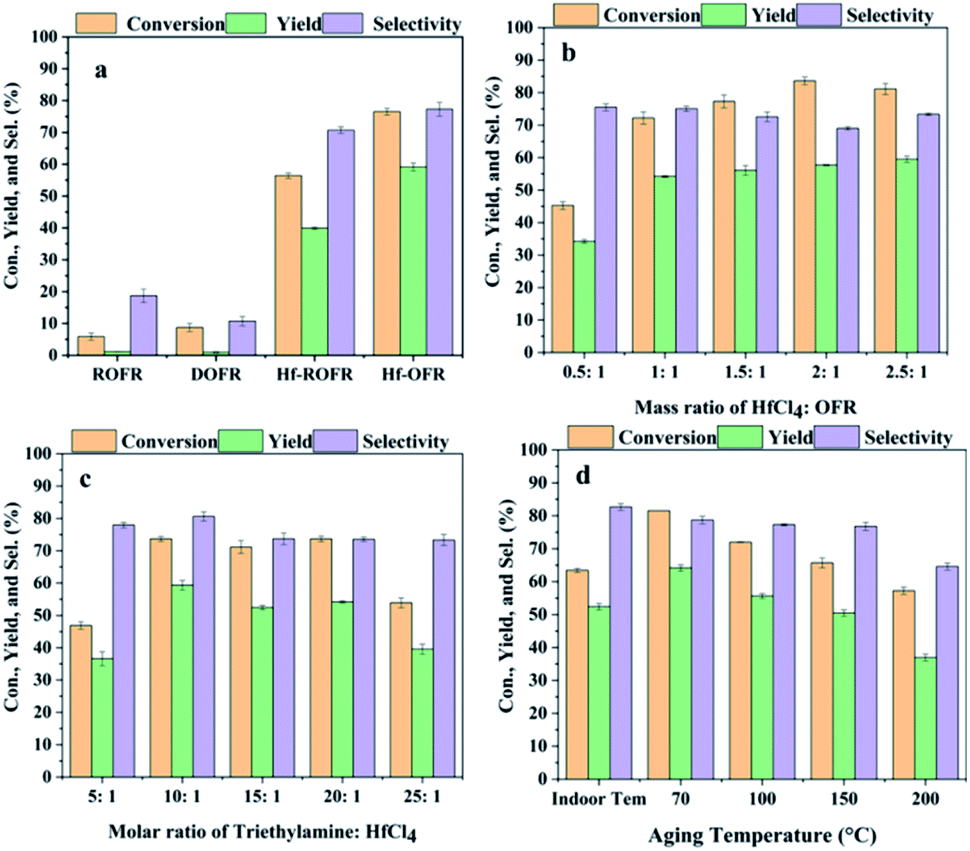
DMF was used as the solvent to prepare the catalyst because the OFR sample could disperse well in DMF. We first investigated the effect of demineralization on Hf-OFR catalyst, and the results are shown in Fig. 2a. The activity of the Hf-OFR catalyst was significantly improved after demineralization. This experimental result was consistent with our previous speculation that more acidic functional groups in ROFR can be exposed by demineralization, thereby coordinating with more Hf4+ to form the effective active site catalytic the hydrogenation reaction. The OFR itself before and after demineralization basically has no catalytic activity. So we later chose DOFR as the ligand to further optimize the preparation conditions of the Hf-OFR catalyst. Different preparation parameters were investigated and optimized, including the mass ratio of Hf precursor to OFR, the dosage of the additive trimethylamine (deprotonate reagent), and the aging temperature of the catalysts. The transfer hydrogenation of ethyl levulinate (EL) into γ-valerolactone (GVL) was used to evaluate the activity of the catalysts. In the process of optimizing the ratio of raw materials, with the increasing of the dosage of Hf precursor, the amount of precipitated precipitate also increased, and the color of the supernatant of the solution became clear, indicating that more OFR interacted with Hf4+ and participated in the formation of the solid catalysts. When the mass ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the supernatant of the solution appeared colorless and transparent. With the further increasing of Hf precursor, the color of the supernatant became turbid again (Fig. S2†). In order to effectively utilize OFR, the catalyst was prepared under the mass ratio of 1:1. The reaction results were given in Fig. 2, as the results shown, under the conditions of HfCl4:DOFR mass ratio of 1:1, the molar ratio of triethylamine to HfCl4 10:1, the aging temperature 70 °C, the prepared catalyst had higher efficiency for the reaction.
:1, the supernatant of the solution appeared colorless and transparent. With the further increasing of Hf precursor, the color of the supernatant became turbid again (Fig. S2†). In order to effectively utilize OFR, the catalyst was prepared under the mass ratio of 1:1. The reaction results were given in Fig. 2, as the results shown, under the conditions of HfCl4:DOFR mass ratio of 1:1, the molar ratio of triethylamine to HfCl4 10:1, the aging temperature 70 °C, the prepared catalyst had higher efficiency for the reaction.
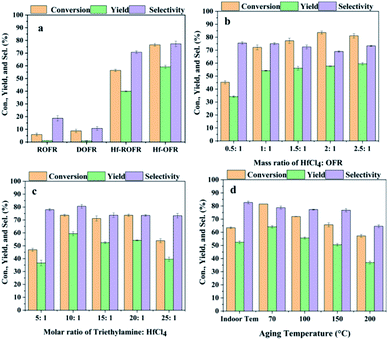 |
| | Fig. 2 Optimize preparation conditions, (a) effect of demineralization on catalytic activity of Hf-OFR catalysts (other preparation conditions: mass ratio of HfCl4:OFR was 1:1, molar ratio of triethylamine:HfCl4 was 10:1, aging temperature was 70 °C), (b) investigate the mass ratio of reactants, (c) investigate the amount of triethylamine added, (d) optimize the aging temperature. Other preparation conditions remain the same when examining any variable, reaction conditions: EL: 1 mmol, isopropanol: 5 mL, decane: 1 mmol, reaction temperature: 150 °C, reaction time: 3 h. | |
We investigated the changes in the specific surface area, pore structure and Hf content of Hf-OFR under different preparation conditions (Fig. S3, S4 and Table S4†). The results showed that the pore size distribution of Hf-OFR was mainly composed of mesopores, and it also contained a small amount of micropores. The specific surface area and pore volume of Hf-OFR decreased significantly as the mass of the Hf precursor input increased. This phenomenon suggested that the introduced Hf4+ may occupy the original pore structure of the OFR. At the same time, as the input of Hf precursor was further increased to 2.5:1, the Hf content in Hf-OFR did not increase proportionally, which indicated that the coordination process had been basically completed when the mass ratio of raw materials was 1:1. When the molar ratio of triethylamine to HfCl4 was 10:1, Hf-OFR had a larger specific surface area and pore volume. At the same time, the Hf-OFR under this preparation condition had better catalytic activity, which may be due to the higher specific surface area, and the large pore volume could increase the mass transfer efficiency between the catalyst and the reaction system. The higher Hf content was also the key factor for high catalytic activity. Finally, the effect of the aging temperature on the specific surface area and pore size of the Hf-OFR was investigated. When the aging temperature reached 200 °C, the specific surface area and pore volume of the catalyst increased significantly, while the yield of the catalyst decreased, and the catalytic activity decreased significantly. These phenomena indicated that too high aging temperature was not conducive to the coordination between Hf precursor and OFR. Next, we conducted a series of characterization analysis on Hf-OFR prepared under the optimal preparation conditions.
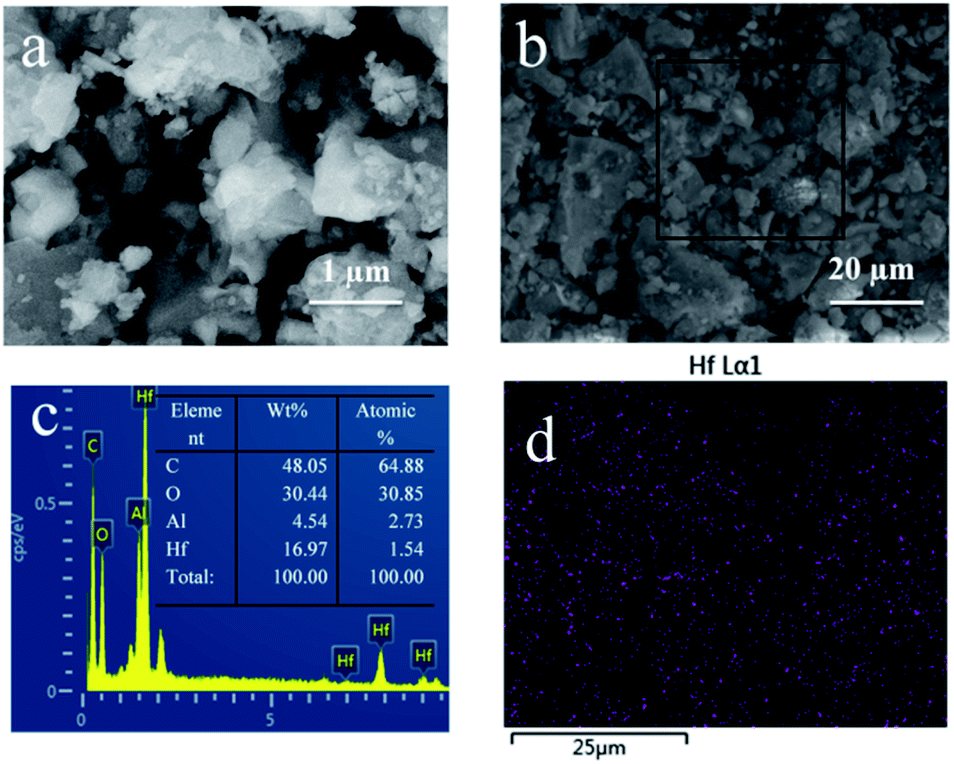
First, the surface morphology and Hf element distribution of Hf-OFR were characterized by SEM-EDS (Fig. 3). The results show that the microstructures of the Hf-OFR catalysts were composed of irregular particles with varied sizes. EDS data shows that the strong Hf element signal appeared for Hf-OFR, in addition, the mapping of Hf shows that Hf element was evenly distributed in the catalyst.
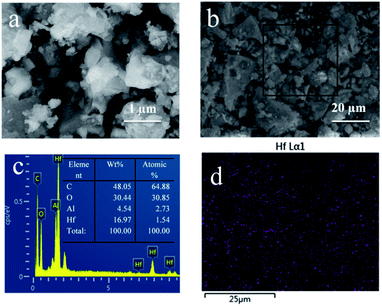 |
| | Fig. 3 Characterization of the Hf-OFR by SEM (a and b), EDS data (c) were obtained from the square area in (b), Hf EDS mappings of Hf-OFR (d). | |
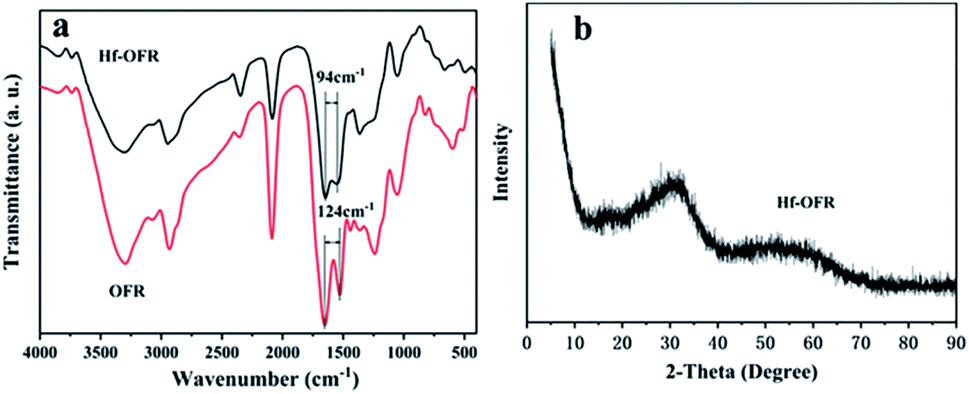
The FTIR spectrum of Hf-OFR catalyst in Fig. 4a exhibited the asymmetric (OFR, 1653 cm−1; Hf-OFR, 1646 cm−1) and symmetric (OFR, 1529 cm−1; Hf-OFR, 1552 cm−1) stretching vibration of carboxylate groups. The split between the symmetric and asymmetric peaks was narrowed from 124 cm−1 for OFR to 94 cm−1 for Hf-OFR, proving that Hf4+ was coordinated with the carboxylate groups.35,36 The bands at 3500–3000 cm−1 and 1400 cm−1 could be attributed to the characteristic stretching vibrations of hydroxyl and bending vibration of hydroxyl groups in water molecules, respectively. The XRD (Fig. 4b) pattern showed one broad diffraction peak, indicating that the obtained catalyst was amorphous.35
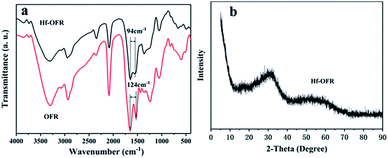 |
| | Fig. 4 FTIR spectra (a) and powder XRD pattern (b) of Hf-OFR catalyst. | |
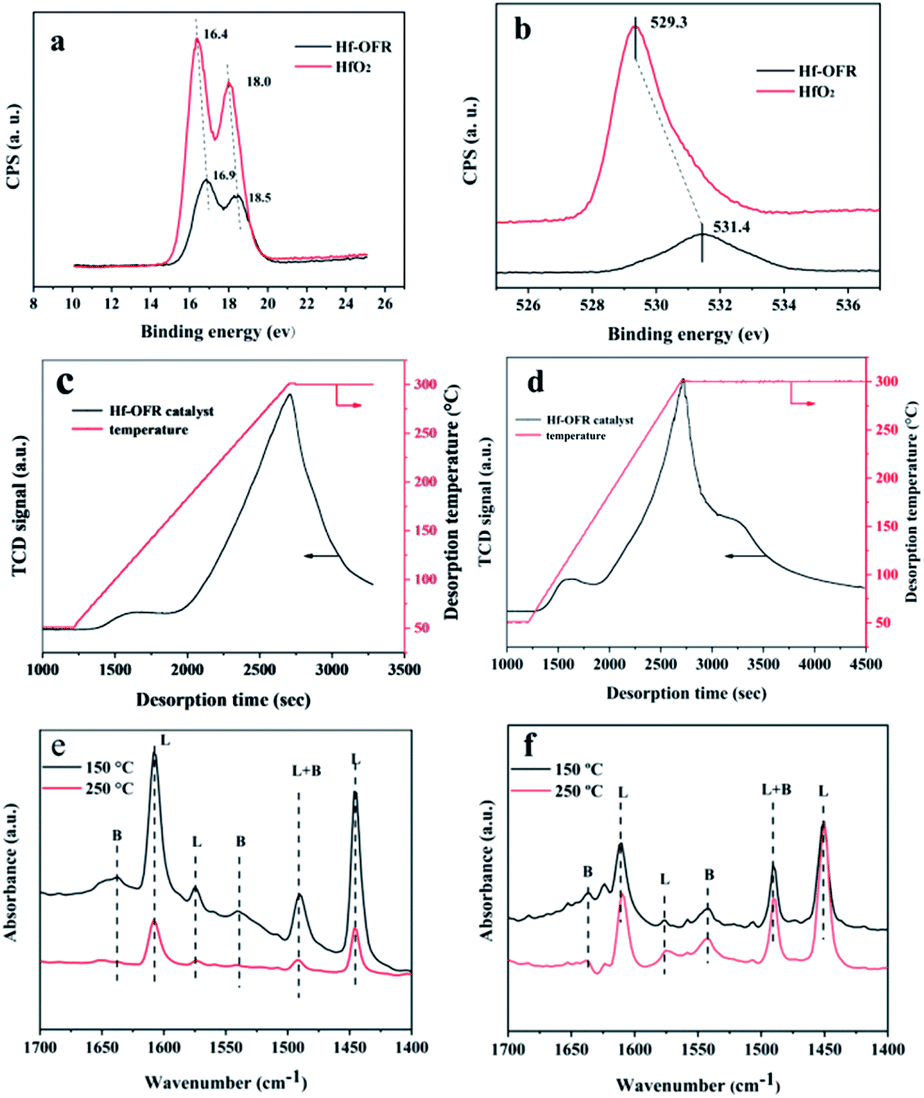
Next we explored the acidic and alkali sites of Hf-OFR. We first used XPS to evaluate the relative binding energy of Hf 4f and O 1s, and compared with HfO2 (Fig. 5a and b). The results show that the hafnium and oxygen species in the synthesized Hf-OFR catalyst had higher binding energy compared to HfO2. Higher binding energy of Hf species indicated the higher positive charges, representing the stronger Lewis acidity.37 The higher binding energy of oxygen species in Hf-OFR corresponded to a lower negative charge, indicating that the basicity of oxygen species in the Hf–O–C framework of Hf-OFR was weaker.38 The increased acidity of Hf species was in favor of the activity of the catalyst. The above analysis of the acidity and alkalinity of Hf-OFR showed that the hybrid Hf-OFR catalyst prepared by OFR had a relatively high acid strength compared to HfO2. At the same time, the alkali strength could be maintained at a relatively low level. Strong acid strength and weak alkali strength could promote the progress of the transfer hydrogenation reaction.39
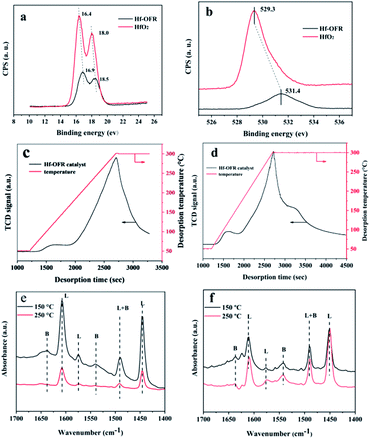 |
| | Fig. 5 XPS spectra of Hf 4f (a), XPS spectra of O 1s (b), CO2-TPD curves (c) and NH3-TPD curves (d) of Hf-OFR catalyst, pyridine-adsorbed FT-IR spectra of OFR (e) and Hf-OFR (f), (L: Lewis acid sites; B: Brønsted acid sites). | |
Specific contents of acidic and basic sites were analyzed by NH3-TPD and CO2-TPD. The TPD temperature ranges were set from 50 °C to 300 °C, and the heating rate was set at 10 °C min−1. The results were shown in Fig. 5c and d. It can be seen that with the increasing of the desorption temperature, a large number of CO2 and NH3 were desorbed, indicating that the prepared catalyst had certain acidic and basic sites (acid site: 1.9 mmol g−1, basic site: 1.6 mmol g−1). The basic sites might result from the presence of N element and oxide anions in DOFR. In order to continue to study the acidity of the catalyst, we used pyridine infrared to analyze the content of Lewis and Brønsted acid contained in the OFR and Hf-OFR under the desorption temperature of 250 °C. In order to compare the influence of the introduction of Hf4+ on the acidity of Hf-OFR, the results are as follows in the Fig. 5e, f and Table S5.† Firstly, the peak positions of 1450 cm−1 and 1610 cm−1 could be attributed to the absorption of pyridine on the Lewis acid sites40 and the peak positions of 1545 cm−1 and 1640 cm−1 were attributed to the absorption of pyridine on the Brønsted acid sites.41 Both OFR and Hf-OFR contain a certain amount of Lewis and Brønsted acidic sites, but the quantitative analysis data shows that the amount of acid in OFR was less than the amount of acid in Hf-OFR under the desorption temperature of 150 °C and 250 °C, which shows that the introduction of Hf4+ may form the Hf–O–C structure with the OFR. This coordination structure can provide Lewis acidic sites for Hf-OFR,40 and at the same time, some unsuccessfully coordinated Hf4+ can also provide Lewis acidic sites for Hf-OFR.40 On the other hand, the Brønsted acidic sites were derived from the protonated carboxyl group and the phenolic hydroxyl in the OFR,41 and the introduced Hf4+ was coordinated with these carboxyl group and the phenolic hydroxyl in the OFR, thereby stabilizing the structure of these oxygen-containing acidic functional groups in the OFR. This may be the main reason for the increase in the amount of Brønsted acid sites in the Hf-OFR. The formation of the alkali site and the presence of the Brønsted acidic site have cooperation effects on the process of hydrogen transfer reaction.42
Optimization of reaction conditions
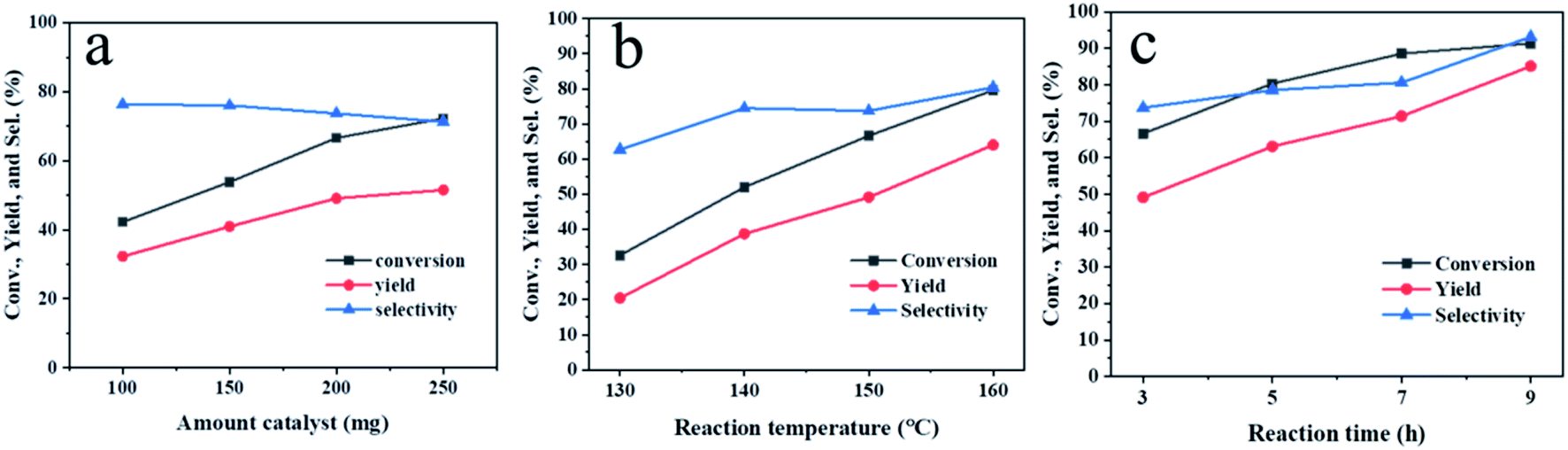
In order to improve the performance of the Hf-OFR catalyst, the reaction conditions were investigated via the hydrogenation reaction of EL into GVL. We first studied the dosage of the catalyst (Fig. 6a). The results showed that both the conversion and the yield increased almost linearly with the increasing of the catalyst dosage in the range of 100 mg to 200 mg under the present reaction condition. Further increasing the amount of catalyst led to poor dispersion of catalyst particles in the reaction system, resulting in the slower increasing rate of the conversion and yield. Therefore, 200 mg was chosen as a suitable dosage under the present reaction condition. Increasing the reaction temperature (Fig. 6b) also had a significant promotion effect on the catalytic reaction. During the process of improving the reaction temperature from 130 °C to 160 °C, the yield of GVL maintained a linear growth trend, but considering the relatively mild reaction conditions can reduce the energy input, the mediate 150 °C was used as the appropriate reaction temperature. Then we extended the reaction time under the dosage of 200 mg and the reaction temperature of 150 °C. When the reaction time was 9 h, the conversion reached 91%, and the yield reached 86%. When the reaction time was extended, the yield remained nearly unchanged. We used GC-MS to analyze the by-products after 9 h. The detection results showed that the by-products were isopropyl levulinate and isopropyl 2-hydroxypentanoate. The byproducts were an intermediate formed by the transesterification of ethyl levulinate and ipropanol.31
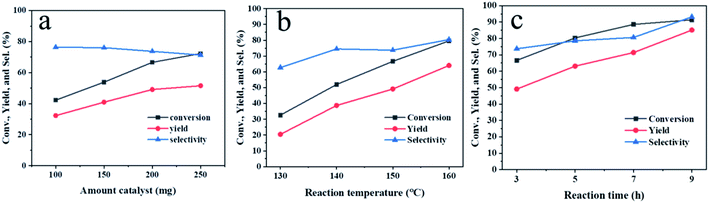 |
| | Fig. 6 The effects of reaction parameters on the performances of Hf-OFR, (a) the amount of catalyst (reaction time: 3 h, reaction temperature: 150 °C), (b) reaction temperature (catalyst amount: 200 mg, reaction time: 3 h), (c) reaction time (catalyst amount: 200 mg, reaction temperature: 150 °C). Typical reaction conditions: EL 1 mmol, 2-PrOH 5 mL. | |
Heterogeneity and reusability of the Hf-OFR
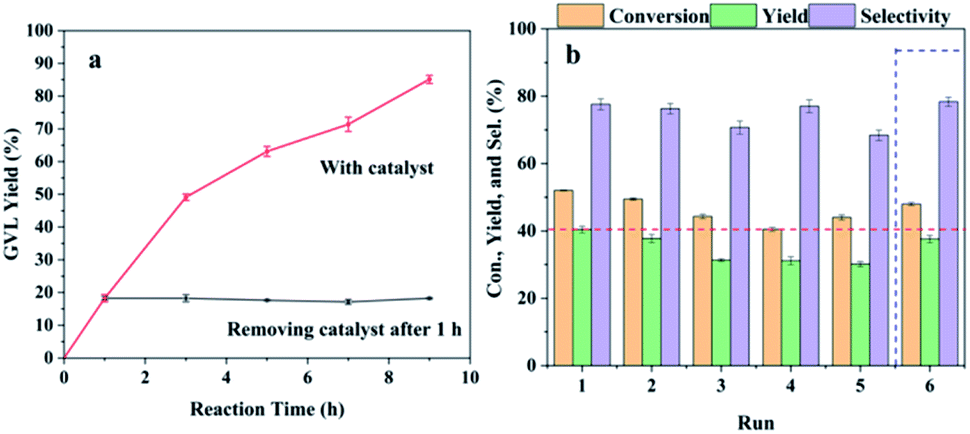
Heterogeneity and reusability are important for the solid catalyst concerning the potential industrial production, so it is necessary to study the reusing stability of the Hf-OFR catalyst (Fig. 7). The conversion and yield were maintained below 50% via controlling the reaction time at 2 h to investigate the real reusability of the catalyst. The heterogeneity of the catalyst was studied by separating the solid catalyst from the reaction system after reaction for 1 h with GVL yield around 20%, and then the reaction solution was allowed to continue to react under the same conditions with no presence of the solid catalyst. From Fig. 7a, it can be seen that the yield of GVL did not further increase after the solid catalyst was removed, indicating that it was the solid catalyst to catalyze the reaction and Hf-OFR was a heterogeneous catalyst. The reusability of the Hf-OFR catalyst was further investigated. As shown in Fig. 7b, the conversion and yield in the first run were 52% and 40%, which decreased to 44% and 30%, respectively, in the fifth run. The recycled catalyst was dried and weighed, and the weight was 152 mg, which was less than the initial catalyst dosage (200 mg). Therefore, the decrease in the conversion and yield was speculated that the physical loss of the catalyst during the recovery process. After the fifth run, the weight of the catalyst was replenished to 200 mg, and the sixth cycle was performed. The results show that the conversion and yield nearly returned to the initial level. The Hf content of the fresh and the recycled catalyst after 5 cycles were analyzed. The result showed that the Hf content only had a slight decrease from 27.3 wt% to 24.2 wt%, which might also contribute to the weak decreasing of the conversion and yield. These results proved that the performance of the Hf-OFR catalyst was stable during the reusing process.
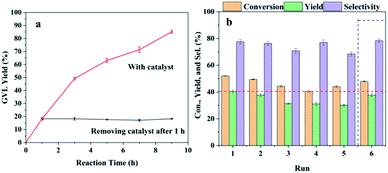 |
| | Fig. 7 Heterogeneity (a) and reusability (b) of the Hf-OFR catalyst. Reaction conditions: EL 1 mmol, 2-PrOH 5 mL, catalyst 200 mg, reaction temperature 150 °C, reaction time 2 h. | |
Characterization of the Hf-OFR catalysts before and after the cycle
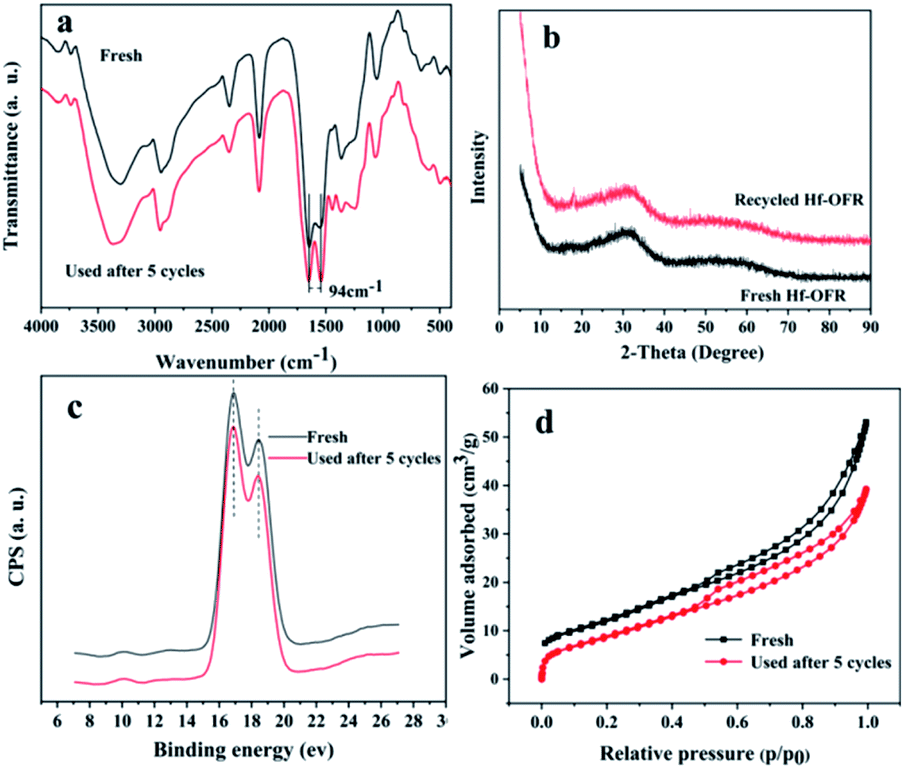
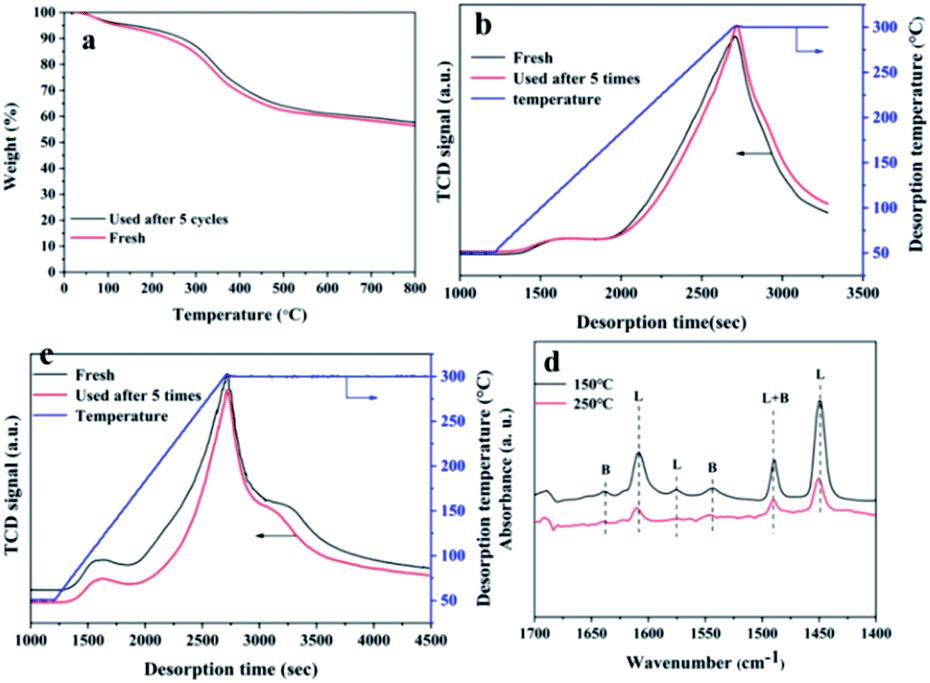
The detailed structural characterization analysis of the catalyst after the fifth cycle was subsequently performed, and compared with the fresh catalyst. FTIR spectra and XRD patterns (Fig. 8a and b) indicated that the characteristic functional groups and crystal structure of the catalyst remain basically unchanged after 5 cycle uses. XPS spectra of Hf 4f showed that the chemical environment of the Hf element did not change significantly before and after recycling. The specific surface area and pore volume of the catalyst have been a slight decreasing before and after recycling (Table S6†). The possible reason is that the adsorption of by-products blocked part of the pore structure. Next, we compared the thermal stability and the acid–base properties of the fresh and the recycled catalysts, as shown in Fig. 9. Fig. 9a showed that the thermal stability of the recycled catalysts had no noticeable difference compared to the fresh catalysts. It could be seen from Fig. 9b that the content of basic sites of the catalyst did not change significantly before and after recycling. However, Fig. 8c showed that the NH3 signal peak of the catalyst after recycling was weaker than the NH3 signal peak of the fresh catalyst, and the content of total acidic sites reduced from 1.90 mmol g−1 to 1.44 mmol g−1 (Table S7†). There may be two main reasons for the decreased in the content of acidic sites. Firstly, some weakly coordinated Hf4+ leached from the catalyst surface after five cycles of reactions; secondly, the adsorption of by-products occupied the original acidic sites. In the infrared analysis of pyridine, it can be seen that the absorption peak positions of the Lewis acid and Brønsted acid. The content of the Lewis acid of the catalyst had not changed significantly before and after recycling, which proved that the Hf–O–C structure was relatively stable. The content of Brønsted acid sites was slightly lost after recycling, decreased from 10.9 μmol g−1 to 8.8 μmol g−1 (desorption temperature 150 °C) and 6.0 μmol g−1 to 3.0 μmol g−1 (desorption temperature 250 °C), which may be due to the partial decomposition of the –OH and –COOH functional group. From the elemental analysis of carbon, it can be seen that the content of carbon increased slightly, which may be caused by the adsorption of the by-products. The adsorbed by-products might occupy the acidic sites and lead to the decreasing of the activity during the reusing process.
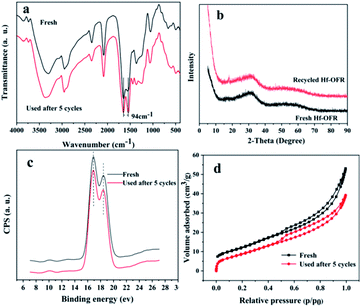 |
| | Fig. 8 Comparison of the freshly prepared and recycled Hf-OFR catalysts after five reuses: (a) FTIR spectra, (b) XRD patterns, (c) XPS spectra of Hf 4f, (d) N2 adsorption–desorption isotherm. | |
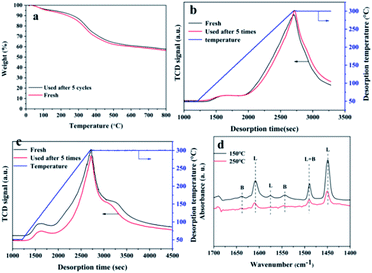 |
| | Fig. 9 Characterization of acid–base sites of catalysts before and after recycling: (a) TG curves, (b) CO2-TPD curves, (c) NH3-TPD curves, (d) pyridine-adsorbed FT-IR spectra (L: Lewis acid, B: Brønsted acid). | |
Possible reaction mechanism for EL transfer hydrogenation
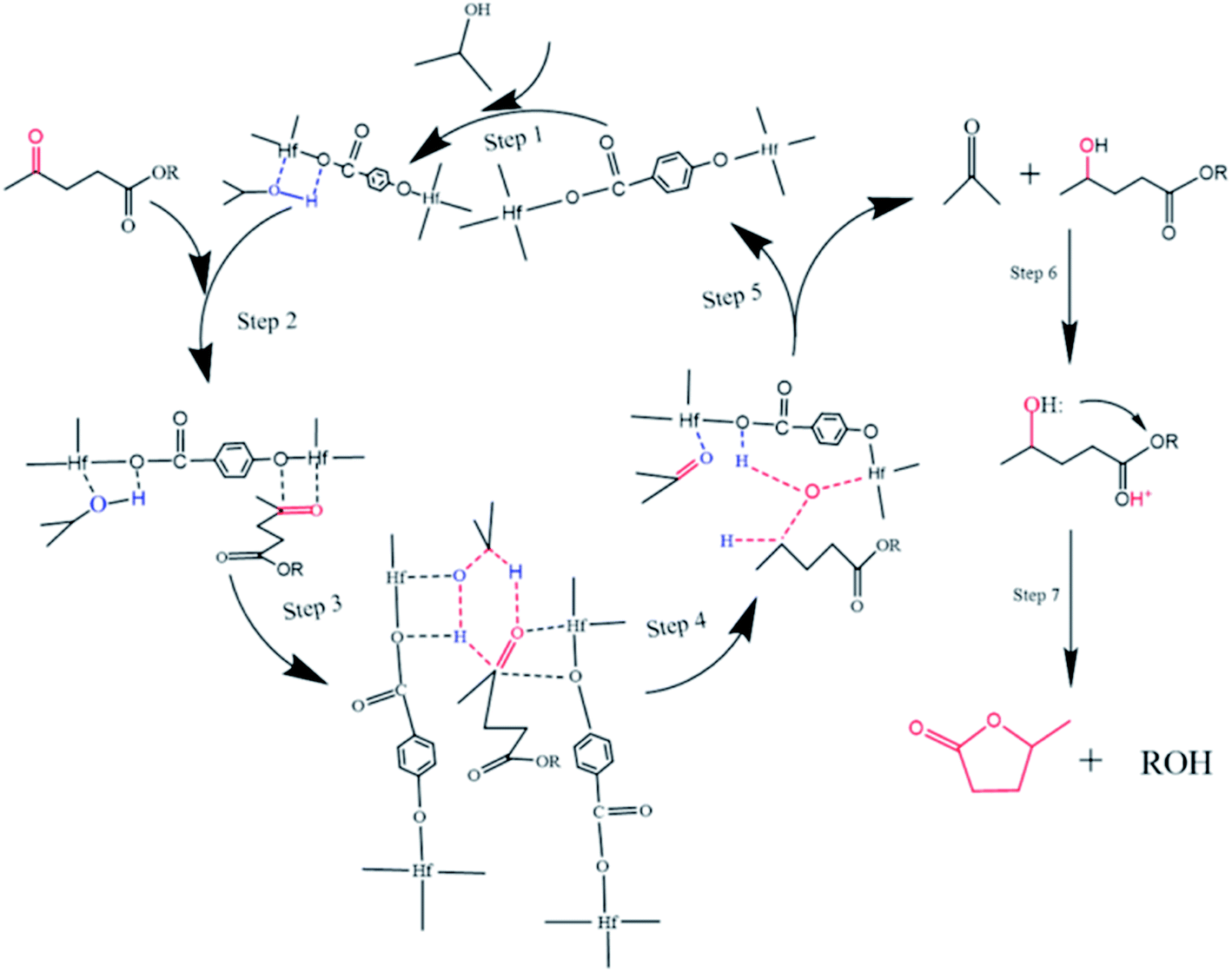
The possible reaction mechanism of Hf-OFR catalyzed transfer hydrogenation of EL was analyzed based on the experimental results and literatures, as shown in Fig. 10. The acidic site Hf4+ and basic site O2− in the catalyst play a vital role in the catalytic process.43 In the first step, the adsorption of isopropanol onto Hf-OFR assisted with Lewis acid–base sites (Hf4+, O2−). Under the synergistic action of the Lewis acid–base sites of the catalyst, isopropanol was fixed and dissociated into isopropoxide and hydride (step 1). Then the carbonyl group in EL under the role of the acidity of the catalyst interacted with the active site Hf4+ and activated by Hf4+ (step 2).44 The dissociated hydride formed a six-membered cyclic intermediate with the activated carbonyl group in the EL (step 3). Then the activated hydrogen atoms in isopropanol were transfer to the activated carbonyl group in EL to form acetone and 4-hydroxypentanoate (step 4 and 5). Finally, 4-hydroxypentanoate was lactonized to form the final product GVL under the cooperation effect of Brønsted acidity and basicity of the catalyst (step 6 and 7).45 Besides, EL could also undergo transesterification reaction with isopropanol to form isopropyl levulinate under the synergistic effect of acidic and basic in the catalyst, and then to isopropyl 2-hydroxypentanoate via transfer hydrogenation, which could be further converted to GVL.45
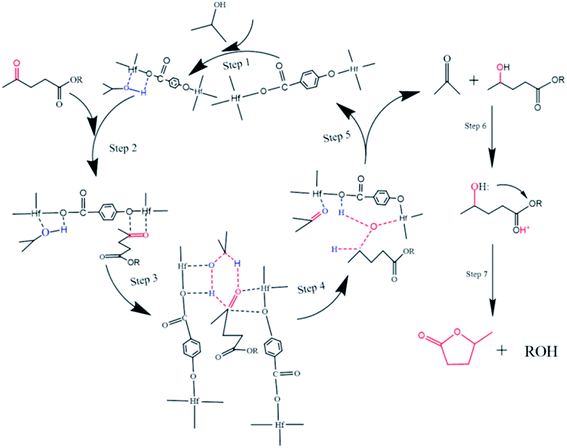 |
| | Fig. 10 Possible catalytic mechanism for the transfer hydrogenation of EL to GVL. | |
Conversion of different carbonyl compounds
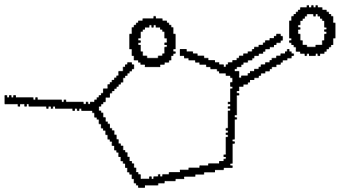
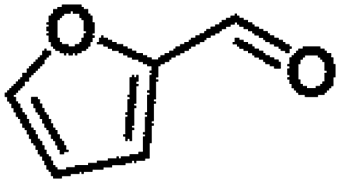
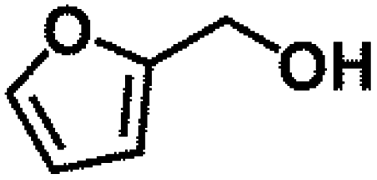
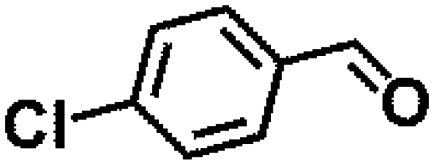
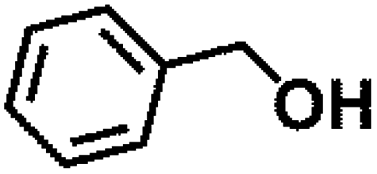
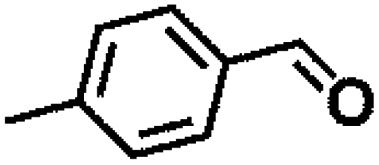
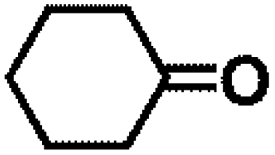
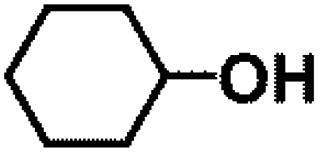
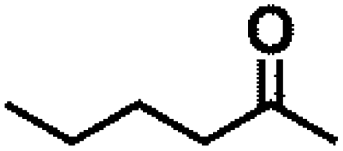
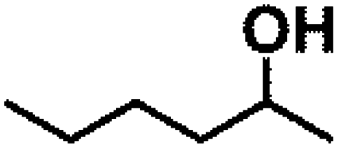
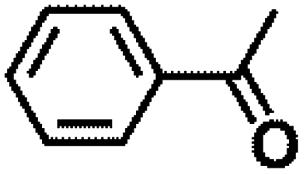
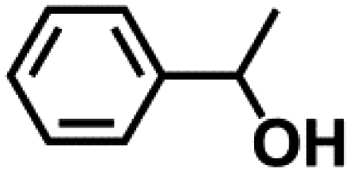
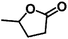
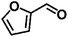
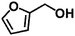
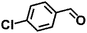
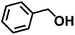
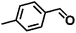
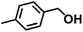
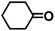
Encouraged by the outstanding performance of Hf-OFR catalyst in the hydrogenation of EL to GVL, we further expanded the scope of the substrate to other carbonyl compounds, to see the general applicability of Hf-OFR in the MPV reaction. As shown in the Table 1, both aliphatic and aromatic carbonyl compounds could be efficiently hydrogenated to the corresponding alcohols under the catalysis of Hf-OFR catalyst.
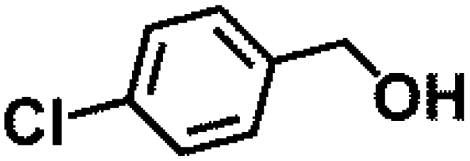
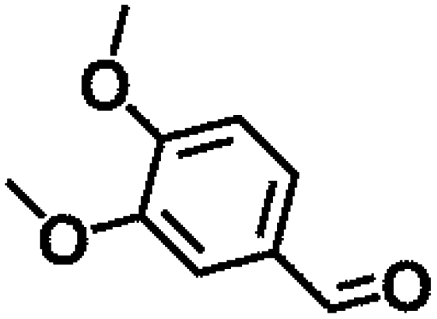
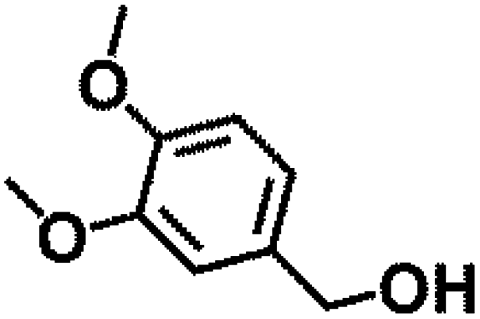
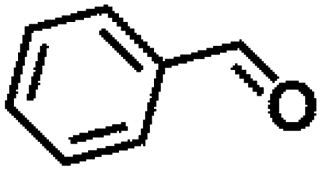
Table 1 MPV reduction of different biomass-derived carbonyl compounds over the Hf-OFR catalyst
| Entry |
Substrate |
Product |
T (°C) |
T (h) |
Conv. (%) |
Yield (%) |
Sel. (%) |
| 1 |
 |
 |
150 |
9 |
91 ± 0.1 |
86 ± 1.3 |
94 ± 1.5 |
| 2 |
 |
 |
80 |
9 |
97 ± 0.1 |
96 ± 0.3 |
99 ± 0.4 |
| 3 |
 |
 |
100 |
6 |
>99 |
>99 |
>99 |
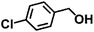
| 4 |
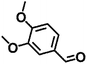 |
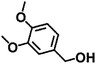 |
110 |
7 |
98 ± 0.7 |
93 ± 1.3 |
94 ± 3.1 |
| 5 |
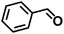 |
 |
110 |
12 |
98 ± 0.6 |
96 ± 0.2 |
98 ± 1.1 |
| 6 |
 |
 |
110 |
10 |
>99 |
>99 |
>99 |
| 7 |
 |
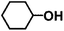 |
100 |
6 |
97 ± 0.3 |
91 ± 2.8 |
94 ± 3.1 |
| 8 |
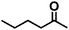 |
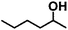 |
150 |
12 |
91 ± 0.1 |
84 ± 1.2 |
93 ± 1.3 |
| 9 |
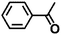 |
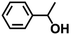 |
110 |
12 |
>99 |
>99 |
>99 |
Conclusions
In summary, we identified that the solid waste from oxytetracycline fermentation broth could be used to construct a novel hybrid Hf-based hydrogenation catalyst through the coordination of Hf4+ with the oxygen-containing acidic functional groups in the solid residue. The prepared catalyst exhibited high activity to MPV reaction for various carbonyl compounds. Moreover, the catalyst could be facilely recycled and reused at least for 5 cycles with no consideration changes in activity and structures. This work suggests new route for the green and valuable utilization of organic solid waste in fermentation industry and the construction of efficient catalyst for biomass conversion.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21968021, 21676149), the Natural Science Foundation of Inner Mongolia (2019MS02025), Program for Grassland Elite in Inner Mongolia, the Innovative and Entrepreneurial Talents Grassland Talents Engineering of Inner Mongolia (Q2017011), CAS “Light of West China” Program, and Local Science and Technology Development Fund Projects Guided by the Central Government (2020ZY0010).
Notes and references
- E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038–1049, 10.1039/B904997P.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870, DOI:10.1021/cr4002269.
- J. L. Martinez, Environ. Pollut., 2009, 157, 2893–2902, DOI:10.1016/j.envpol.2009.05.051.
- S. Ren, X. Guo, A. Lu, X. Guo, Y. Wang, G. Sun, W. Guo, C. Ren and L. Wang, Bioresour. Technol., 2018, 265, 155–162, DOI:10.1016/j.biortech.2018.05.087.
- G. Zhang, C. Li, D. Ma, Z. Zhang and G. Xu, Bioresour. Technol., 2015, 192, 257–265, DOI:10.1016/j.biortech.2015.05.014.
- Y. Liu, X. Zhu, X. Wei, S. Zhang, J. Chen and Z. Ren, Chem. Eng. J., 2018, 334, 1101–1107, DOI:10.1016/j.cej.2017.11.033.
- Z. Li, M. Li, X. Liu, Y. Ma and M. Wu, Sci. Total Environ., 2014, 493, 481–486, DOI:10.1016/j.scitotenv.2014.06.005.
- M. Wang, H. Liu, X. Cheng, B. Zhang, C. Cai and J. Wang, Bioresour. Technol., 2019, 271, 143–149, DOI:10.1016/j.biortech.2018.09.106.
- W. Zhong, Z. Li, J. Yang, C. Liu, B. Tian, Y. Wang and P. Chen, Bioresour. Technol., 2014, 151, 436–440, DOI:10.1016/j.biortech.2013.10.100.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547, 10.1039/C3GC41492B.
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46, DOI:10.1016/S0960-8524(01)00118-3.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802, 10.1039/B703294C.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549, 10.1039/B707343G.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793, 10.1039/C0GC00401D.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978, 10.1039/C000075B.
- F. Martel, B. Estrine, R. Plantier-Royon, N. Hoffmann and C. Portella, Top. Curr. Chem., 2010, 294, 79–115, DOI:10.1007/128_2010_54.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599, DOI:10.1021/cr900354u.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617, 10.1039/C1CS15124J.
- J. R. Ruiz and C. Jimenez-Sanchidrian, Curr. Org. Chem., 2007, 11, 1113–1125, DOI:10.2174/138527207781662500.
- Z. Yang, Y. B. Huang, Q. X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328–5330, 10.1039/C3CC40980E.
- K. Yan, J. Liao, X. Wu and X. Xie, RSC Adv., 2013, 3, 3853–3856, 10.1039/C3RA22158J.
- R. Cohen, C. R. Graves, S. T. Nguyen, J. M. L. Martin and M. A. Ratner, J. Am. Chem. Soc., 2004, 126, 14796–14803, DOI:10.1021/ja047613m.
- F. Wang, N. Ta and W. Shen, Appl. Catal., A, 2014, 475, 76–81, DOI:10.1016/j.apcata.2014.01.026.
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Zeng, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827–834, DOI:10.1016/j.apcatb.2013.10.021.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233–12235, 10.1039/C1CC14748J.
- J. Wang, K. Okumura, S. Jaenicke and G.-K. Chuah, Appl. Catal., A, 2015, 493, 112–120, DOI:10.1016/j.apcata.2015.01.001.
- J. Song, B. Zhou, H. Zhou, L. Wu, Q. Meng, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2015, 54, 9399–9403, DOI:10.1002/anie.201504001.
- H. Li, T. Yang and Z. Fang, Appl. Catal., B, 2018, 227, 79–89, DOI:10.1016/j.apcatb.2018.01.017.
- C. Xie, J. Song, B. Zhou, J. Hu, Z. Zhang, P. Zhang, Z. Jiang and B. Han, ACS Sustainable Chem. Eng., 2016, 4, 6231–6236, DOI:10.1021/acssuschemeng.6b02230.
- Y. Sha, Z. Xiao, H. Zhou, K. Yang, Y. Song, N. Li, R. He, K. Zhi and Q. Liu, Green Chem., 2017, 19, 4829–4837, 10.1039/C7GC01925D.
- F. Li, L. J. France, Z. Cai, Y. Li, S. Liu, H. Lou, J. Long and X. Li, Appl. Catal., B, 2017, 214, 67–77, DOI:10.1016/j.apcatb.2017.05.013.
- F. Verpoort, T. Haemers and P. Roose, Appl. Spectrosc., 1999, 53, 1528–1534, DOI:10.1366/0003702991946262.
- A. N. A. Aryee, F. R. van de Voort and B. K. Simpson, Process Biochem., 2009, 44, 401–405, DOI:10.1016/j.procbio.2008.12.004.
- J. Hao, L. Han, Y. Sha, X. Yu, H. Liu, X. Ma, Y. Yang, H. Zhou and Q. Liu, Fuel, 2019, 239, 1304–1314, DOI:10.1016/j.fuel.2018.11.129.
- L. Peng, J. Zhang, J. Li, B. Han, Z. Xue and G. Yang, Chem. Commun., 2012, 48, 8688–8690, 10.1039/C2CC34416E.
- J. Song, L. Wu, B. Zhou, H. Zhou, H. Fan, Y. Yang, Q. Meng and B. Han, Green Chem., 2015, 17, 1626–1632, 10.1039/C4GC02104E.
- B. Tang, W. Dai, X. Sun, G. Wu, N. Guan, M. Hunger and L. Li, Green Chem., 2015, 17, 1744–1755, 10.1039/C4GC02116A.
- H. Li, Z. Fang, J. He and S. Yang, ChemSusChem, 2017, 10, 681–686, DOI:10.1002/cssc.201601570.
- H. Li, X. Liu, T. Yang, W. Zhao, S. Saravanamurugan and S. Yang, ChemSusChem, 2017, 10, 1761–1770, DOI:10.1002/cssc.201601898.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2012, 5, 1812–1819, DOI:10.1002/cssc.201200072.
- J. Jiménez-Jiménez, P. Maireles-Torres, P. Olivera-Pastor, E. Rodriguez-Castellón, A. Jiménez-López, D. J. Jones and J. Rozière, Adv. Mater., 1998, 10, 812–815, DOI:10.1002/(SICI)1521-4095(199807)10:10<812::AID-ADMA812>3.0.CO;2-A.
- L. Bui, H. Luo, W. R. Gunther and Y. Roman-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022–8025, DOI:10.1002/anie.201302575.
- Y. Lin, Q. Bu, J. XU, X. Liu, X. Zhang, G. Lu and B. Zhou, Mol. Catal., 2021, 502, 2468–8231, DOI:10.1016/j.mcat.2021.111405.
- N. A. S. Ramli and N. A. S. Amin, Appl. Catal., B, 2015, 163, 487–498, DOI:10.1016/j.apcatb.2014.08.031.
- M. Sudhakar, V. V. Kumar, G. Naresh, M. L. Kantam, S. K. Bhargava and A. Venugopal, Appl. Catal., B, 2016, 180, 113–120, DOI:10.1016/j.apcatb.2015.05.050.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01738a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Runxia He*a and
Quansheng Liua
*a,
Runxia He*a and
Quansheng Liua