
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting gas selectivity in organic ionic plastic crystals by free energy calculations
Vinay S. Kandagal,
Jennifer M. Pringle ,
Maria Forsyth and
Fangfang Chen*
,
Maria Forsyth and
Fangfang Chen*
Institute for Frontier Materials, ARC Centre of Excellence for Electromaterials Science, Deakin University, Burwood, VIC 3125, Australia. E-mail: chenf@deakin.edu.au
First published on 1st June 2021
Abstract
Organic ionic plastic crystals (OIPCs) are molecularly disordered solids, and their potential for the development of gas separation membranes has recently been demonstrated. Here, the gas absorption capability of the OIPC, diethyl(methyl)(isobutyl)phosphonium hexafluorophosphate ([P122i4][PF6]), for four gases is predicted through potential of mean force (PMF) calculations based on two methods – average force method and adaptive biasing force method. Both methods correctly predicted the different trends of adsorption and absorption of these gases across the OIPC–gas interface. The distinct energy barriers of the PMF profiles of CO2 and N2 near the interface directly reflect the good selectivity of OIPC to these two gases. However, the selectivity of CH4 and O2 cannot be accurately reflected by the PMF curve near the interface, because the relative energy varies greatly at different positions inside the OIPC. Thus the average free energy change should be calculated over the entire OIPC box to evaluate the difference in selectivity between the two gases. This also suggests that gas absorption in OIPCs is greatly affected by the structural order and chemical environment. The adaptive biasing force method overall outperforms the average force method. The method should be able to provide a prediction of gas selectivity for a wider range of organic ionic plastic crystals and other solid materials.
Introduction
Greenhouse gas removal, especially the reduction of carbon dioxide, is a critical global challenge of the 21st century, having had a profound impact on natural ecology and human society. The advanced materials used to reduce industrial gas is a core research topic in the field of greenhouse gas control. Over the past two decades, ionic liquids (ILs) have received widespread attention for their potential as carbon dioxide absorbing materials, since the first report by Blanchard et al. in 1999.1 ILs consist of weakly interacting cations and anions, which usually have a low melting point and maintain liquid state at ambient temperature. The CO2 absorption mechanism of an IL can be physical and/or chemical absorption, depending on the chemical structure of the cations and anions. Physical absorption originates from various interactions between gas and ion molecules, including electrostatic, van der Waals interactions and hydrogen bonding, all of which affect the solubility of gas in ILs.2–5 Chemically altering the structure of ILs by introducing amino or other functional groups that can interact with CO2 is another design strategy to promote chemical absorption.6 Meanwhile, the use of ILs to capture other industrial exhaust gases such as SO2, H2S and NH3 has also been reported.7However, ILs have discernible disadvantages for gas separation because of their fluidity. They are usually incorporated with other porous materials, such as metal–organic frameworks (MOFs) and polymers, to make membranes for gas separation with enhanced gas separation capabilities.8,9 Even so, maintaining good stability of the supported IL membrane (SILM) during long-term use is still a challenge as the IL can blow off the support, particularly at high gas pressure. Thus, researchers have looked at solid analogues of ILs, such as polymerized ILs, which show a good mechanical stability for CO2 separation membranes, and demonstrate acceptable CO2/CH4 and CO2/N2 permselectivities.10–13 However, the drop in both gas permeability and diffusivity is significant compared to their IL analogues in a SILM.14
Another solid analogue of ILs exists in a disordered crystalline state, known as organic ionic plastic crystals (OIPCs), which have recently been studied for their potential in CO2 separation. The ideal CO2/N2 selectivity of 30 at 35 °C was reported for the OIPC, diethyl(methyl)(isobutyl)phosphonium hexafluorophosphate ([P122i4][PF6]), supported on a polyvinylidene difluoride (PVDF) membrane.15 OIPCs have a similar chemical composition to ILs, however some ion chemistries and cation/anion combinations can result in plastic phases and crystallization temperatures above room temperature. As a result of the chemical similarities, many gas absorption mechanisms and the knowledge learnt from ILs could also apply to OIPCs.
OIPCs are usually soft materials that undergo temperature-driven solid–solid plastic crystal phase transitions, resulting from internal degrees of freedom such as rotational and translational motions of the ions or parts of the ions. The phase transitions change the short range order and the internal free volume of the OIPC, which could affect the gas intake as previously reported.15 Gas uptake in different OIPC phases was also studied in [P122i4][PF6] through molecular dynamics (MD) simulations.16 [P122i4][PF6] exhibits three solid–solid-phase transitions, at 303, 353, and 393 K, before it starts to melt at around 418 K.17 There are two aspects to the effect of increasing temperature, from low to intermediate temperature, on CO2 uptake in an OIPC. Increasing temperature will reduce CO2 adsorption on the surface of the OIPC, but due to the increase in free volume in the higher temperature OIPC phase, e.g. 313 K (40 °C) in the case of [P122i4][PF6], the CO2 absorption increases significantly in the bulk phase.16
To compare the absorption capability of an OIPC for different gases, one can calculate the number density of the gases permeating into the bulk OIPC phase after a period of simulation time, through molecular dynamics (MD) simulations. However, the simulation time can affect the accuracy of this method. If the absorption process is slow, it may not be accessible via MD simulations within a reasonable time scale. In this case, the selectivity of the gas can be predicted by directly calculating the potential of mean force (PMF) of the gas molecules permeating into the OIPC. Herein, two computational methods, including the average force method and the adaptive biasing force (ABF) method,18,19 were used to calculate the PMF. These methods have previously been applied to study the energy barrier associated with CO2 crossing the IL interface.20–22 Our work demonstrates that both methods can qualitatively evaluate the gas uptake capability in an OIPC. A comparison of the two methods and their advantages and limitations are discussed.
Methods and computational details
In order to estimate the feasibility of the transport of a gas molecule into an OIPC, free energy can be calculated through the average force method, wherein a solute molecule (e.g. a gas molecule) is forced into the solvent (e.g. OIPC) and results in an energy cost of the process.22,23 The free energy surface along the transport pathway is referred to as the PMF. The PMF is obtained by integrating the total force acting on a gas molecule along a specified path from A (gas phase) to B (OIPC phase) (in our simulation, this path is in the longest z-axis direction of the MD simulation box). A gas molecule is applied with a biasing force, such as a harmonic spring force, to constrain its movement along the specified path AB. The total force from the surrounding molecules is calculated, and the actual force required to calculate the PMF is then obtained by subtracting the biasing force from the total force. This procedure is repeated at various positions along the path AB, with the accuracy of the method depending on sufficient sampling.An alternative technique, the ABF method, does not use a predefined constraining force. Instead, the constraining force is calculated and applied during the simulation, based on the magnitude of the energy barrier encountered, hence the biasing force is adaptive. This method can flatten the energy landscape to allow gas molecules to diffuse faster in a short simulation time, so that it is easier to access different minimum energy states separated by high energy barriers, and this largely improves the sampling efficiency when exploring the potential energy surface through MD simulations.24 Both methods have been applied and are discussed in this work.
Another way to investigate the free energy profile Δμex connected with moving the gas molecule from the gas phase into the OIPC phase is through the equilibrium gas concentrations, based on eqn (1):23,25
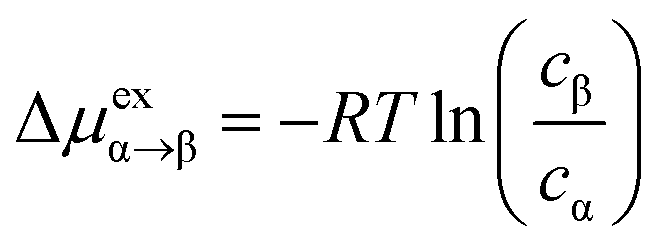 | (1) |
Computational details
The MD simulation procedure follows our previous studies with detailed description of the force field.16,26 The CHARMM force field functional was adopted for the OIPC with the parameters described in the work by Chen et al. (atomic charges27& refined parameters for others28). The CO2, N2, and O2 were modeled as rigid molecules. The parameters for both CO2 and N2 were from Potoff and Siepmann.29 The parameters for O2 and CH4 were adopted from Vujić and Lyubartsev30,31 and Sun et al.,31 respectively. The modelling OIPC system is a 2 × 2 × 8 supercell generated from a unit cell obtained by X-ray diffraction at 123 K,17 with 256 [P122i4][PF6] ion pairs and has a longer z-side, around 12–13 nm. The OIPC system is heated gradually from 123 K to 275 K and 325 K at a temperature step of 50 K in the isothermal-isobaric (NPT) ensemble and 1 atm pressure. Long equilibration calculations of 4–10 ns are conducted at 275 K and 325 K, respectively, in a NPT ensemble. Two vacuum regions are then introduced to both sides of OIPC with the z-length extended by at least 15 nm to ensure a sufficient separation so that the periodic condition can be applied. The system is equilibrated again at 275 K and 325 K in a canonical ensemble (NVT), until no further structural changes were observed in the solid. Four types of gases: CO2, N2, O2, and CH4 are introduced individually to the vacuum region at a pressure of 40 atm so that a quick gas permeation is allowed. The program large-scale atomistic and molecular massively parallel simulator or LAMMPS (version 2015) was used for all MD calculations.32 A cutoff of 1.5 nm was used for both Lennard–Jones and real space Coulomb potentials. The long-range electrostatic energies were treated using a particle–particle particle–mesh technique as implemented in LAMMPS. A time step of 2 fs was used. All calculations were carried out with periodic boundary condition imposed in all three directions. The velocity-Verlet integration algorithm is adopted by LAMMPS. The Nosé–Hoover barostat and thermostat were applied to control the pressure and temperature during simulations. The temperature (or pressure) during thermostatting (or barostatting) was relaxed every 2 ps.The free energy calculations were conducted at both 275 K and 325 K. In an average force method, a single gas molecule is constrained by a spring force along a reaction coordinate, perpendicular to the OIPC/gas interfacial XY plane, and the actual force acting on the gas molecule is calculated by subtracting the spring force from the total force of surrounding molecules on the gas molecule. The actual force is then integrated along the reaction coordinate to obtain the PMF.
The calculations involving the ABF method were carried out using the Colvars module33 implemented in the LAMMPS. Two reference positions were chosen: the center of mass of a single gas molecule and the center of mass of the OIPC solid. The difference in the z coordinates of these two systems form the reaction coordinate along which the PMF is defined. Calculations are performed by constraining the test gas molecule within a window of certain Z length (equal to 10 or 15 Å). This is done by applying an artificial potential barrier of magnitude 20 kcal mol−1 at the end of this window. The simulation time for each window was at least 15 ns and the energies were calculated every 0.1 Å along the reaction coordinate.
Results and discussion
In order to detect the number of CO2 molecules inside the bulk phase of the OIPC [P122i4][PF6] (absorption), or at the OIPC–gas interface (adsorption), the number density of CO2 was calculated together with the number density of P2 and P1 (the phosphorus atoms in [P122i4]+ and [PF6]−, respectively, see insert of the Fig. 1b). These are presented in Fig. 1a within a z-distance of 35 Å across the interface (z = 0). The green map shows the distribution of CO2, which has a higher population at the interface of the OIPC and the gas. The oscillation in density distribution of P1 and P2 indicates the ordered arrangement of the cations and anions in the OIPC.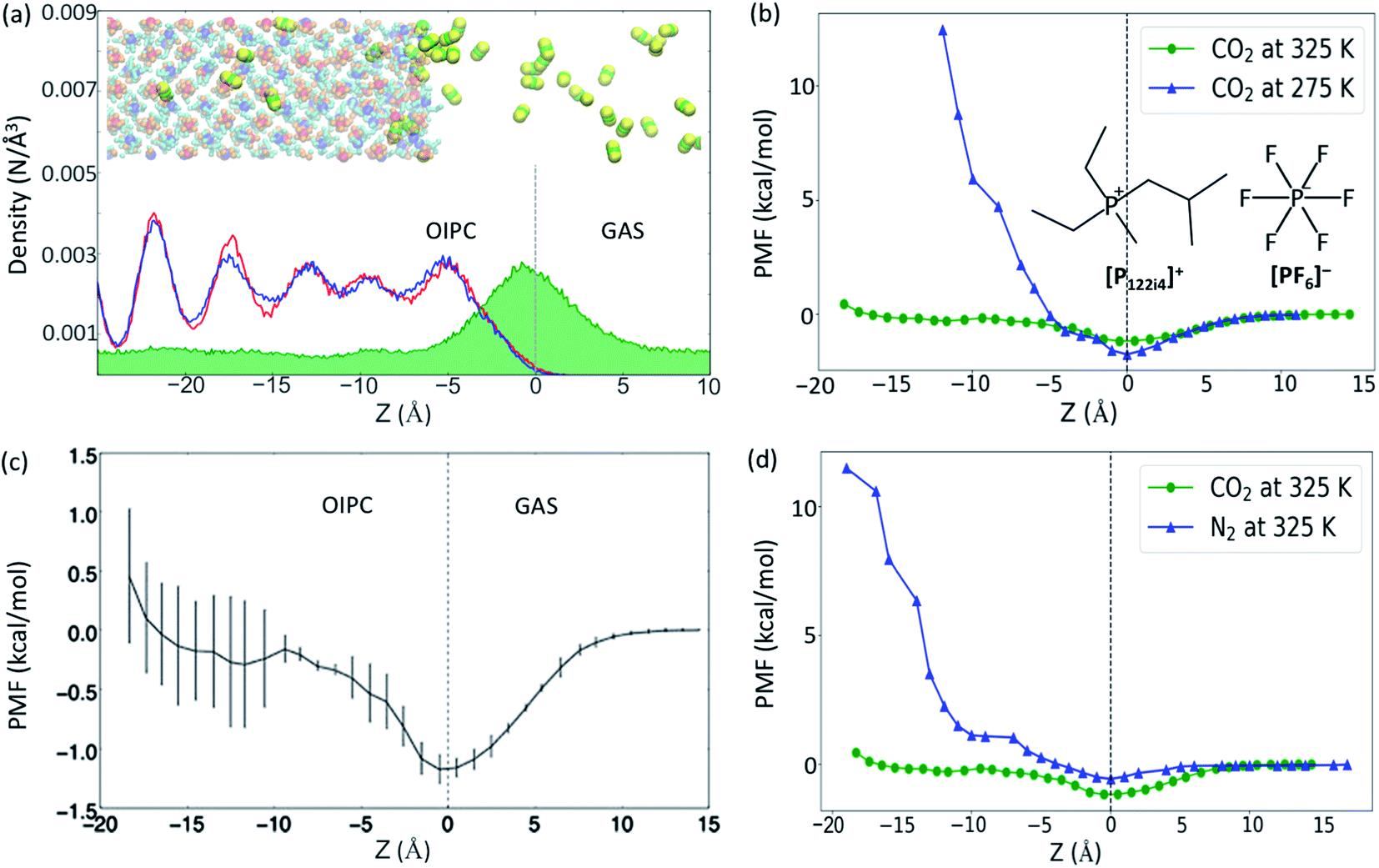 | ||
| Fig. 1 (a) Atomic number density profiles of CO2 (green) and OIPC cations (blue) and anions (red) calculated using the carbon atom in CO2 and the phosphorous atom of the cation and anion, respectively, at 325 K. The inset snapshot shows the OIPC/CO2 model at 325 K. (b) PMF plots of CO2 across the OIPC/gas interface at both 275 K and 325 K, and the inset shows the chemical structure of P122i4 and PF6 (c) Average PMF values acquired at 325 K from 3 independent simulations, showing the associated standard deviation error bars for CO2 across the OIPC/gas interface and (d) PMF plots of both CO2 and N2 at 325 K across the OIPC/gas interface. The PMF results here are all based on the average force method. | ||
The PMF was calculated first to quantitatively evaluate the absorption and adsorption capability of the OIPC at different temperatures, of 275 and 325 K, using the average force method. At 325 K, the OIPC [P122i4][PF6] is partially disordered, with higher free volume compared to the lower temperature of 275 K where the structure of the OIPC is more ordered.16 When moving a CO2 molecule from the gas phase into the OIPC phase, the PMF decreases initially as the CO2 molecule approaches the OIPC interface, as shown in Fig. 1b, and it reaches a negative minimum before it increases after the gas enters the OIPC solid. The negative minimum corresponds to the position of the OIPC–gas interface, where gas adsorption occurs, which appears as a peak in the CO2 density distribution profile in Fig. 1a. The decrease in the free energy indicates that the adsorption of gas at the interface is a spontaneous process, and the more negative change suggests an easier adsorption process.
Previous studies on gas uptake in different OIPC phases have shown the different behavior of gas uptake at low and high temperatures both experimentally and computationally.15,16 At selected temperatures, CO2 absorption is more limited at 275 K than at 325 K, with the opposite trend observed for CO2 adsorption. This is due to the change of free volume and the result of entropic effects. At higher temperatures, the increase in free volume due to expansion of the OIPC and the more disordered interface allows more gas molecules to be accommodated in the OIPC, whereas the high temperature lowers the stability of adsorption on the solid surface. Following this initial result, the free energy changes for CO2 across the OIPC interface at the same two temperatures were also compared in Fig. 1b, which confirms these conclusions from an energy perspective. The PMF at the OIPC–gas interface (z = 0) is more negative at 275 K than at 325 K, indicating that CO2 adsorption at the OIPC surface is easier at 275 K, which results in a higher CO2 surface concentration. This is consistent with the results of the previous MD simulation, but at a different initial gas pressure of 26 atm, where the CO2 surface concentration was calculated to be 3.6 ± 0.04 and 1.4 ± 0.06 μmol m−2 at 275 and 325 K, respectively.15,16 However, the PMF significantly increases after the CO2 reaches the bulk phase of the OIPC, where at a distance of around 11 Å from the interface the energy barrier is as large as 12 kcal mol−1 at 275 K. This is significantly higher than that at 325 K, suggesting more difficult absorption of CO2 at 275 K, and this is also consistent with the previous results.15,16
To assess the variability in PMF profiles acquired using the average force method, three independent 3 ns simulation runs were acquired and Fig. 1c demonstrates the average values and associated standard deviation highlighted by the error bars. Obviously, the standard deviation becomes greater further into the OIPC compared with that near the interface and in the gas phase. This may be due to the difference in the OIPC structure near the interface and inside the bulk phase, while the latter is more ordered (see inset picture of a snapshot of the OIPC/CO2 model at 325 K) and the OIPC near interface is more disordered. The free energy for gas absorption could change significantly due to different chemical environments at different positions. More precise results would require the average over longer simulation times, owing to the high solid density and subsequent slow dynamics, and/or an increase in the number of independent simulation runs.
To our knowledge, this is the first report of PMF calculations for gases in OIPCs. However, comparisons can be made with previous studies to model gas solubility in chemically similar ionic liquid systems. Dang et al.20 calculated the PMF for CO2 absorption in two ILs containing the cation 1-butyl-3-methylimidazolium [bmim]+ and two different anions, tetrafluoroborate [BF4]− and hexafluorophosphate [PF6]−, through MD simulations. While these results cannot be directly compared with results of this work due to different simulation conditions, changes in the PMF diagrams show similar behaviors when a CO2 molecule is moved from the gas phase to the IL phase, with a negative minimum also appearing at the IL/gas interface. The PMF curves of two ILs oscillate between −2 and 0.25 kcal mol−1 at 350 K. Compared with ionic liquids, the free energies may be higher in the OIPC system since the ILs normally have larger free volumes. Our results in Fig. 1c show that the highest PMF value is around 0.5 kcal mol−1 at 325 K.
To assess selectivity of the OIPC for CO2 and N2, a comparison of the gas intake behaviors was assessed at 325 K (Fig. 1d). Consistent with our previous simulation that there are more CO2 than N2 molecules both on the OIPC surface (5.99 ± 0.04 vs. 3.8 ± 0.06 μmol m−2) and in the bulk OIPC (287 ± 8 vs. 0 mol m−3).16 The PFM of the CO2 gas is lower both near z = 0 and in the whole range of z < 0 than that of the N2. These results suggest good selectivity of this OIPC for CO2 over N2, which is supported by our previous simulation and experimental results.15,16 The dramatic difference in selectivity between the two gases is attributed to the different interaction strengths between the gas molecules and the OIPC ions. CO2 interacts with OIPC ions more strongly than N2 does, as suggested by our previous density functional theory calculation of their binding energy [(Eb(P122i4–CO2) = −3.64, Eb(P122i4–N2) = −1.81; Eb(PF6–CO2) = −3.29, Eb(PF6–N2) = −1.11, in kcal mol−1)].26 Therefore, this facilitates the absorption of CO2 over N2, as supported by the low free energy change of the CO2 absorption.
In our previous MD study, we also compared gas absorption of CO2, CH4, O2 and N2 in the OIPC [P122i4][PF6]16 in order to study its gas separation capability. Different gas absorption capabilities were identified following the order of CO2 > CH4 > O2 > N2, and the last three gases showed relatively low absorption compared to CO2. Here, we have calculate the free energy change of intake of these four gases using the ABF method to assess the method in predicting gas absorption and selectivity. Due to the use of more adequate data for averaging, the method is expected to achieve more accurate results, which will be beneficial for capturing subtle differences in the PMFs, such as between CH4 and O2. The PMF profiles were calculated at 325 K for a z-distance of 30 Å (from 50 to 80 Å with the interface at 72 Å) across the interface. In addition, the change in free energy was also calculated from the previously obtained gas concentration profiles at the same temperature across the whole OIPC box, based on eqn (1). The results are presented in Fig. 2a, and are compared with the ABF results for the same z-range of 50–80 Å near the interface in Fig. 2b.
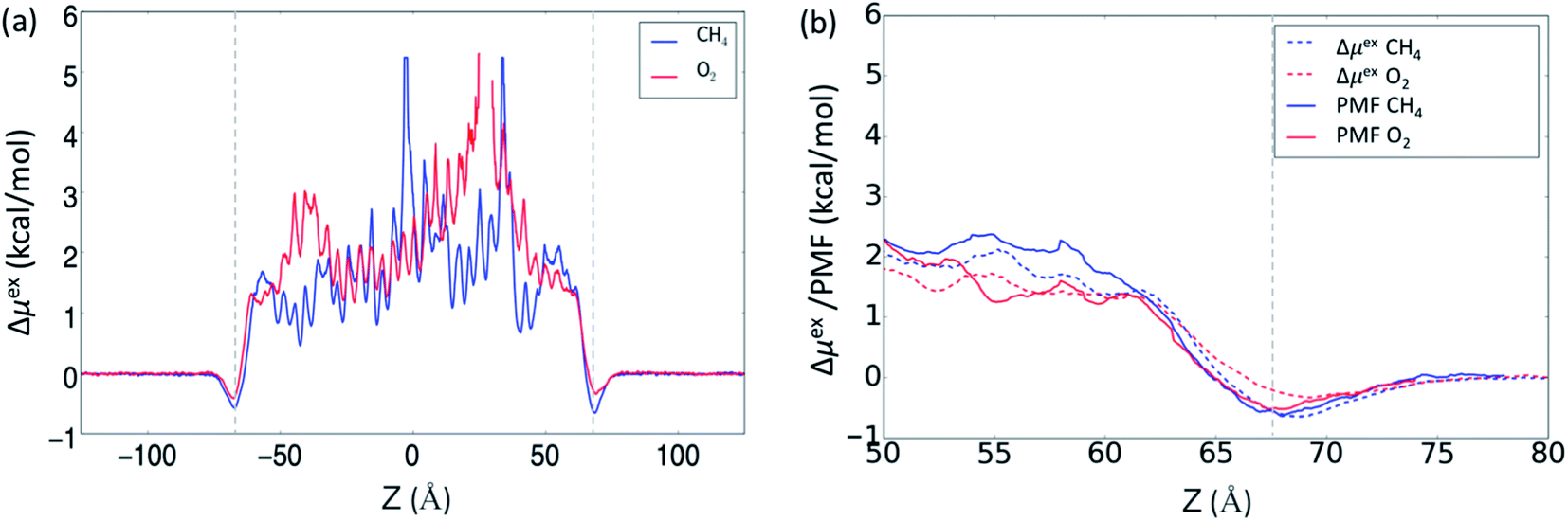 | ||
| Fig. 2 (a) The change in free energy (Δμ) for CH4 and O2 at 325 K calculated using the gas concentration across the whole OIPC z-range described by eqn (1); (b) comparison of the free energy change calculated using the ABF method with the method based on eqn (1), in a z-range of 50 to 80 Å with the interface at z = 72 Å. | ||
In Fig. 2b, the PMF profile of CH4 is slightly lower than that of O2 at the interface (marked by the vertical dotted line in Fig. 2a), and this is consistent with the previous calculated surface gas concentration: 2.5 ± 0.1 and 1.7 ± 0.1 μmol m−2 for CH4 and O2, respectively.16 When the gas molecules enter the OIPC phase, the free energy is very similar for both gases, before becoming slightly higher for CH4 compared to O2 (by about 1 kcal mol−1) around 12 Å from the interface. This difference is also observed when the free energy is calculated from the gas concentration profile, as shown in Fig. 2b. The lower energy barrier suggests that O2 absorption near the OIPC interface is more favorable compared to that of CH4. However, in the previous MD investigation, a higher bulk phase gas concentration of 105 ± 9 mol m−3 was reported for CH4 compared to 62 ± 4 mol m−3 for O2. This suggests that the gas absorption capability of those two gases will change further inside the OIPC bulk phase, where O2 could encounter a higher energy barrier. This inference is supported by the evaluation of free energy profiles based on gas concentration profiles in the whole OIPC range in Fig. 2a. For example, O2 shows higher energy barriers than CH4 in the two different z-ranges. The average free energy changes of Δμex calculated throughout the whole solid, from Fig. 2b, are 2.2 and 1.8 kcal mol−1 for O2 and CH4, respectively, confirming the overall lower absorption of O2 in a broader range.
These results are significant because they suggest that the relative PMF between two gases are not always consistent across the OIPC solid: it can be affected by the structure order between the interface area and the bulk phase area or different chemical environments that are encountered through the solid. The outer OIPC layers near the interface are more disordered, and the PMF barrier of O2 is lower than that of CH4, whereas the opposite situation could be present in the more ordered bulk phase of the OIPC. Furthermore, since the OIPC has a layered ordered structure, different layers of the ionic composition could also have divergent effects on the absorption of different gases. These factors all require further investigation. Nevertheless, these results indicate that it is sometimes inaccurate to assess different gas absorption capabilities based only on the PMF curve close to the interface, especially when the difference in PMF profiles between two gases is not distinct. In this case, the calculation should be extended to a longer distance into the bulk phase or use the entire range of the OIPC model in the MD simulation.
Lastly, in Fig. 3 we compare the ABF and average force methods for the low gas solubility cases, i.e. CO2 at 275 K and N2 at 325 K. Fig. 3a shows the PMF profiles for CO2 at 275 K, where the benefits of the ABF method are demonstrated by its ability to capture the details of small energy barriers, and these undulations are not present in the trace obtained using the average force method. Fig. 3b show the large variance between the PMF profiles from two methods for the N2 case at 325 K, which may be attributed to insufficient sampling of the phase space when using the average force method. The study of O2 and CH4 demonstrated that the free energy profiles obtained from the ABF method were more consistent with the method based on gas concentration, which could indicate that the ABF method is a more accurate reflection of the gas absorption. Nevertheless, both methods adopted in this work have been demonstrated to qualitatively reflect the difference in gas absorption and adsorption between different gases.
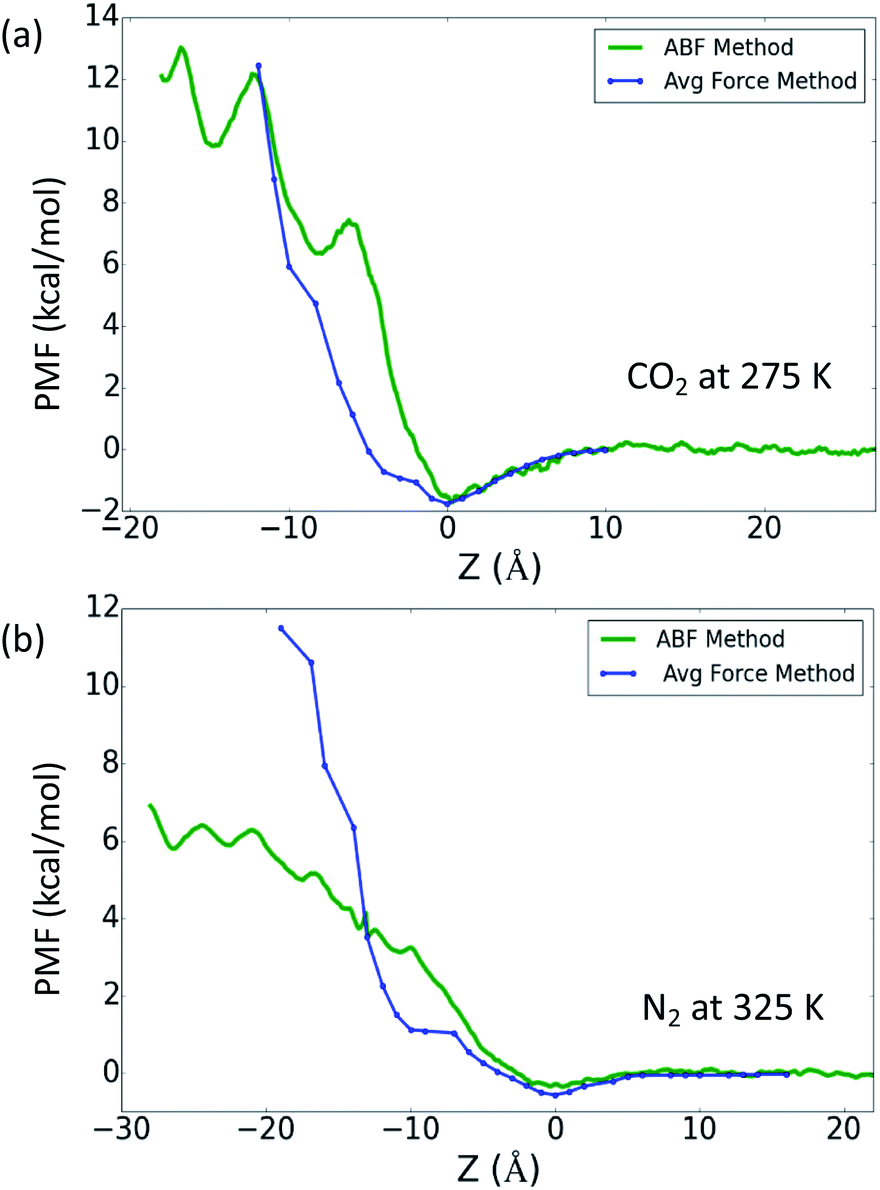 | ||
| Fig. 3 PMF profiles for (a) CO2 at 275 K and (b) N2 at 325 K, calculated from both the ABF and average force methods. | ||
Conclusions
Two free energy methods, namely average force and adaptive biasing force, were used to calculate the potential of mean force of intake of four different gases: CO2, N2, CH4 and O2 across the gas/OIPC interface, in order to evaluate gas adsorption, absorption and selectivity in an OIPC [P122i4][PF6]. The free energy results qualitatively support the differences in gas absorptivity obtained from previous experimental analysis and MD simulations. The results show that the gas adsorption on the OIPC surface is a spontaneous process and that low temperature is beneficial to this process. The difference in the free energy barrier inside the OIPC bulk phase accurately distinguishes the difficulty of absorption of different gases into the OIPC, as well as reflects the effect of temperature and the different OIPC phase structures. When comparing performance of the two methods for predicting gas selectivity in OIPCs, the performance of the ABF method was overall superior to the average force method, especially in providing the more discreet details relating to small energy barriers. Our results also suggest that the evaluation of different gas absorption capabilities through the PMF calculations should sometimes be conducted across the entire OIPC phase, because the difference in structural order of the OIPC between the bulk phase and the interface, as well as the different compositions of ion layers, could affect the gas absorption differently. These PMF and ABF methods were demonstrated to be successful in comparing the intake process of different gases into an OIPC, and they should also be useful to study other solid materials for gas separation applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the funding support from Australian Research Council (ARC) through CE140100012 and FL110100013. The simulation work was undertaken with the assistance of resources provided at the NCI National Facility systems at the Australian National University through the National Computational Merit Allocation Scheme supported by the Australian Government.References
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Green processing using ionic liquids and CO2, Nature, 1999, 399(6731), 28–29 CrossRef.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, Why Is CO2 So Soluble in Imidazolium-Based Ionic Liquids?, J. Am. Chem. Soc., 2004, 126(16), 5300–5308 CrossRef CAS PubMed.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Solvent–solute interactions in ionic liquids, Phys. Chem. Chem. Phys., 2003, 5(13), 2790–2794 RSC.
- S. G. Kazarian, B. J. Briscoe and T. Welton, Combining ionic liquids and supercritical fluids: ATR-IR study of CO dissolved in two ionic liquids at high pressures, Chem. Commun., 2000,(20), 2047–2048 RSC.
- K. Dong, S. Zhang, D. Wang and X. Yao, Hydrogen Bonds in Imidazolium Ionic Liquids, J. Phys. Chem. A, 2006, 110(31), 9775–9782 CrossRef CAS PubMed.
- H. Gao, L. Bai, J. Han, B. Yang, S. Zhang and X. Zhang, Functionalized ionic liquid membranes for CO2 separation, Chem. Commun., 2018, 54(90), 12671–12685 RSC.
- D. Shang, X. Liu, L. Bai, S. Zeng, Q. Xu, H. Gao and X. Zhang, Ionic liquids in gas separation processing, Curr. Opin. Green Sustain. Chem., 2017, 5, 74–81 CrossRef.
- M. Zia ul Mustafa, H. bin Mukhtar, N. A. H. Md Nordin, H. A. Mannan, R. Nasir and N. Fazil, Recent Developments and Applications of Ionic Liquids in Gas Separation Membranes, Chem. Eng. Technol., 2019, 42(12), 2580–2593 CrossRef CAS.
- H. Li, L. Tuo, K. Yang, H.-K. Jeong, Y. Dai, G. He and W. Zhao, Simultaneous enhancement of mechanical properties and CO2 selectivity of ZIF-8 mixed matrix membranes: Interfacial toughening effect of ionic liquid, J. Membr. Sci., 2016, 511, 130–142 CrossRef CAS.
- J. E. Bara, S. Lessmann, C. J. Gabriel, E. S. Hatakeyama, R. D. Noble and D. L. Gin, Synthesis and Performance of Polymerizable Room-Temperature Ionic Liquids as Gas Separation Membranes, Ind. Eng. Chem. Res., 2007, 46(16), 5397–5404 CrossRef CAS.
- J. E. Bara, C. J. Gabriel, E. S. Hatakeyama, T. K. Carlisle, S. Lessmann, R. D. Noble and D. L. Gin, Improving CO2 selectivity in polymerized room-temperature ionic liquid gas separation membranes through incorporation of polar substituents, J. Membr. Sci., 2008, 321(1), 3–7 CrossRef CAS.
- J. Tang, W. Sun, H. Tang, M. Radosz and Y. Shen, Enhanced CO2 Absorption of Poly(ionic liquid)s, Macromolecules, 2005, 38(6), 2037–2039 CrossRef CAS.
- J. Tang, H. Tang, W. Sun, H. Plancher, M. Radosz and Y. Shen, Poly(ionic liquid)s: a new material with enhanced and fast CO2 absorption, Chem. Commun., 2005,(26), 3325–3327 CAS.
- L. C. Tomé and I. M. Marrucho, Ionic liquid-based materials: a platform to design engineered CO2 separation membranes, Chem. Soc. Rev., 2016, 45(10), 2785–2824 RSC.
- J. L. McDonald, D. R. MacFarlane, M. Forsyth and J. M. Pringle, A novel class of gas separation membrane based on organic ionic plastic crystals, Chem. Commun., 2016, 52(88), 12940–12943 RSC.
- V. S. Kandagal, F. Chen, J. M. Pringle and M. Forsyth, Atomistic Simulation of Gas Uptake and Interface-Induced Disordering in Solid Phases of an Organic Ionic Plastic Crystal, J. Phys. Chem. B, 2018, 122(34), 8274–8283 CrossRef CAS PubMed.
- L. Jin, K. M. Nairn, C. M. Forsyth, A. J. Seeber, D. R. MacFarlane, P. C. Howlett, M. Forsyth and J. M. Pringle, Structure and Transport Properties of a Plastic Crystal Ion Conductor: Diethyl(methyl)(isobutyl)phosphonium Hexafluorophosphate, J. Am. Chem. Soc., 2012, 134(23), 9688–9697 CrossRef CAS PubMed.
- E. Darve and A. Pohorille, Calculating free energies using average force, J. Chem. Phys., 2001, 115(20), 9169–9183 CrossRef CAS.
- E. Darve, D. Rodríguez-Gómez and A. Pohorille, Adaptive biasing force method for scalar and vector free energy calculations, J. Chem. Phys., 2008, 128(14), 144120 CrossRef PubMed.
- L. X. Dang and C. D. Wick, Anion Effects on Interfacial Absorption of Gases in Ionic Liquids. A Molecular Dynamics Study, J. Phys. Chem. B, 2011, 115(21), 6964–6970 CrossRef CAS PubMed.
- C. D. Wick, T.-M. Chang and L. X. Dang, Molecular Mechanism of CO2 and SO2 Molecules Binding to the Air/Liquid Interface of 1-Butyl-3-methylimidazolium Tetrafluoroborate Ionic Liquid: A Molecular Dynamics Study with Polarizable Potential Models, J. Phys. Chem. B, 2010, 114(46), 14965–14971 CrossRef CAS PubMed.
- M. E. Perez-Blanco and E. J. Maginn, Molecular Dynamics Simulations of CO2 at an Ionic Liquid Interface: Adsorption, Ordering, and Interfacial Crossing, J. Phys. Chem. B, 2010, 114(36), 11827–11837 CrossRef CAS PubMed.
- R. Vácha, P. Slavíček, M. Mucha, B. J. Finlayson-Pitts and P. Jungwirth, Adsorption of Atmospherically Relevant Gases at the Air/Water Interface: Free Energy Profiles of Aqueous Solvation of N2, O2, O3, OH, H2O, HO2, and H2O2, J. Phys. Chem. A, 2004, 108(52), 11573–11579 CrossRef.
- J. Comer, J. C. Gumbart, J. Hénin, T. Lelièvre, A. Pohorille and C. Chipot, The Adaptive Biasing Force Method: Everything You Always Wanted To Know but Were Afraid To Ask, J. Phys. Chem. B, 2015, 119(3), 1129–1151 CrossRef CAS PubMed.
- A. Ben-Naim and Y. Marcus, Solvation thermodynamics of nonionic solutes, J. Chem. Phys., 1984, 81(4), 2016–2027 CrossRef CAS.
- V. S. Kandagal, F. Chen, E. Jónsson, J. M. Pringle and M. Forsyth, Molecular simulation study of CO2 and N2 absorption in a phosphonium based organic ionic plastic crystal, J. Chem. Phys., 2017, 147(12), 124703 CrossRef PubMed.
- F. Chen, L. Jin, S. W. de Leeuw, J. M. Pringle and M. Forsyth, Atomistic simulation of structure and dynamics of the plastic crystal diethyl(methyl)(isobutyl)phosphonium hexafluorophosphate, J. Chem. Phys., 2013, 138(24), 244503 CrossRef CAS PubMed.
- F. Chen, J. M. Pringle and M. Forsyth, Insights into the Transport of Alkali Metal Ions Doped into a Plastic Crystal Electrolyte, Chem. Mater., 2015, 27(7), 2666–2672 CrossRef CAS.
- J. J. Potoff and J. I. Siepmann, Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen, AIChE J., 2001, 47(7), 1676–1682 CrossRef CAS.
- B. Vujić and A. P. Lyubartsev, Transferable force-field for modelling of CO2, N2, O2and Ar in all silica and Na+exchanged zeolites, Modell. Simul. Mater. Sci. Eng., 2016, 24(4), 045002 CrossRef.
- Y. Sun, D. Spellmeyer, D. A. Pearlman and P. Kollman, Simulation of the solvation free energies for methane, ethane, and propane and corresponding amino acid dipeptides: a critical test of the bond-PMF correction, a new set of hydrocarbon parameters, and the gas phase-water hydrophobicity scale, J. Am. Chem. Soc., 1992, 114(17), 6798–6801 CrossRef CAS.
- S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys., 1995, 117(1), 1–19 CrossRef CAS.
- G. Fiorin, M. L. Klein and J. Hénin, Using collective variables to drive molecular dynamics simulations, Mol. Phys., 2013, 111(22–23), 3345–3362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |