
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoclusters as prospective carriers and detectors of pramipexole†
Nguyen Thanh Si *ab,
Nguyen Thi Ai Nhungc,
Thanh Q. Buic,
Minh Tho Nguyend and
Pham Vu Nhat*e
*ab,
Nguyen Thi Ai Nhungc,
Thanh Q. Buic,
Minh Tho Nguyend and
Pham Vu Nhat*e
aComputational Chemistry Research Group, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: nguyenthanhsi@tdtu.edu.vn
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cDepartment of Chemistry, University of Sciences, Hue University, Hue, Vietnam
dInstitute for Computational Science and Technology (ICST), Ho Chi Minh City, Vietnam
eDepartment of Chemistry, Can Tho University, Can Tho, Vietnam. E-mail: nhat@ctu.edu.vn
First published on 5th May 2021
Abstract
Pramipexole (PPX) is known in the treatment of Parkinson's disease and restless legs syndrome. We carried out a theoretical investigation on pramipexole–Au cluster interactions for the applications of drug delivery and detection. Three AuN clusters with sizes N = 6, 8 and 20 were used as reactant models to simulate the metallic nanostructured surfaces. Quantum chemical computations were performed in both gas phase and aqueous environments using density functional theory (DFT) with the PBE functional and the cc-pVDZ-PP/cc-pVTZ basis set. The PPX drug is mainly adsorbed on gold clusters via its nitrogen atom of the thiazole ring with binding energies of ca. −22 to −28 kcal mol−1 in vacuum and ca. −18 to −24 kcal mol−1 in aqueous solution. In addition to such Au–N covalent bonding, the metal–drug interactions are further stabilized by electrostatic effects, namely hydrogen-bond NH⋯Au contributions. Surface-enhanced Raman scattering (SERS) of PPX adsorbed on the Au surfaces and its desorption process were also examined. In comparison to Au8, both Au6 and Au20 clusters undergo a shorter recovery time and a larger change of energy gap, being possibly conducive to electrical conversion, thus signaling for detection of the drug. A chemical enhancement mechanism for SERS procedure was again established in view of the formation of nonconventional hydrogen interactions Au⋯H–N. The binding of PPX to a gold cluster is expected to be reversible and triggered by the presence of cysteine residues in protein matrices or lower-shifted alteration of environment pH. These findings would encourage either further theoretical probes to reach more accurate views on the efficiency of pramipexole–Au interactions, or experimental attempts to build appropriate gold nanostructures for practical trials, harnessing their potentiality for applications.
1. Introduction
In recent years, nanomaterials have become more and more widely used in biomedical fields as a promising approach for the design of drug delivery systems, diagnosis agents, imaging and biosensors.1,2 Numerous nanostructures are applicable in medicine thanks to their peculiar properties that greatly differ from those observed in fine particles or bulk materials. Generally, nanomaterials relevant for biomedical applications can be categorized as either those with organic origins including liposomes,3–5 dendrimers,6,7 and polymers,8,9 or inorganic backgrounds such as noble nanoparticles (NPs),10 iron oxide NPs,11 and other inorganic compounds.12–14 In terms of chemical stability, environmental compatibility and high mechanical strength, the inorganic groups are likely to prevail over the organic counterparts.15–17Of the common inorganic nanostructures, those consisting of gold have peaked much interest as they exhibit valuable impacts and clear-cut advantages.18 Firstly, gold nanoparticles (AuNPs) are highly compatible with a variety of bimolecular systems and exhibit a much lower inherent toxicity to humans than many others.19 They also pose a remarkable stability and are willing to be functionalized by a number of biological systems, such as drugs, genes and targeting ligands.20 Moreover, AuNPs with various sizes/shapes, i.e. spherical, rod-like, cage-like forms and so on, can be easily synthesized.21 Such particles enhance optical properties, distinctive surface and macroscopic quantum tunneling effects, along with surface plasmon resonance (SPR) phenomenon,22 and are able to penetrate through the cell membrane without creating pores on the cell membrane.23 As a result, AuNPs become one of the most efficient materials for various biomedical applications including bio-sensing, molecular imaging, drug delivery, etc.24–26 The presence of gold particles in drugs results in several beneficial outcomes, i.e. enhancing the therapeutic effect of the drug,27 allowing an effective drug delivery28 in increasing the therapeutic retention time in the circulation,29 and improving the target specificity of treatments.19,30
Numerous theoretical and experimental studies have been carried out to validate the advantages of NPs by exploring the nature of their interactions with drugs and biomolecules, as well as to elucidate inherent physicochemical properties.31 At the molecular scale, metallic cluster models are commonly used to examine the adsorption/desorption mechanism of a molecule on the nanoparticle surface. In a recent study,32 we demonstrated that gold nanoclusters exhibit promising characteristics of bio-sensing and targeted drug delivery of some thione-containing drugs. The mercaptopurine and thioguanine binding on small gold clusters was found to be reversible processes with adsorption energies ∼34 to 40 kcal mol−1 in vacuum and ∼28 to 32 kcal mol−1 in aqueous solution. The drugs are in addition able to detach from the gold surface due to either a slight change of pH in tumor cells, or a presence of cysteine residues in protein matrices. Besides, the gold clusters undergo a rather short recovery time and a large change of energy gap, which could be conducive to electrical signaling conversion for selective detection of the drugs. In addition, gold nanoclusters typically provide a suitable surface for loading various therapeutic agents, such as small biomolecules, peptides, proteins, and nucleic acids.14 El-Mageed and co-workers33 also examined the interactions of D-penicillamine with some small gold clusters (Aun, n = 2, 4, and 6) and observed rather strong interactions between them which are governed by the anchoring Au–O/S/N bond, unconventional hydrogen Au⋯H–S bonding, and electrostatic Au⋯H–C coupling. Likewise, a variety of drugs and biomolecules such as methimazole,34 tamoxifen,35 amino acids,36 DNA bases,31,37,38 curcumin,39 pectin,40 and others31 have also been found to strongly be adsorbed on the surface of gold nanoclusters with significant electronic responses.
Pramipexole (PPX), whose structural formula is shown in Fig. 1, is commonly commercialized under the Mirapex brand name along with several other ingredients to treat the Parkinson's disease and restless legs syndrome.41 However, the therapeutic intake of this compound may result in many undesirable effects such as nausea, headache, fatigue, including heart failure and hypotension.42–44 Use of this drug during pregnancy and lactation is of unclear safety.45 Therefore, it is of importance to find a suitable carrier to deliver the drug where it is needed, with the aim to improve its therapeutic efficacy. In addition, design of simple and powerful sensors for selective detection of PPX is also of great interest. In this context we set out to investigate in the present theoretical work using quantum chemical calculations as an attempt to reveal the potentiality of using gold nanostructures for detection and delivery of PPX. Small AuN clusters including N = 6, 8, and 20 are considered as reactant models to simulate the surfaces of gold nanoparticles. Structural features, energetic properties, along with spectroscopic and electronic properties of the resulting complexes were determined by DFT calculations and analyzed in detail. Effects of aqueous solution are evaluated using the continuum IEF-PCM model. The triggering factors for drug release are also investigated. In particular, a chemical enhancement mechanism for the surface-enhanced Raman scattering (SERS) spectroscopy is again demonstrated on the basis of the collected results in an effort to provide us with deeper insights into the surface enhanced Raman phenomenon.
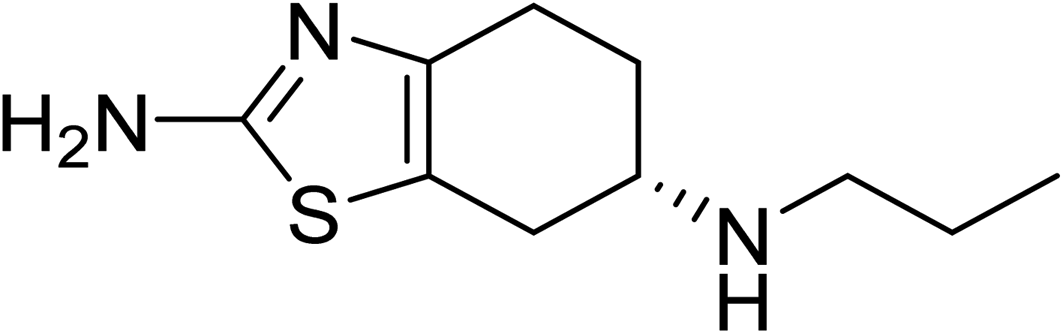 | ||
| Fig. 1 The chemical structure of PPX molecule. | ||
2. Computational methods
Quantum chemical calculations for all studied compounds are performed using the Gaussian 16 package.46 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation method47 is mainly used throughout this work in conjunction with a mixed basis set of the effective core potential (ECP) cc-pVDZ-PP48 for gold atoms and the cc-pVTZ for PPX. The PBE functional and the cc-pVDZ-PP/cc-pVTZ basis sets have been shown to describe properly the interactions between gold atoms and the sulfur-containing molecules.32,49 The effect of solvent, i.e. an aqueous solution in this study, is simulated using the continuum model known as the Integral Equation Formalism-Polarizable Continuum Model (IEF-PCM).50 To carry out the electronic density of states (DOS) simulations, the GaussSum program51 is used. Concerning the dispersion correction, the Grimme-style D3 is not used in this study as a sufficient convergence can be reached by the current level of theory. Otherwise, hitherto no experimental evidence is available to confirm either the optimized structures or the calculated energies, thus no experiment–theory comparison can be made so far.The value of binding energies (Eb), which is defined as the total energy difference between that of AuN–PPX complexes and those of isolated species, is computed using the following expression (1):
| Eb = EAuN–PPX − (EAuN + EPPX) | (1) |
The enthalpy and Gibbs free energy of the interaction are calculated as follows:
| ΔH° = (E + Hcorr)AuN–PPX − [(E + Hcorr)AuN + (E + Hcorr)PPX] | (2) |
| ΔG° = (E + Gcorr)AuN–PPX − [(E + Gcorr)AuN + (E + Gcorr)PPX] | (3) |
Energies of frontier orbitals HOMO/LUMO, the HOMO–LUMO gap (Eg), and the change of Eg upon the binding of the drug to gold clusters are also determined to evaluate the effect of interacting species on each other. The energy gap Eg is a useful parameter for determining the kinetic reactivity of materials,53 and its change upon the adsorption process can be used to evaluate the sensitivity of an adsorbent to an adsorbate. For the frontier orbital energy gaps and excitation energy predictions, we employ the long-range correction LC-BLYP functional, which composes of the Becke 1988 (B88) exchange and Lee–Yang–Parr correlation (BLYP)54–56 instead of using the pure GGA PBE functional. Previous studies proved that DFT approaches within the GGA framework, such as the PBE, tend to significantly underestimate the HOMO–LUMO gaps in many chemical systems and in solids as well.57,58 Such gaps are not internally consistent within different methods and greatly depend on the amount of the Hartree–Fock exchange.59 Some recently developed functionals with corrections for long-range interactions have been found to perform much better than the traditional GGA approaches in predicting the orbital energies.55,60 In fact, inclusion of long-range exchange effects in the LC-BLYP functional was demonstrated to improve the calculated orbital energies in noble clusters.32,58
3. Results and discussion
3.1 Structures and energetics
Muthu and co-workers61 carried out in 2013 a combined computational and experimental study on structural and spectroscopic properties of PPX, confirming that the molecule prefers a twisted form t-PPX as shown in Fig. 2. However, these authors were likely to overlook an important configuration that could be more stable. In vacuum, the straight conformation PPX (Fig. 2) is located to be around 1.2 kcal mol−1 above t-PPX, computed at either PBE/cc-pVTZ or B3LYP/6-311G(d,p) level of theory. The energy difference is slightly reduced to 1.0 kcal mol−1 in water solution. Henceforth, the former tautomer will be considered for most calculations and discussion in this study. For further information, some basic molecular properties of PPX such as vibrational (IR, Raman) spectra, energies of frontier orbitals, band gaps, proton affinities are also investigated and presented in Table S1 and Fig. S1 of the ESI file.†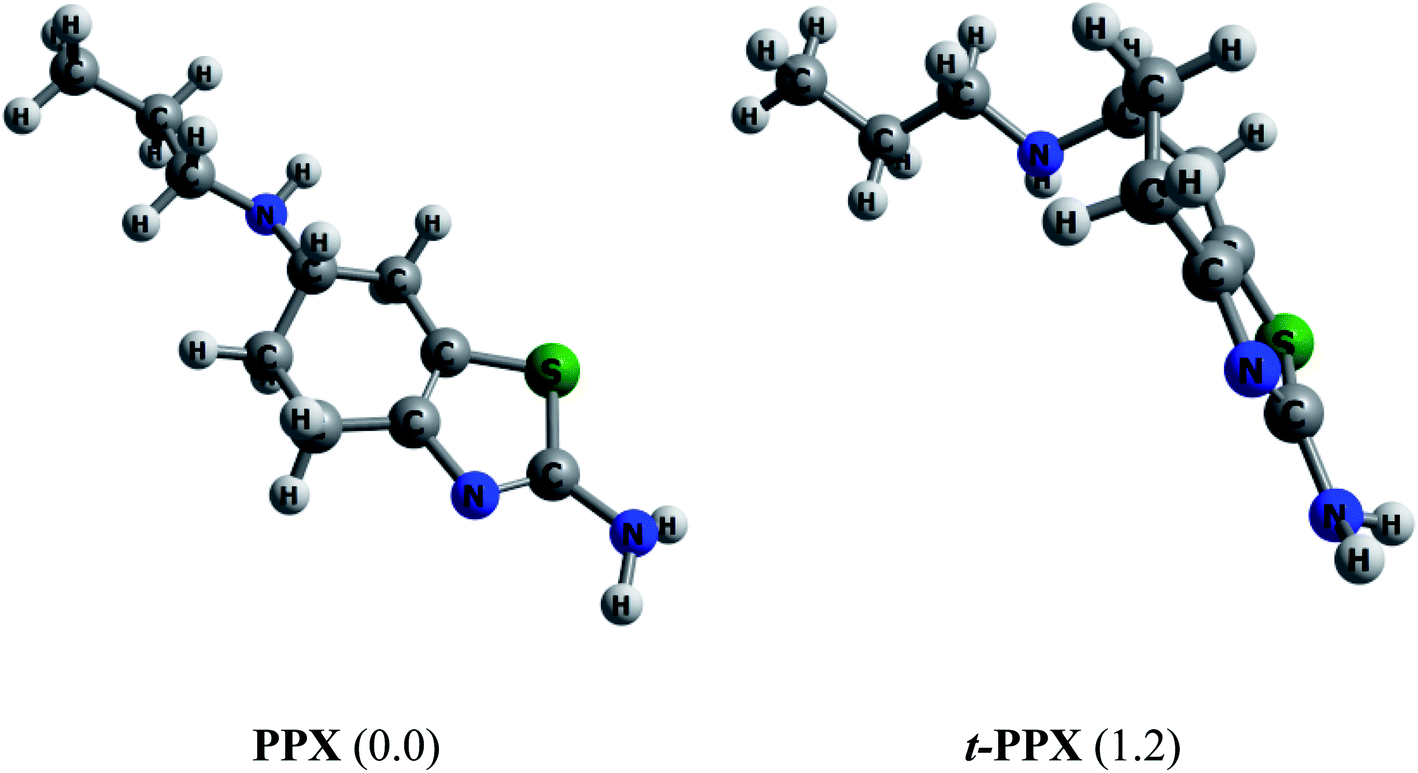 | ||
| Fig. 2 Equilibrium structures of two PPX lowest-energy conformations. Values in brackets are their relative energies (kcal mol−1) in gas phase (PBE/cc-pVTZ). | ||
The ground state structures of Au6 and Au8 are regular triangle (D3h) and square planar (D4h), respectively, while that of Au20 is a pyramidal structure with Td symmetry (Fig. 3).62 The gold clusters are expected to interact with PPX molecule through positions with high electron density, aka. sulfur and nitrogen atoms. It was found that both forward or backward donation are mostly important factors governing the interactions between S-containing drugs and gold clusters.32 Accordingly, gold clusters are not only able to receive electrons from heteroatom lone pairs but also able to donate back some negative charges to the antibonding orbitals of the PPX molecule. In addition, they can play an important role as a proton acceptor by forming a nonconventional Au⋯H–N hydrogen bond.63 An analysis of NBO charge (Fig. 3) for AuN clusters at the same theoretical level shows that the cornered Au atoms (green color) contain a more positive charge than those of others (red color). Therefore, the former is likely to be more suitable for nucleophilic attacks.
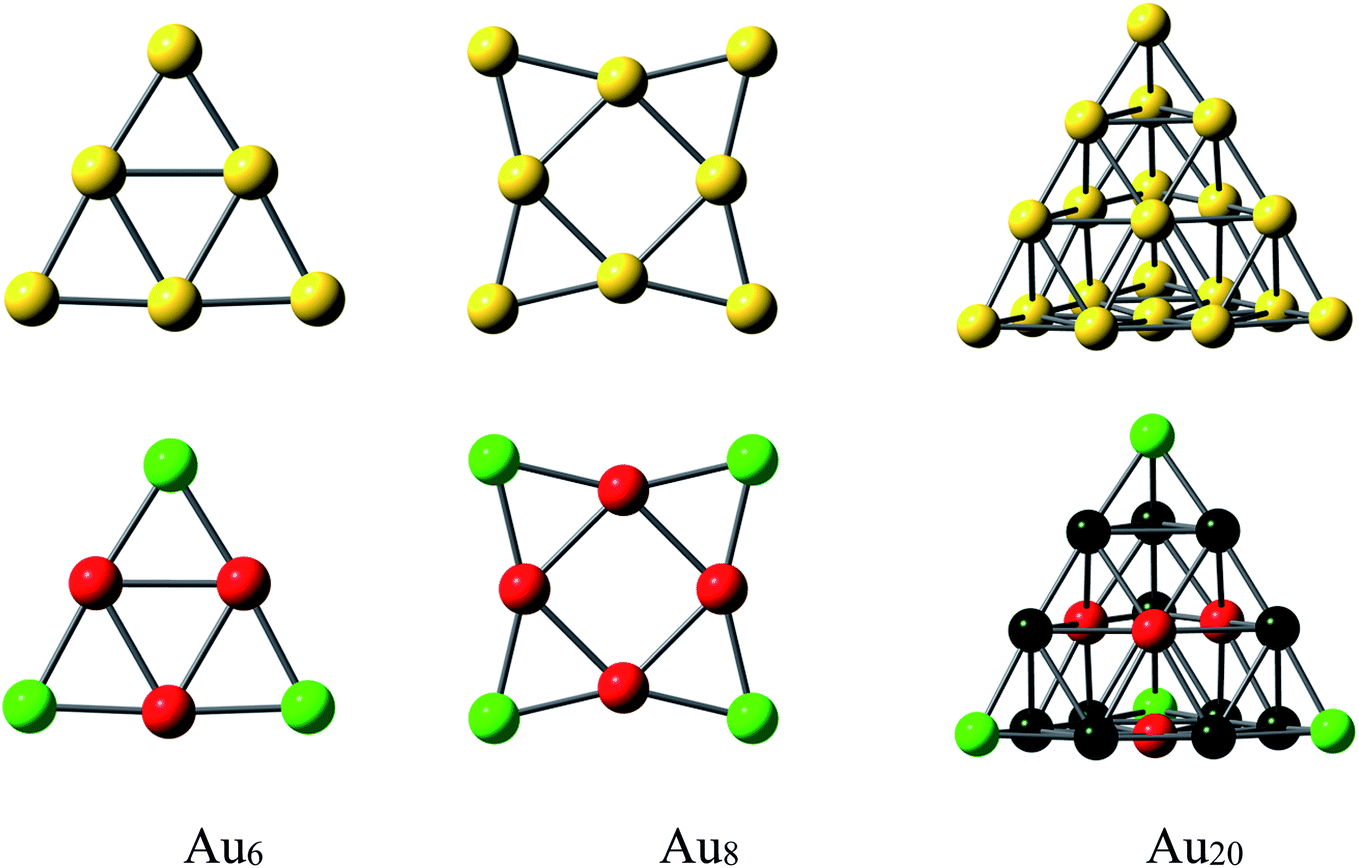 | ||
| Fig. 3 Equilibrium geometries and NBO charges of AuN clusters considered (N = 6, 8, 20). Color range in a.u.: green, more positive than 0.07; red, more negative than −0.07; black, less positive than 0.02. | ||
Lower-lying structures located for complexes of PPX and t-PPX tautomers with three AuN clusters are presented in Fig. 4–6. They are denoted as AuN–PPX_Z and AuN–t-PPX_Z, respectively, with N = 6, 8, 20 and Z = 1–4 being the number of isomers with increasing relative energy (kcal mol−1). For interaction with PPX tautomer, the Au6 cluster tends to anchor perpendicularly on the nitrogen atom of the thiazole ring, forming the most stable complex Au6–PPX_1 with a binding energy ca. −25 kcal mol−1. This is followed by Au6–PPX_2, whose the corresponding configuration is of direct binding the Au6 to the N atom of the secondary amine group. The energy gap between these two complexes is insignificant, being only ∼3 kcal mol−1 (PBE value). Another isomer Au6–PPX_3, which is constructed by anchoring the Au6 on the S atom of the thiazole, is computed to hold ∼7 kcal mol−1 higher in energy than Au6–PPX_1. The remaining isomer (Au6–PPX_4) with an Au6–NH2 bond is much less stable, indicated by the energy of ∼11 kcal mol−1 above that of the most stable form Au6–PPX_1. Similarly, the t-PPX tautomer also prefers to anchor on Au6 via the N atom of the thiazole giving rise to most stable complex Au6–t-PPX_1. Other lower-lying conformations are lying from 3–11 kcal mol−1 above Au6–t-PPX_1 (Fig. 4).
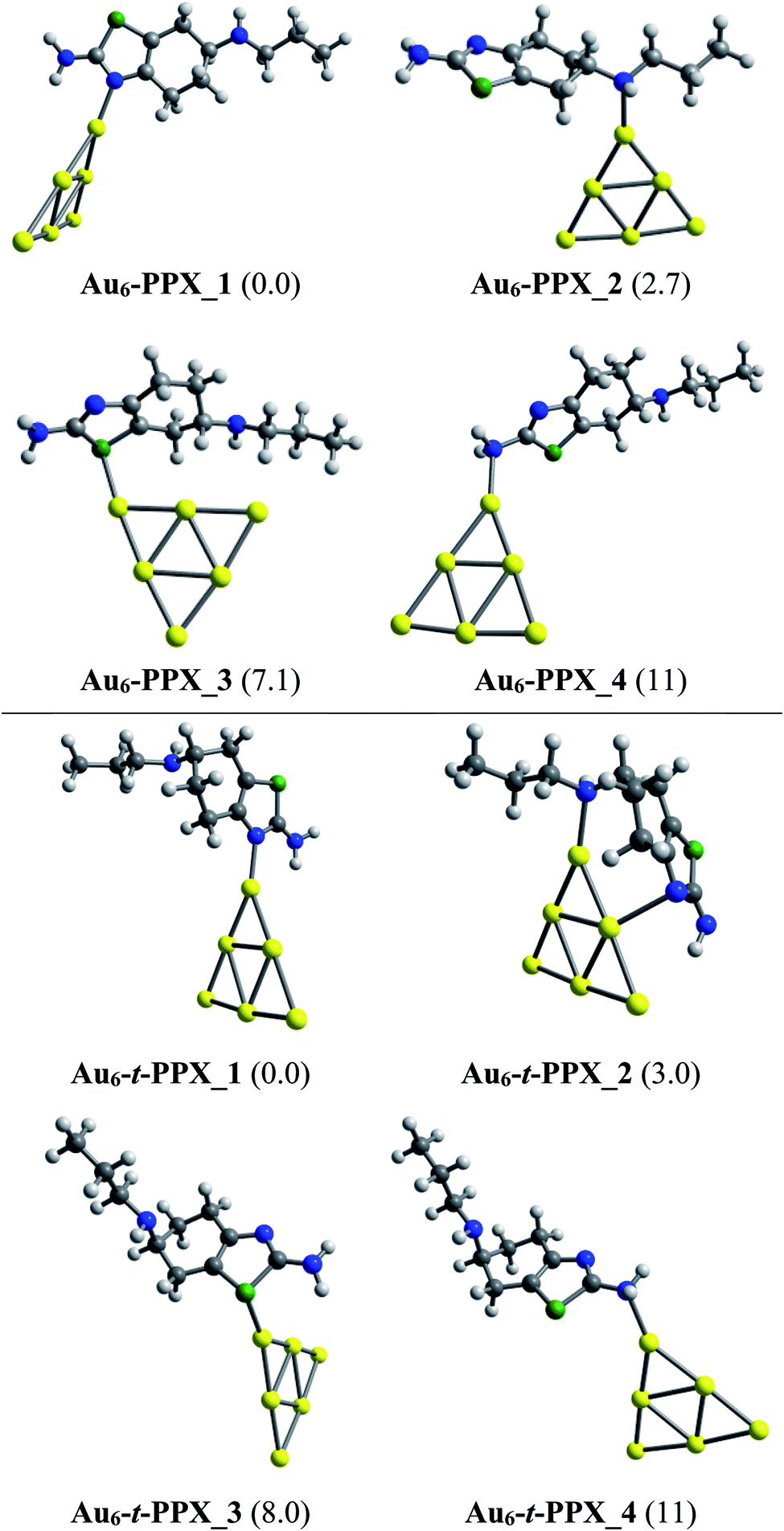 | ||
| Fig. 4 Lower-lying structures located for the Au6–PPX (above) and Au6–t-PPX (below) complexes in gas phase. Relative energies (kcal mol−1) to the lowest-energy isomer are given in parentheses. | ||
As in Au6–PPX, the nitrogen atoms of the thiazole ring and the secondary amine group are also the most active sites for the Au8–PPX interaction. As shown in Fig. 5, the lowest-energy Au8–PPX_1 and Au8–PPX_2 isomers have the same structural arrangements as observed from Au6–PPX_1 and Au6–PPX_2. At PBE/cc-pVDZ-P/cc-pVTZ level, the energy of Au8–PPX_2 is computed to be ∼3 kcal mol−1 above the corresponding figure for Au8–PPX_1. Other conformations, i.e. Au8–PPX_3 and Au8–PPX_4, which contain the Au6–S and Au6–NH2 bonds, are predicted to be around 8–12 kcal mol−1 higher in energy than that of the most stable form Au8–PPX_1. Contrary to the mono-dentate interactions of Au8–PPX complexes, the t-PPX tautomer tends to play as a bidentate ligand when binding to Au8. Four local minima located for Au8–t-PPX are also presented in Fig. 5.
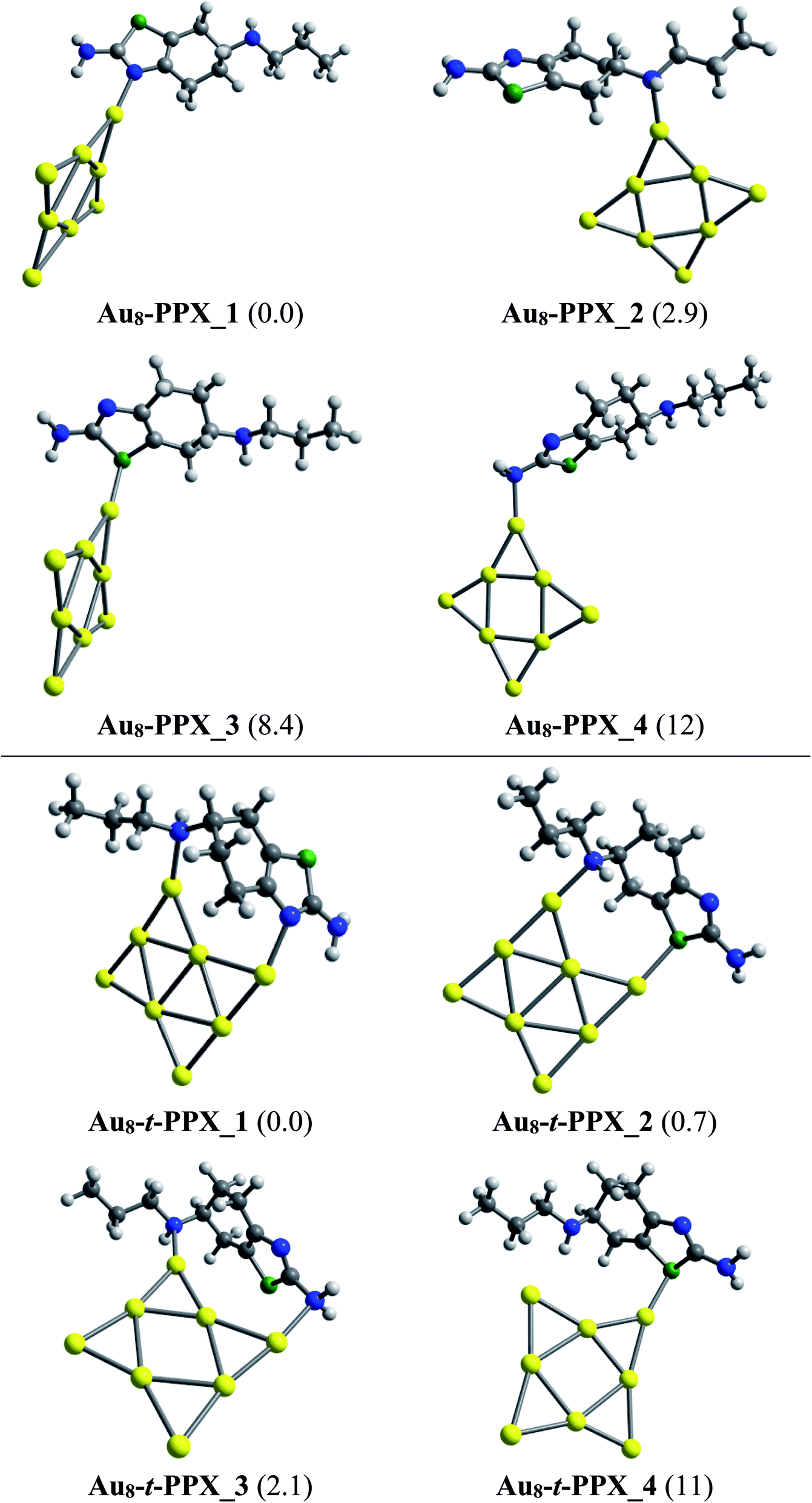 | ||
| Fig. 5 Lower-lying structures located for the Au8–PPX (above) and Au8–t-PPX (below) complexes in gas phase. Relative energies (kcal mol−1) to the lowest-energy isomer are given in parentheses. | ||
Regarding the interactions of Au20 cluster with the drug, four stable complexes are presented in Fig. 6. In particular, Au20–PPX_1 registers a binding energy of −23 kcal mol−1. Similar to observations obtained from Au6–PPX and Au8–PPX, the Au20 binding to the secondary nitrogen atom results in the second most stable Au20–PPX_2 complex. This form is calculated at ca. 3 kcal mol−1 less stable. Finally, two local minima Au20–PPX_3 and Au20–PPX_4, in which the PPX molecule is adsorbed from its sulfur atom and amino head on a vertex gold atom of Au20, are ca. 8–11 kcal mol−1 (PBE value) energetically above the normalized ground state. Overall, the most energetically preferred site for gold binding is the nitrogen atom of the thiazole cycle, while the amino group is predicted as the least favored position. PBE calculated results consistently demonstrate that complexes with Au–Nthiazole bond are ∼2–12 kcal mol−1 lower in energy than those with other interactions. In addition, we find a predominance of mono-dentate interactions in the low-energy populations of the Au20–PPX complex. However, a strong competition between both mono-dentate and bidentate conformations emerges for the binding of Au20 to the t-PPX tautomer. The energy difference between these structural motifs amounts to only around 0.7 kcal mol−1 (Fig. 6).
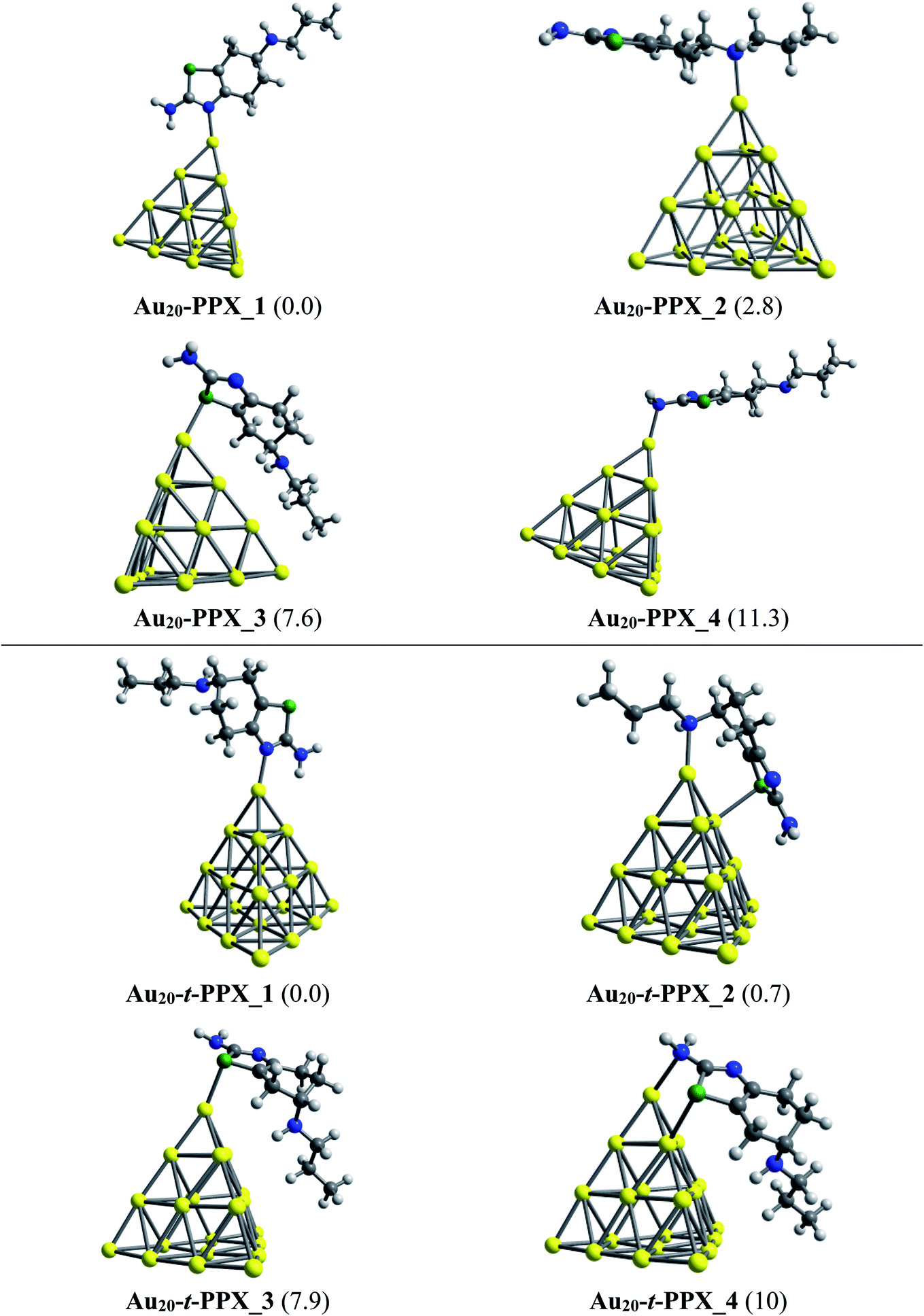 | ||
| Fig. 6 Lower-lying structures located for the Au20–PPX (above) and Au20–t-PPX complexes in gas phase. Relative energies (kcal mol−1) to the lowest-energy isomer are given in parentheses. | ||
Binding energies, changes of enthalpy and Gibbs free energy for the adsorption processes, which are denoted as Eb, ΔH and ΔG, respectively, allow the strength of interactions between the drug and its targeted gold cluster-structures to be evaluated. Numerical data for these energetic properties of AuN–PPX_1 complexes are given in Table 1, whereas those of AuN–t-PPX_1 counterparts are given in Table S1.† Overall, both PPX and t-PPX tautomers exhibit a comparable affinity to gold clusters. For the most stable AuN–PPX_1 complexes, the binding energies are calculated to be about −25, −28 and −23 kcal mol−1 for N = 6, 8 and 20, respectively, whereas the corresponding Gibbs energies decline to −14, −18, and −13 kcal mol−1 (in vacuum). The smaller negative values of Gibbs energies as compared to those of the binding energies are no doubt due to the entropic effect, i.e. the entropy is decreased upon adsorption processes. As expected the higher reactivity toward the drug molecules of Au8 in comparison with Au6 can be explained by its smaller HOMO–LUMO gap and lower stability.62
| Complex | In gas phase | In water | ||||||
|---|---|---|---|---|---|---|---|---|
| Eb | ΔH° | ΔG° | rAu–X | Eb | ΔH° | ΔG° | rAu–X | |
| Au6–PPX_1 | −24.5 | −23.1 | −14.2 | 2.16 | −21.5 | −20.1 | −11.1 | 2.15 |
| Au6–PPX_2 | −21.8 | −20.0 | −11.0 | 2.27 | −20.9 | −18.9 | −8.6 | 2.24 |
| Au6–PPX_3 | −17.4 | −17.2 | −6.0 | 2.50 | −15.2 | −15.0 | −3.8 | 2.50 |
| Au6–PPX_4 | −13.4 | −11.7 | −3.7 | 2.29 | −11.8 | −9.9 | −0.7 | 2.27 |
| Au8–PPX_1 | −28.0 | −26.7 | −17.5 | 2.14 | −24.7 | −23.3 | −13.6 | 2.13 |
| Au8–PPX_2 | −25.2 | −23.3 | −13.2 | 2.24 | −24.1 | −22.1 | −12.7 | 2.22 |
| Au8–PPX_3 | −19.6 | −18.4 | −9.5 | 2.48 | −16.3 | −15.7 | −5.2 | 2.47 |
| Au8–PPX_4 | −16.2 | −14.4 | −5.0 | 2.25 | −14.4 | −12.5 | −3.5 | 2.23 |
| Au20–PPX_1 | −22.8 | −21.5 | −13.2 | 2.18 | −18.5 | −17.7 | −9.4 | 2.18 |
| Au20–PPX_2 | −20.0 | −8.2 | −9.9 | 2.29 | −18.4 | −17.1 | −8.3 | 2.27 |
| Au20–PPX_3 | −15.2 | −14.7 | −5.2 | 2.53 | −12.7 | −12.1 | −2.5 | 2.53 |
| Au20–PPX_4 | −11.5 | −9.8 | −2.7 | 2.34 | −9.4 | −7.6 | 0.7 | 2.31 |
When the effects of water solvent are included, these thermodynamic parameters become overall less negative, even though a similar tendency to gas phase processes is still registered. In particular, the Eb and ΔG values for Au20–PPX_1 are now computed to be −19 and −9 kcal mol−1, in comparison to the corresponding values of −23 and −13 kcal mol−1 in gas phase. Besides, the PPX interacting with Au6 and Au8 also turns out to be more breakable in water. Numerical data obtained for interactions of PPX and AuN (N = 6, 8, 20) clusters in aqueous environment are presented in Table 1, which shows that the Au8 cluster also exhibits the highest affinity with PPX molecule among the three clusters investigated.
The equilibrium distances of Au–N bond in Au6–PPX_1, Au8–PPX_1 and Au20–PPX_1 are 2.16, 2.14 and 2.18 Å, respectively (Table 1). In principle, it can be expected that the shorter the bond length equates the stronger interaction. As found in Table 1, the interaction energy correlates well with the interaction distance when the same atoms are considered. Note that such a correlation does not hold true for AuN–PPX_3 complexes which contain an Au–S bond, rather than an Au–N as in other species. In addition, distances for the anchoring bonds Au–N in these complexes are slightly shorter than the sum of the covalent radii of nitrogen (0.75 Å) and gold (1.44 Å) atoms.64 For Au–N interactions, the Au–S bond lengths Au6–PPX_1, Au8–PPX_1 and Au20–PPX_1 correspond to 2.50, 2.48 and 2.53 Å, which are apparently longer than the sum of covalent radii 2.46 Å of Au (1.44 Å) and S (1.02 Å).64 Therefore, the results indicate elevated effectiveness of the interactions between the thiazole N atom and metallic surface.
3.2 Electronic properties
To obtain deeper insights into the nature of interactions, we further examine the energies of frontier orbitals, i.e. HOMO and LUMO, of the PPX molecule, gold clusters and their resulting complexes. The LC-BLYP functional is used to compute such parameters since long-range correction approaches are normally more reliable than pure GGA functional in predicting orbital energies.32,65,66 The change of HOMO–LUMO band gap (ΔEg) is then calculated by the following expression (4):
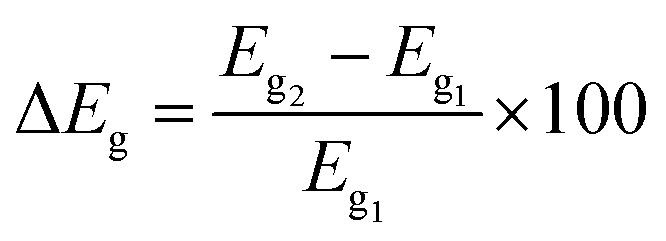 | (4) |
As found in Table 2, the relatively larger HOMO–LUMO gap of Au6 correlates well with its lower reactivity toward the drug molecules, as compared to Au8. However, this does not hold true for PPX binding to Au20. In fact, Au6 is more likely to react with the drug than Au20, even though the former has a larger energy gap than the latter. Thus it can be expected that the affinity of gold clusters towards PPX tends to decrease with respect to the numbers of gold atoms. Information from frontier orbitals clarifies the mechanism of bonding between the drug and cluster. The most important combinations relate to the HOMO and the LUMO states of the drug and those of gold clusters. Depending on their symmetry and energy gap, the forward or backward donation registers the dominance.
| Species | In gas phase | In water | ||||||
|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | Eg | ΔEg | HOMO | LUMO | Eg | ΔEg | |
| Au6 | −8.80 | −1.29 | 7.51 | — | −8.36 | −0.49 | 7.87 | — |
| Au8 | −8.54 | −1.84 | 6.70 | — | −8.12 | −1.13 | 6.99 | — |
| Au20 | −7.89 | −1.71 | 6.18 | — | −7.42 | −1.01 | 6.41 | — |
| PPX | −8.39 | 2.20 | 10.6 | — | −8.50 | 2.12 | 10.6 | — |
| Au6–PPX_1 | −7.81 | −0.56 | 7.25 | −3.5% | −7.97 | −0.43 | 7.54 | −4.2% |
| Au8–PPX_1 | −7.81 | −1.09 | 6.72 | 0.3% | −7.93 | −0.84 | 7.09 | 1.4% |
| Au20–PPX_1 | −7.35 | −1.40 | 5.95 | −3.7% | −7.15 | −1.02 | 6.13 | −4.4% |
The energy differences between the HOMO of PPX and LUMO of AuN are in the range of 6.6 to 7.1 eV, which are significantly smaller than the values of 10–11 eV between the HOMO of AuN and LUMO of PPX. Hence, the forward donation (PPX → AuN) should be more effective and the nature of these interactions is mainly characterized by an electron transfer from the PPX to gold atoms. In other words, gold clusters tend to serve as electron acceptors, and the drug is likely to play a role of an electron donor. This charge-flow tendency, from organic molecules to metallic surfaces, is also consistent with previous reports.67,68 The nature of Au–PPX bond is basically analogous to that of gold clusters interacting with lone-pair ligands69 and mostly dominated by a σ bond with the distribution of unshared electrons in the HOMO of PPX molecule and the LUMO of gold clusters. As illustrated in Fig. 7, an overlap between the LUMO of Au6 with the HOMO of PPX gives rise to an σ-type bonding orbital. In addition, the LUMO of PPX can also interact with the HOMO of Au6, forming further π-type bonding orbital. However, the forward donation still plays a much more important role than the back transfer, and there exists some charge flow from the drug to Au metals.
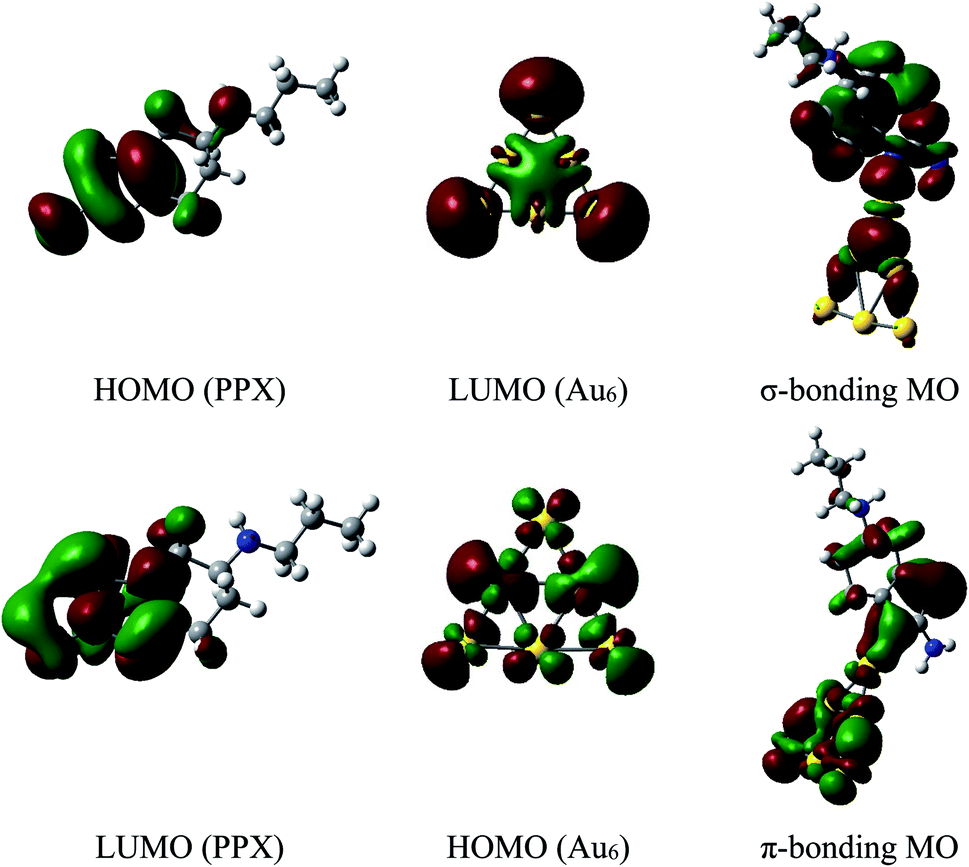 | ||
| Fig. 7 Shapes of frontier orbitals of Au6 cluster and PPX molecule before and after interaction (isovalue for the orbital rendering is 0.02). | ||
Since an overlap between the frontier orbitals of gold clusters and PPX molecule is normally followed by a charge transfer, we further examine the NBO charge distribution of systems to gain more insight into the problem. It is found that the Au8 moiety in Au8–PPX_1 is likely to gain more negative charge from PPX molecule, as compared to Au6 and Au20 counterparts in Au6–PPX_1 and Au20–PPX_1, respectively. Indeed, the net charge of Au8 in Au8–PPX_1 is computed to be −0.21 au, which is marginally more negative than the corresponding value of −0.20 au in Au6–PPX_1. A more effective charge transfer in Au8–PPX complex corresponds to a better overlap between frontier orbitals of Au8 and PPX, resulting in a stronger interaction.
The above observation can further be confirmed by analysis of the density of states (DOS) of relevant species. The DOS plots for PPX and AuN clusters before and after interaction are presented in Fig. 8. Overall, the HOMO and LUMO energies of both PPX and AuN species change significantly due to the complexation. First, the disappearance of the peak corresponding to the PPX lone pair at around −8.4 eV in the DOS of the resulting complexes is most profound. This clearly indicates that the drug is adsorbed on gold clusters by partly donating electron densities to the metals. Regarding Au6–PPX_1 and Au20–PPX_1 complexes, both HOMO and LUMO levels of clusters shift to a more positive region, and the former registers a more significant shift. Consequently, the Eg values of Au6 and Au20 are decreased by ∼4% (LC-BLYP value). In terms of Au8–PPX_1, both HOMO and LUMO levels shift rather equally to the higher-energy region. Thus, the energy gap of Au8 remains almost unchanged following interaction. The change in the energy gap due to drug adsorption may induce an adjustment of fluorescence emission, which in turn can help detect the drug by relevant spectrophotometers.
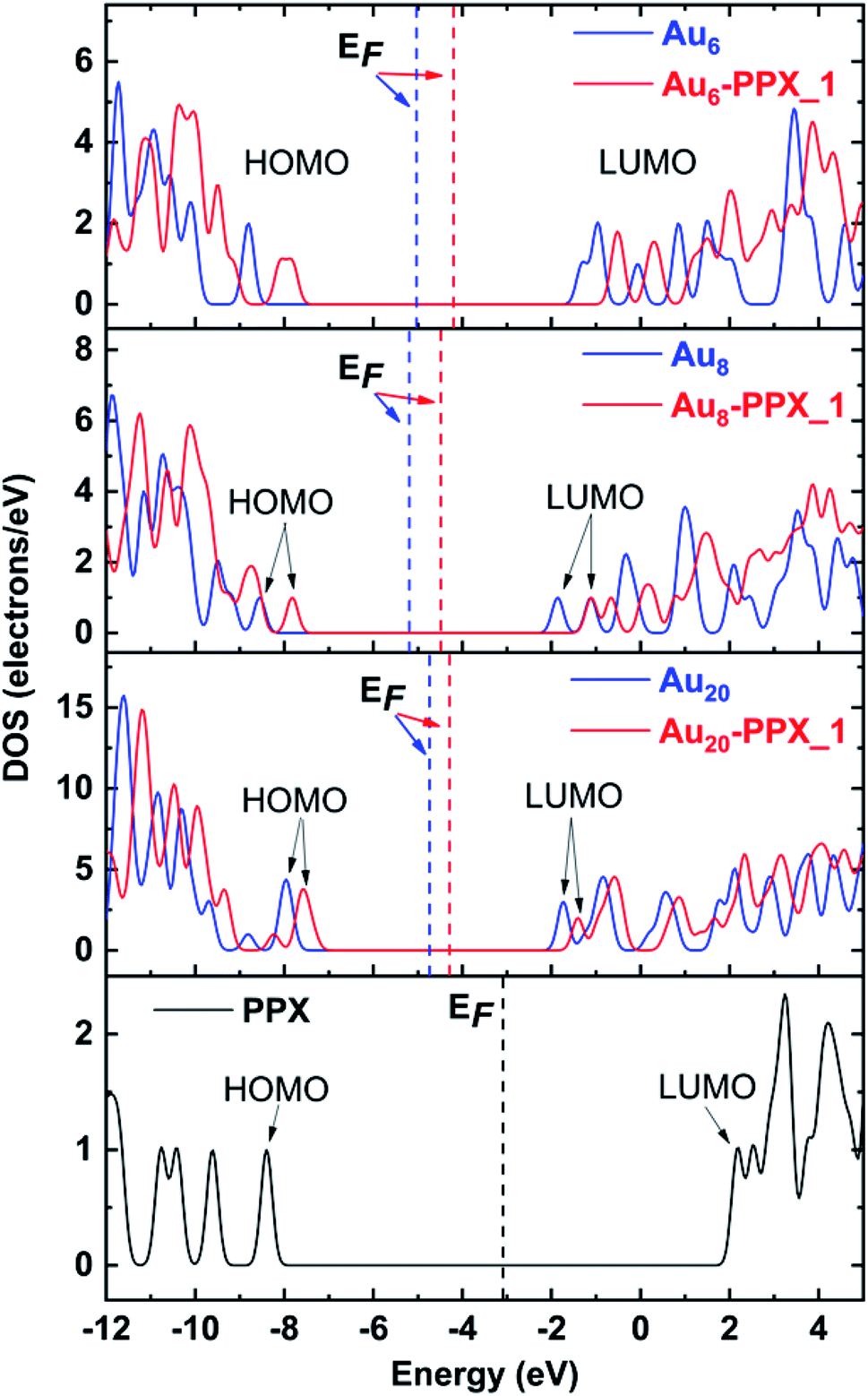 | ||
| Fig. 8 Density of states of stable complexes. Orbitals shown are HOMO and LUMO of complexes. The Fermi levels are denoted as vertical dash lines. | ||
Due to their tendency to give unbalanced electron correlations, most available DFT functionals tend to provide large errors for energies of both occupied and unoccupied levels.57,60 Therefore, the band gaps have been often evaluated by subtracting the vertical electron affinities (vEA) from the vertical ionization energies (vIE), i.e., vIE–vEA.32 The band gap in Au20 is experimentally measured to be >5 eV.70 Calculations using the PBE functional yield a very small band gap of 1.8 eV which is smaller by ∼3 eV as compared to the value of 4.6 eV obtained by subtracting its vEA from the vIP. On the other hand, the LC-BLYP values of 6.3 and 5.8 eV for HOMO–LUMO and vIE–vEA gaps, respectively, are comparable to each other, but both rather overestimate the experimental result. In general, such an energy gap is greatly modified with respect to different functionals depending on the amount of Hartree–Fock exchange included.59,71,72 Thus, it is hard to predict the insulating behavior of these systems merely relying on the band gap computed from a single functional.
It was well established in recent literature that the change of band gap can be a proper indicator for recognizing the presence and attachment of the drug to the clusters.73 Indeed, the Eg value is closely related to the electrical conductivity (σ) of the material through the three-parameter expression (5):74
| σ = AT3/2e−Eg/2kBT | (5) |
It can be argued that a close relationship between the sensitivity and properties exists, especially the band gap. According to eqn (5), the electrical conductivity of the cluster will exponentially increase with respect to a decrease of Eg. Such a change then can be converted to an electrical signal, helping to identify the drug. In addition, the change in Eg computed from eqn (4) can be employed to evaluate the electrical conductivity of clusters. With PPX adsorption, the changes in energy gaps (ΔEg) of Au6 and Au20 are predicted to decline around 4% in both vacuum and water environments. However, such a change obtained for Au8 appears to be slightly different, i.e. increasing by 0.3% (vacuum) and 1.4% (water).
3.3 SERS spectra of PPX on gold surfaces
Analysis of vibrational features and SERS spectra of the drug is of great interest as they can be reproduced in experimental studies, and serve as an important role in the rationalization of adsorption mechanism of the drug on metal surfaces. In principle, Raman spectroscopy detects vibrational modes of functional groups of molecules. However, for detection of molecules at trace concentrations, this approach is inherently restricted due to the very weak Raman intensity. Therefore, the surface-enhanced Raman scattering (SERS) techniques are often employed to amplify the Raman intensity.75 Of the broad variety of biological and spectroscopic approaches established to detect biomolecules, SERS methods have attracted much attention from scientific community and have been extensively applied to biosensors.76,77When discussing the SERS phenomenon, two important mechanisms are frequently put forward, including the electromagnetic and chemical enhancements.78 Indeed, in the former mechanism, some Raman modes of a molecule in close vicinity of a metallic surface get remarkably enhanced because the Raman intensity is proportional to the squared local electromagnetic field intensity. Moreover, the Raman-scattered light induces an additional enhancement when the Raman mode overlaps with a plasmonic resonance.79 The latter mechanism, on the contrary, primarily refers to a chemical interaction or/and a charge transfer between nanoparticles and molecules.80 For interpretation of Raman spectral features as well as the SERS chemical enhancement mechanism of organic molecules adsorbed on metal surfaces, quantum chemical calculations provide us with appropriate information with the use of small metallic clusters as models to simulate the nanoparticle surfaces. Although the normal Raman spectrum of PPX could be found in the literature,61 its SERS procedures remain ambiguous. To support further experimental studies, we now perform some analysis of the SERS spectra of PPX molecules adsorbed on Au6, Au8 and Au20 clusters. The spectra of free PPX molecule and its complexes with gold clusters in wavelength range of 1000 to 4000 cm−1 are generated in Fig. 7, and their highly intense Raman signals are given in Table 3.
| Modes | PPX | Au20–PPX_1 | PPX | Au20–PPX_1 | ||||
|---|---|---|---|---|---|---|---|---|
| Position | Intensity | Position | Intensity | Position | Intensity | Position | Intensity | |
| In vacuum | In water | |||||||
| a νas – asymmetric stretching; νs – symmetric stretching; β – in-plane bending. | ||||||||
| νas (NH2) | 3579 | 14 | 3596 | 16 | 3582 | 26 | 3589 | 62 |
| νs (NH2) | 3471 | 63 | 3393 | 445 | 3476 | 90 | 3443 | 794 |
| νas (CH3) | 3037 | 32 | 3040 | 56 | 3034 | 63 | 3025 | 131 |
| νas (CH2) | 3014 | 29 | 3027 | 35 | 2988 | 278 | 3023 | 108 |
| νs (CH3) | 3028 | 32 | 2965 | 54 | 3024 | 67 | 2961 | 475 |
| νs (CH2) | 2995 | 63 | 2970 | 106 | 2945 | 233 | 2944 | 179 |
| β (NH2) | 1587 | 52 | 1582 | 57 | 1579 | 154 | 1573 | 1400 |
| ν (NC) | 1529 | 36 | 1505 | 436 | 1519 | 50 | 1499 | 173 |
| ν (CC) | 1563 | 68 | 1598 | 451 | 1563 | 184 | 1597 | 1100 |
| β (CH3) | 1357 | 27 | 1362 | 32 | 1350 | 65 | 1350 | 82 |
| β (CH2) | 1289 | 33 | 1263 | 46 | 1286 | 78 | 1221 | 44 |
The normal Raman spectrum of PPX (Fig. 9) consists of three low-intensity bands in the N–H stretching region at near 3400 cm−1, which correspond to the asymmetric and symmetric stretching modes of NH2 and NH groups. Previously, the stretching mode of NH group was also detected at 3379 cm−1 in the experimental study by Muthu and co-workers.61 In aqueous solution, these stretching modes are slightly shifted to the lower-energy region but with higher intensity. The C–H stretching modes of CH2 and CH3 groups are computed vibrating in the range of 2827–3037 cm−1 in vacuum and between 2840–3035 cm−1 in water solution, which are comparable to experimental data of 2925–3027 cm−1.61 It is rather difficult to clearly assign other intense peaks in the range of 1250–1600 cm−1 as they arise from a mixing of several vibrations such as C–X stretching and XH2 (X = N, C) bending modes. When taking the effects of water solvent into accounts, the positions of these vibrational modes remain almost unchanged but their intensities consistently become much stronger.
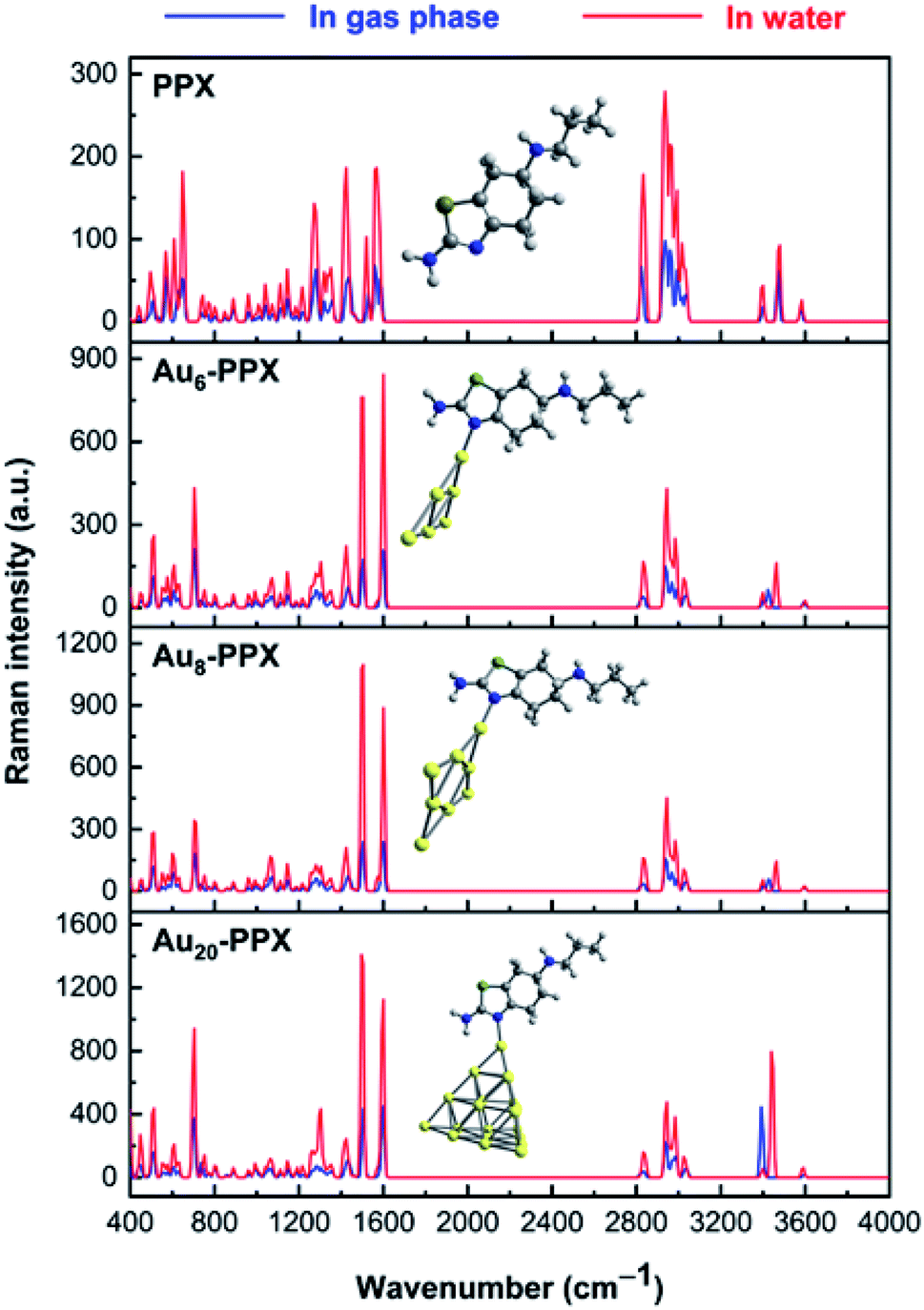 | ||
| Fig. 9 Calculated Raman spectrum of PPX and SERS spectra of the most stable complexes AuN–PPX_1 (N = 6, 8, 20). | ||
In the computed SERS spectra of PPX on gold clusters, some significant modifications as compared to the normal Raman spectrum could be observed (Fig. 9). Accordingly, the NH2 bending and C–N stretching modes turn out to be the highest intensity bands, rather than the overlapped bands around 3000–3200 cm−1 arising from C–H stretching modes as in free PPX. The intensities of signals associated with C–N stretching and NH2 bending vibrations, that are centered at ∼1500 and 1590 cm−1, represent the most significant increase. This can be understood by the fact that these bonds directly relate to the adsorption of PPX molecules on the gold surfaces. Indeed, while the nitrogen atom of the CN moiety is the preferable site of PPX to bind to gold metals, a weak Au⋯H–N coupling exists between the amine head with metallic surface. Other significant enhancements are found for signals associated with C–S and H–NH stretching vibrations, which are located near 700 and 3400 cm−1, respectively, in the AuN–PPX_1 complexes.
The cluster size also contributes differently to the SERS chemical enhancement. Although the positions of distinctive signals are almost similar, the relative intensities in the SERS spectrum of PPX on Au20 cluster see significant changes, in comparison to those of smaller clusters. From the SERS spectrum of Au20–PPX_1, a particularly characteristic peak emerges either at 3394 cm−1 in vacuum or at 3444 cm−1 in water, which is produced from the symmetric stretching of the amine group. The intensity of this peak in the SERS spectrum of Au20–PPX_1 is much more enhanced than that of Au6–PPX_1 and Au8–PPX_1. Such a phenomenon can be explained by formation of a stronger Au⋯H–N bonding in Au20–PPX_1, which is less significant in Au6–PPX_1 and Au8–PPX_1. Formation of such nonconventional hydrogen interaction, along with an N–AuN covenant bond, is likely the main factor leading to a SERS chemical enchantment of PPX on gold nanostructured surfaces.
3.4 The recovery time and drug release
When exposed to light, a drug desorption process may occur. According to the transition state theory,81 the recovery time τ for the drug desorption from the gold surfaces can be calculated as eqn (6):| τ = ν−1e−Eb/kBT | (6) |
The time for the desorption process of PPX molecules from the gold surfaces at 298 K are given in Table 4. Overall, the recovery time of in the gas phase is consistently much longer than in water environment. For example, if an attempt frequency of ν = 6.0 × 1014 Hz (500 nm) is applied, the recovery of PPX molecules from the Au6 surface is about 1.5 × 103 seconds in gas phase, in comparison to a corresponding value of 10 seconds in aqueous medium. Similarly, the recovery time amounts to in the range of 82 seconds (in vacuum) to 0.06 seconds (in water) for Au20–PPX_1 complex. On the contrary, due to its higher affinity with the drug, Au8 undergoes much longer recovery times (Table 4).
| Attempt frequency | In gas phase | In water | ||||
|---|---|---|---|---|---|---|
| Au6–PPX_1 | Au8–PPX_1 | Au20–PPX_1 | Au6–PPX_1 | Au8–PPX_1 | Au20–PPX_1 | |
| 3.0 × 1016 Hz (10 nm) | 31 | 12 × 103 | 2 | 0.2 | 45 | 1.2 × 10−3 |
| 7.5 × 1014 Hz (400 nm) | 1233 | 5.0 × 105 | 65 | 7.8 | 1818 | 0.05 |
| 6.0 × 1014 Hz (500 nm) | 1541 | 6.0 × 105 | 82 | 9.7 | 2273 | 0.06 |
| 4.3 × 1014 Hz (700 nm) | 2150 | 8.5 × 105 | 114 | 14 | 3171 | 0.09 |
We now consider the ability to release the drug from the carrier in target cells, which is one of the most vital steps in drug delivery process. To detach the drug from the carrier, either external stimuli or internal stimuli operating within a biologically controlled manner such as the pH change or amino acid residues such as glutathione and cysteine can be used.83 As a result of the excessive lactic production, a tumor cell is typically more acidic than normal cells. In fact, tumor cells usually have lower pH (below 6) as compared to the blood (pH ≈ 7.35–7.45).84 In this context, we examine the effect of protons on the stability of the AuN·PPX complexes. The protons can attack any rich-electron site of the drug, but such an attack to the thiazole nitrogen atom may affect the drug release more significantly. Based on the computed proton affinities (Table S1 of the ESI†), the thiazole nitrogen atom is the most preferable site for protonation. The binding of PPX to gold clusters is a reversible process. Thus, the molecule can be found in an isolated form at any moment in solution. In addition, it seems that even after interacting with Au, this N-atom remains the most negatively charged site open for protonation. Therefore, we consider the effects of protons on the stability of AuN·PPX by protonation of the N atom of the thiazole ring, and then perform the optimization for resulting AuN·PPXH+ products. The effects of water solvent are also considered using the IEF-PCM model.
With the presence of protons, interactions between the PPX drug and gold clusters becomes more easily breakable as it is now characterized by weaker H-bonds as illustrated in Fig. 11, rather than the covalent bond as in AuN·PPX. The binding energies of PPX on gold clusters are substantially reduced to ∼−9 kcal mol−1, as compared to the value from −23 to −28 kcal mol−1 in neutral condition. Therefore, it is expected that in an acidic environment the PPX drug molecule binding to the carrier is getting weaker, thereby able to be released faster.
Another factor contributing crucially to drug release is an internal force related to amino acids present in the protein matrices.83 Some amino acid constituents containing sulfur element such as cysteine and methionine are found to be the most preferred binding sites for noble metals.85,86 At physiological pH, cysteine mainly exists as the deprotonated forms by proton cleavage of either the carboxyl group (pKa = 1.7) or thiol group (pKa = 8.3),84 as shown in Fig. 10. At the PBE/cc-pVTZ level, two forms Cys(−H+)_1 and Cys(−H+)_2 are nearly iso-energetic, with a marginal energy difference of 2.0 kcal mol−1. However, Cys(−H+)_2 tends to interact much more strongly with gold clusters than Cys(−H+)_1 due to formation of a hydrogen bond in the resulting complexes. To probe further into the drug release from gold nanostructured surfaces, we examine the following ligand interchange reaction (7):
| AuN·PPX(aq) + Cys(−H+)(aq) → Au6·Cys(−H+)(aq) + PPX(aq) | (7) |
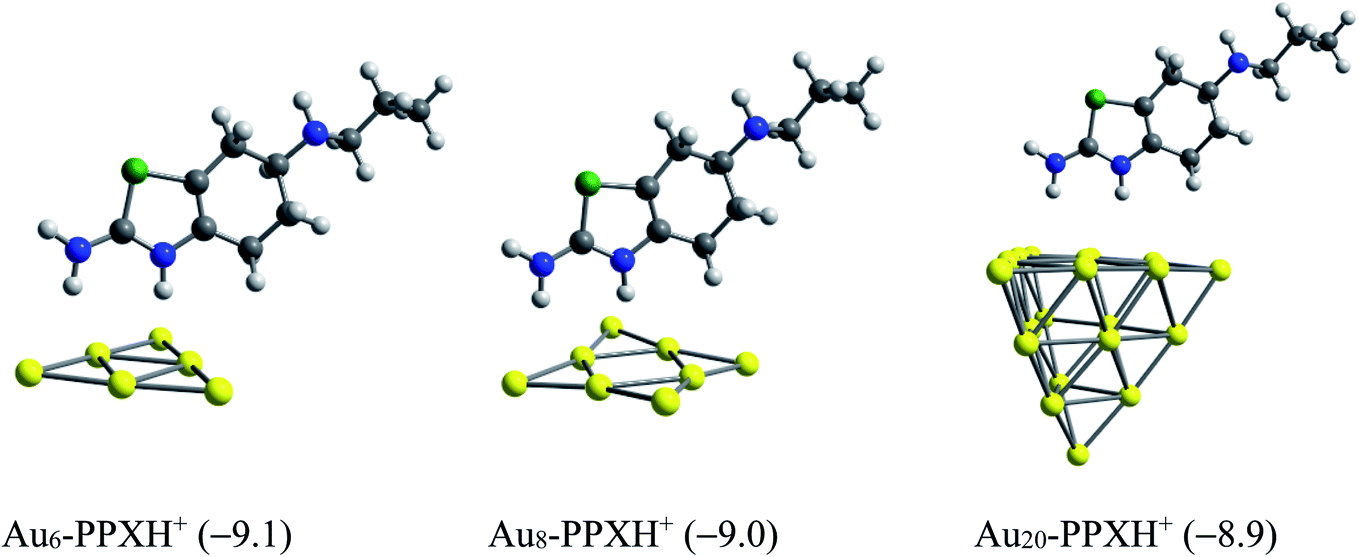 | ||
| Fig. 10 Optimized structures of AuN–PPXH+ complexes. Values in parentheses are binding energies (kcal mol−1, PBE/cc-pVTZ/cc-pVDZ-PP + ZPE). | ||
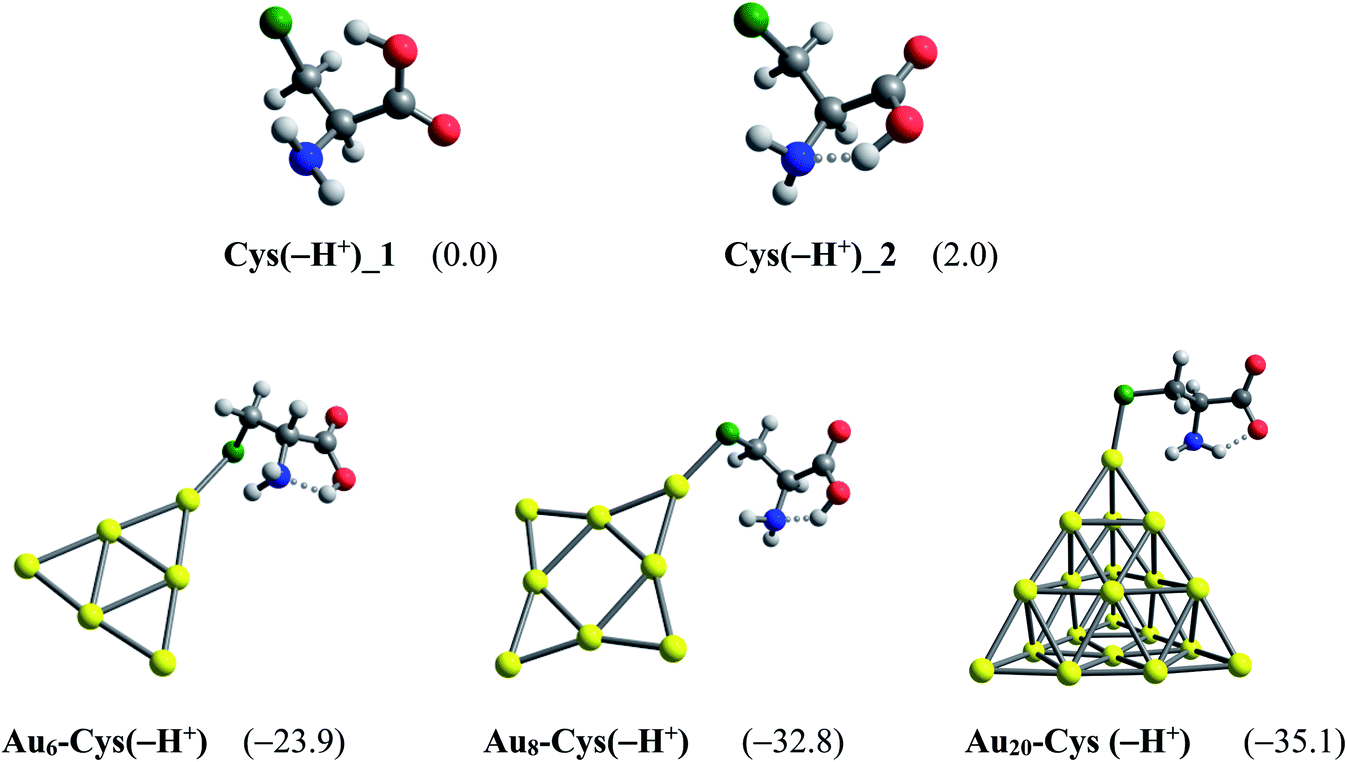 | ||
| Fig. 11 Global minima located for the deprotonated cysteine and its resulting complexes with AuN (N = 6, 8, 20) clusters. Values in parentheses are binding energies (kcal mol−1, PBE/cc-pVTZ/cc-pVDZ-PP + ZPE). | ||
In agreement with a recent report,49 the cysteine molecule prefers binding to AuN clusters via its S-atom of the thiolate group in aqueous solution with the binding energies of −39, −43 and −35 kcal mol−1 for N = 6, 8 and 20, respectively. These values are much more negative than the binding energies of PPX on gold clusters, being in the range of −18 to −25 kcal mol−1 (Table 1). Therefore, it can be concluded that gold nanoparticles have much higher affinity with cysteine residues than with PPX and the drug releasing from the gold surface in target cells is likely to ensue as a result of this internal force. The Gibbs energies for the ligand interchange reaction above are up to −14 kcal mol−1, indicating that these processes are spontaneous.
4. Concluding remarks
The most favored state is the interaction between the nitrogen of the thiazole ring and the positively charged atoms of gold clusters with binding energies in the range of ca. −23 to −28 kcal mol−1 in vacuum and ca. −18 to −25 kcal mol−1 in aqueous continuum. Adsorption processes occur spontaneously with rather negative Gibbs energies, ca. −13 to −18 kcal mol−1 in vacuum and ca. −4 to −14 kcal mol−1 in water, respectively. In the resulting complexes, the Au–Nthiazole bond length is getting shorter, while the Au–S bonds are somewhat longer than the sum of covalent radii of binding atoms, indicating the elevated interactive effectiveness regarding the former. The energy gap of clusters are also modified due to the drug attachment that may result in a change of optical properties. Analysis on the basis of frontier orbitals suggests the forward donation from the HOMO of PPX to LUMO of AuN as primary contribution for PPX–Au bonding formation. While the normal Raman spectrum of PPX exhibits several main peaks related to C–H stretching vibrations, C–X stretching and XH2 (X = N, C) bending modes, a significantly-enhanced shaped peak of SERS spectrum of PPX on Au20 corresponds to the stretching vibrations of N–H bonds. This lends support for applicability of such nanostructures for drug detection at trace concentration. Regarding drug release mechanism, the triggering factors could be referred to either the presence of cysteine residues (usually found in protein matrices) or low pH (often reported for those of cancerous tissues). The results altogether propose gold-surface nanostructures of high potentiality for design of drug carriers and detectors, especially in cancer-tissue physiological mediums. This study represents a preliminary but necessary contribution to applications of gold nanoparticles for PPX delivery and detection. We would expect that relevant experimental work could soon be carried out to verify further the gold nanocluster model.Funding
This work was funded by VinGroup (Vietnam) and supported by VinGroup Innovation Foundation (VinIF) under project code VINIF.2020.DA21.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
NTS is grateful to the Mississippi Center for Supercomputing Research, USA for providing computing resources.References
- S. Akhter, I. Ahmad, M. Z. Ahmad, F. Ramazani, A. Singh, Z. Rahman, F. J. Ahmad, G. Storm and R. J. Kok, Curr. Cancer Drug Targets, 2013, 13, 362 CrossRef CAS PubMed.
- N. A. C. Lah, R. Gray and S. Trigueros, Microb. Cell Fact., 2021, 20, 1–11 CrossRef PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- H. Daraee, A. Eatemadi, E. Abbasi, S. Fekri Aval, M. Kouhi and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 410–422 CrossRef CAS PubMed.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2012, 64, 102–115 CrossRef.
- M. Kalomiraki, K. Thermos and N. A. Chaniotakis, Int. J. Nanomed., 2016, 11, 1 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- D. R. Vogus, V. Krishnan and S. Mitragotri, Curr. Opin. Colloid Interface Sci., 2017, 31, 75–85 CrossRef CAS.
- L. Wei, J. Lu, H. Xu, A. Patel, Z.-S. Chen and G. Chen, Drug Discovery Today, 2015, 20, 595–601 CrossRef CAS PubMed.
- K. Hola, Z. Markova, G. Zoppellaro, J. Tucek and R. Zboril, Biotechnol. Adv., 2015, 33, 1162–1176 CrossRef CAS PubMed.
- C. S. Kim, G. Y. Tonga, D. Solfiell and V. M. Rotello, Adv. Drug Delivery Rev., 2013, 65, 93–99 CrossRef CAS PubMed.
- S. Kwon, R. K. Singh, R. A. Perez, E. A. Abou Neel, H.-W. Kim and W. Chrzanowski, J. Tissue Eng., 2013, 4, 1–35 Search PubMed.
- X. Sun, Z. Feng, T. Hou and Y. Li, Comput. Pharm., 2015, 149 CrossRef CAS.
- Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS.
- M. Auffan, J. Rose, J.-Y. Bottero, G. V. Lowry, J.-P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634–641 CrossRef CAS PubMed.
- H.-C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, J. Controlled Release, 2011, 155, 344–357 CrossRef CAS PubMed.
- S. Borker, M. Patole, A. Moghe and V. Pokharkar, Gold Bull., 2017, 50, 235–246 CrossRef CAS.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2005, 79, 248–253 CrossRef PubMed.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, Nano Res., 2015, 8, 1771–1799 CrossRef CAS.
- G. Ajnai, A. Chiu, T. Kan, C.-C. Cheng, T.-H. Tsai and J. Chang, Int. J. Clin. Exp. Med., 2014, 6, 172 CrossRef CAS.
- A. Kumar, X. Zhang and X.-J. Liang, Biotechnol. Adv., 2013, 31, 593–606 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- C. Wang, H. Zhang, D. Zeng, L. San and X. Mi, Chin. J. Chem., 2016, 34, 299–307 CrossRef CAS.
- I. Khalil, N. M. Julkapli, W. A. Yehye, W. J. Basirun and S. K. Bhargava, Materials, 2016, 9, 406 CrossRef PubMed.
- F.-Y. Kong, J.-W. Zhang, R.-F. Li, Z.-X. Wang, W.-J. Wang and W. Wang, Molecules, 2017, 22, 1445 CrossRef PubMed.
- M. Demurtas and C. C. Perry, Gold Bull., 2014, 47, 103 CrossRef CAS.
- L. A. Austin, M. A. Mackey, E. C. Dreaden and M. A. El-Sayed, Arch. Toxicol., 2014, 88, 1391 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813 CrossRef CAS PubMed.
- M. Demurtas and C. C. Perry, Gold Bull., 2014, 47, 103–107 CrossRef CAS.
- M. Ramezanpour, S. Leung, K. Delgado-Magnero, B. Bashe, J. Thewalt and D. Tieleman, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1688–1709 CrossRef CAS PubMed.
- P. V. Nhat, N. T. Si, N. T. T. Tram, L. V. Duong and M. T. Nguyen, J. Comput. Chem., 2020, 1289–1299 CAS.
- H. Abd El-Mageed, F. Mustafa and M. K. Abdel-Latif, Struct. Chem., 2020, 31, 781–793 CrossRef.
- M. Muniz-Miranda, F. Muniz-Miranda and A. Pedone, Phys. Chem. Chem. Phys., 2016, 18, 5974–5980 RSC.
- R. A. Teixeira, A. A. Lataliza, N. R. Raposo, L. A. S. Costa and A. C. Sant'Ana, Colloids Surf., B, 2018, 170, 712–717 CrossRef CAS PubMed.
- M. D. Ganji, H. T. Larijani, R. Alamol-Hoda and M. Mehdizadeh, Sci. Rep., 2018, 8, 1–13 Search PubMed.
- S. G. Harroun, ChemPhysChem, 2018, 19, 1003–1015 CrossRef CAS PubMed.
- H. Farrokhpour, S. Abedi and H. Jouypazadeh, Colloids Surf., B, 2019, 173, 493–503 CrossRef CAS PubMed.
- B. Khodashenas, M. Ardjmand, M. S. Baei, A. S. Rad and A. A. Khiyavi, J. Inorg. Organomet. Polym. Mater., 2019, 29, 2186–2196 CrossRef CAS.
- B. Khodashenas, M. Ardjmand, M. S. Baei, A. S. Rad and A. Akbarzadeh, Appl. Organomet. Chem., 2020, 34, e5609 CrossRef CAS.
- P. Schwerdtfeger, Angew. Chem., Int. Ed., 2003, 42, 1892–1895 CrossRef CAS PubMed.
- E.-K. Tan and W. Ondo, Arch. Neurol., 2000, 57, 729–732 CrossRef CAS PubMed.
- J. M. Bostwick, K. A. Hecksel, S. R. Stevens, J. H. Bower and J. E. Ahlskog, Mayo Clin. Proc., 2009, 84, 310–316 CrossRef CAS.
- T. J. Moore, J. Glenmullen and D. R. Mattison, JAMA Intern. Med., 2014, 174, 1930–1933 CrossRef PubMed.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. J. E. MontgomeryPeralta Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099 CrossRef CAS.
- P. V. Nhat, P. T. N. Nguyen and N. T. Si, J. Mol. Model., 2020, 26, 1–8 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- N. M. O'boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef PubMed.
- J. W. Ochterski, Thermochemistry in Gaussian, see: E-mail: help@gaussian.com, 2000 Search PubMed.
- N. L. Hadipour, A. Ahmadi Peyghan and H. Soleymanabadi, J. Phys. Chem. C, 2015, 119, 6398 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540 CrossRef CAS.
- T. Tsuneda and K. Hirao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 375 CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757 RSC.
- T. Tsuneda, J. Comput. Chem., 2019, 40, 206 CrossRef CAS PubMed.
- J. N. Harvey, Struct. Bonding, 2004, 151–184 CrossRef CAS.
- T. Tsuneda, J.-W. Song, S. Suzuki and K. Hirao, J. Phys. Chem., 2010, 133, 174101 CrossRef PubMed.
- S. Muthu, J. U. Maheswari and S. Srinivasan, Spectrochim. Acta, Part A, 2013, 115, 64–73 CrossRef CAS PubMed.
- P. V. Nhat, N. T. Si, J. Leszczynski and M. T. Nguyen, Chem. Phys., 2017, 493, 140 CrossRef CAS.
- A. H. Pakiari and Z. Jamshidi, J. Phys. Chem. A, 2007, 111, 4391 CrossRef CAS PubMed.
- A. M. James and M. P. Lord, Macmillan's chemical and physical data, Macmillan, 1992 Search PubMed.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 16187 RSC.
- T. Tsuneda, R. K. Singh and A. Nakata, J. Comput. Chem., 2017, 38, 2020 CrossRef CAS PubMed.
- G. Yao, Z. Zhai, J. Zhong and Q. Huang, J. Phys. Chem. C, 2017, 121, 9869–9878 CrossRef CAS.
- M. V. Cañamares, F. Pozzi and J. R. Lombardi, J. Phys. Chem. C, 2019, 123, 9262–9271 CrossRef.
- T. Rajsky and M. Urban, J. Phys. Chem. A, 2016, 120, 3938–3949 CrossRef CAS PubMed.
- J. V. Koppen, M. Hapka, M. M. Szczęśniak and G. Chałasiński, J. Chem. Phys., 2012, 137, 114302 CrossRef PubMed.
- D. M. A. Smith, M. Dupuis and T. P. Straatsma, Mol. Phys., 2005, 103, 273–278 CrossRef CAS.
- H. Hirao, D. Kumar, W. Thiel and S. Shaik, J. Am. Chem. Soc., 2005, 127, 13007–13018 CrossRef CAS PubMed.
- A. Ahmadi Peyghan, N. L. Hadipour and Z. Bagheri, J. Phys. Chem. C, 2013, 117, 2427 CrossRef CAS.
- A. Ahmadi, N. L. Hadipour, M. Kamfiroozi and Z. Bagheri, Sens. Actuators, B, 2012, 161, 1025 CrossRef CAS.
- A. Mohammadi, D. L. Nicholls and A. Docoslis, Sensors, 2018, 18, 3404 CrossRef PubMed.
- S. J. Bauman, Z. T. Brawley, A. A. Darweesh and J. B. Herzog, Sensors, 2017, 17, 1530 CrossRef PubMed.
- E. Cordero, F. Korinth, C. Stiebing, C. Krafft, I. W. Schie and J. Popp, Sensors, 2017, 17, 1724 CrossRef PubMed.
- D. Cialla, S. Pollok, C. Steinbrücker, K. Weber and J. Popp, Nanophotonics, 2014, 3, 383–411 CAS.
- T. Itoh, K. Yoshida, V. Biju, Y. Kikkawa, M. Ishikawa and Y. Ozaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 085405 CrossRef.
- L. Jensen, C. M. Aikens and G. C. Schatz, Chem. Soc. Rev., 2008, 37, 1061–1073 RSC.
- S. Peng, K. Cho, P. Qi and H. Dai, Chem. Phys. Lett., 2004, 387, 271 CrossRef CAS.
- Y. Yong, H. Cui, Q. Zhou, X. Su, Y. Kuang and X. Li, RSC Adv., 2017, 7, 51027–51035 RSC.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307 CrossRef CAS PubMed.
- P. Swietach, R. D. Vaughan-Jones, A. L. Harris and A. Hulikova, Philos. Trans. R. Soc., B, 2014, 369, 20130099 CrossRef PubMed.
- X. Le Guével, B. Hötzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955 CrossRef.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables and figures display computed characteristic properties of pramipexole and its interactions with gold clusters. See DOI: 10.1039/d1ra02172a |
| This journal is © The Royal Society of Chemistry 2021 |