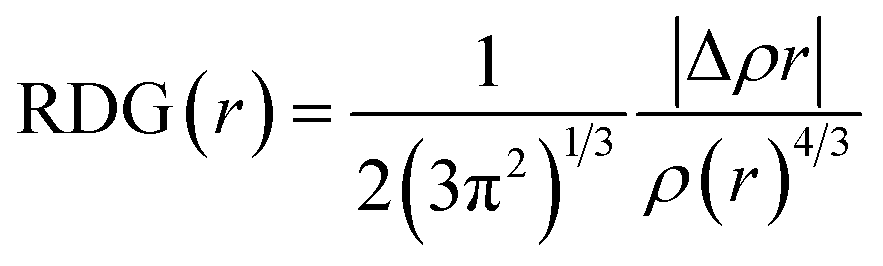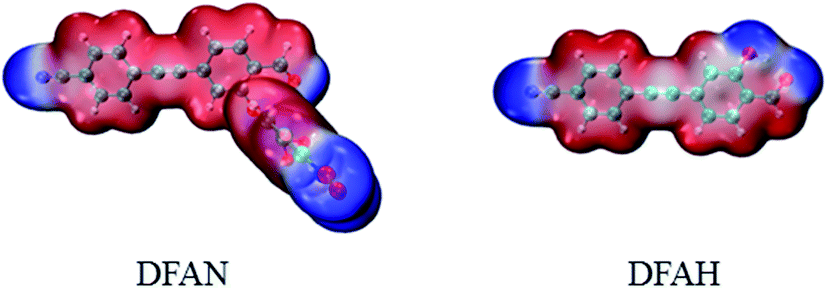DOI:
10.1039/D1RA02511B
(Paper)
RSC Adv., 2021,
11, 22214-22220
Sensing mechanism of a new fluorescent probe for hydrogen sulfide: photoinduced electron transfer and invalidity of excited-state intramolecular proton transfer†
Received
30th March 2021
, Accepted 2nd June 2021
First published on 23rd June 2021
Abstract
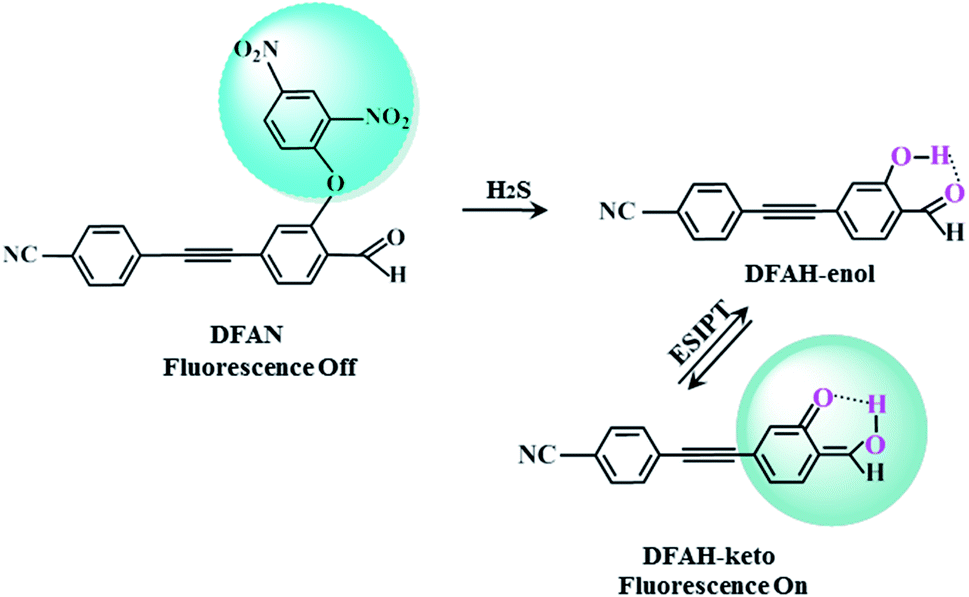
It is of great significance for biological research to develop efficient detection methods of hydrogen sulfide (H2S). When DFAN reacts with H2S, 2,4-dinitrophenyl ether group acting as an electron acceptor generates a hydroxyl-substituted 2,4-dinitrophenyl ether group, resulting in the disappearance of photoinduced electron transfer (PET), and the new formed DFAH can be observed, while being accompanied by a significant fluorescence. In the present study, the PET sensing mechanism of probe DFAN and the excited state intramolecular proton transfer (ESIPT) process of DFAH have been explored in detail based on the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) methods. Our theoretical results show that the fluorescence quenching of DFAN is caused by the PET mechanism, and the result of ESIPT mechanism is not due to the large Stokes shift fluorescence emission of DFAH. We also optimized the geometric structure of the transition state of DFAH. The frontier molecular orbitals and potential barrier show that the ESIPT process does not easy occur easily for DFAH. The enol structure of DFAH is more stable than that of the keto structure. The absence of the PET process resulted in the enol structure emitting strong fluorescence, which is consistent with the single fluorescence in the experiment. Above all, our calculations are sufficient to verify the sensing mechanism of H2S using DFAN.
1. Introduction
H2S, the smallest member among intracellular sulfur species, following nitric oxide (NO) and carbon monoxide (CO), has been identified as being the momentous gaseous signaling molecule.1,2 H2S can be biosynthesized from 3-mercaptopyruvate thiotransferase,3 and it plays critical roles in numerous biological processes such as the anti-inflammatory action,4 mitochondrial energy production, relaxation of blood vessels and the suppression of oxidative stress.5 Importantly, an imbalance of hydrogen sulfide in living systems causes a variety of diseases such as Huntington's,7 liver cirrhosis,8 low intelligence,6 Down's syndrome9 and Alzheimer's diseases.10 The amphoteric effects of H2S make it very important to monitor the concentration of H2S in vitro and in vivo. Therefore, the real-time detection and analysis of H2S are of considerable significance for disease diagnosis in the related biomedicine.
To date, numerous analysis approaches for tracing and measuring H2S have been employed, including metal-induced precipitation,11,12 electrochemical analysis,13,14 gas chromatography15,16 and fluorescence spectrometry.17–21 In particular, due to high sensitivity,22,23 noninvasive property,24 real-time spatial imaging26 and high throughput,27,28 fluorescent techniques are extremely attractive, which have been considered to be a reliable detection technique. It is different from traditional analytical methods; the advances in fluorescent probes can sense H2S inside the living cells, particularly, transduce the nanoscale biochemical processes into optical signals. In the biology field, based on the nucleophilic reaction, good reducibility towards azide compounds29,30 and high binding affinity towards copper ions,31–33 fluorescent probes for hydrogen sulfide are used. Moreover, it is difficult to design fluorescent probes to sense H2S from other biological mercaptans (biological nucleophile). It is worth noting that H2S has the smaller steric hindrance than other biological mercaptans and is a stronger nucleophile under physiological conditions. For instance, comparing 100 μM H2S with 1 mM GSH or cysteine, the selectivity of 7-azidocoumarin to H2S was obviously better.34
ESIPT, which takes place through intramolecular hydrogen bond X–H⋯Y, refers to a photochemical process in which a class of photoinduced enol–keto tautomerization occurs (Fig. S1†). Upon optical excitation, a new hydrogen bond X⋯H–Y is formed, after H transfers from X to Y.49–51 Due to the significant Stokes shift emission, ESIPT has attracted considerable attention in the fields of solar energy capture, optical storage,36 dual band emission37 and green fluorescent proteins.38 There are numerous fluorescent compounds with ESIPT properties, such as 2,6-diazabindole,4′-dimethylaminobrassinol,1,3-bis(2-pyridylimino)isoindoline and 3-hydroxylflavone derivatives.39,41,42 Recently, the hydrogen bond enhancement theory in the excited sate was proposed by Han et al.47,48 Based on the hydrogen bond strengthening theory and PET (Fig. S2†),45 a new novel fluorescent probe with the weak fluorescence was synthesized.54 The 2,4-dinitrophenyl ether group masks the cyan-appended benzyne unit and causes fluorescence quenching. After sensing H2S, the 2,4-dinitrophenyl ether group of DFAN was replaced by the hydroxyl group, forming a new compound DFAH and restoring the fluorescence characteristics (Scheme 1). In the experiment, the fluorescence quenching mechanism of the probe and the causes of the fluorescence emission of DFAH were not proposed.54 Thus, in this study, based on DFT and TD-DFT, we will investigate the fluorescence quenching of DFAN (probe) and the ESIPT of DFAH (reaction product). We have studied the variation of the hydrogen bond strength by analyzing the geometric parameters of hydrogen bond, infrared vibration spectrum (IR) and reduced density gradient (RDG). In order to study the proton transfer (PT) process, the potential energy curves of the ground and excited states of DFAH are constructed. The charge distribution is analyzed by frontier molecular orbital (FMOs),25 Mulliken charge analysis, and electrostatic potential diagram (MEPs), which provide strong evidence for the occurrence of the PET process and ESIPT reaction. The transition analysis and FMOs demonstrate that the fluorescence quenching is induced by the PET process. In order to detect H2S, this study will provide a reasonable sensing mechanism to facilitate the development of new fluorescent probes.
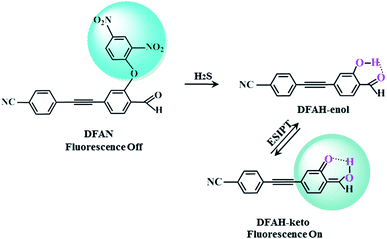 |
| | Scheme 1 Sensing mechanism of H2S by probe DFAN. | |
2. Computational details
In this study, theoretical calculations were conducted via DFT/TDDFT and Gaussian 16 program suite.35 The geometric structures and reaction pathways are calculated without constraint using B3LYP function (Hybrid Becke, 3-parameter, with 20% Hartree–Fock exchange energy) with the TZVP basis set.52,53 In order to accurately grasp the hydrogen bonding behavior, the dispersion term is also considered. The integral equation formalism (IEF) version of the polarisable continuum model (PCM) is used to simulate the experimental environment.46 In order to determine the local minimum of the ground state (S0) and the first excited state (S1), the vibration frequency of the optimized structure is analyzed. The potential energy curves are scanned in S0 and S1 states by fixing the bond length of the hydroxyl group (H2–O3) in the DFAH molecule as a series of values. We have used the Multiwfn software to analyze Mulliken's charge situation and to study the type of hydrogen bond interaction by applying RDG function using the Multiwfn software.40,44 The formula is as follows:
The weak interactions depend on the λ2 of eigenvalue and the ρ of electron density in view of Bader's atoms-in-molecules (AIM) theory. The relationship between them is as follows:
In order to ascertain B3LYP/TZVP, we have used other functionals and basis set to achieve the electronic spectrum information, and the details are listed in Table 1.
Table 1 Comparing theoretical absorption and emission data (corresponding oscillator strengths) for DFAN-abs, DFAH(N)-abs, DFAH(N)-flu forms based on different functionals (B3LYP, CAM-B3LYP, M06-2X, B1B95, PBEPBE, and WB97XD)
| |
B3LYP |
CAM-B3LYP |
M06-2X |
B1B95 |
PBEPBE |
WB97XD |
Exp. |
| DFAN-abs |
382(0.006) |
325(0.003) |
338(0.001) |
347(0.014) |
520(0.002) |
324(0.003) |
350 |
| DFAH(N)-abs |
362(1.223) |
318(1.193) |
313(1.266) |
345(1.253) |
429(0.226) |
314(1.105) |
405 |
| DFAH(N)-flu |
437(1.829) |
398(1.832) |
390(1.866) |
420(1.843) |
494(0.535) |
392(1.825) |
510 |
3. Results and discussion
3.1 Geometric structures, IR vibrational spectra and RDG isosurfaces
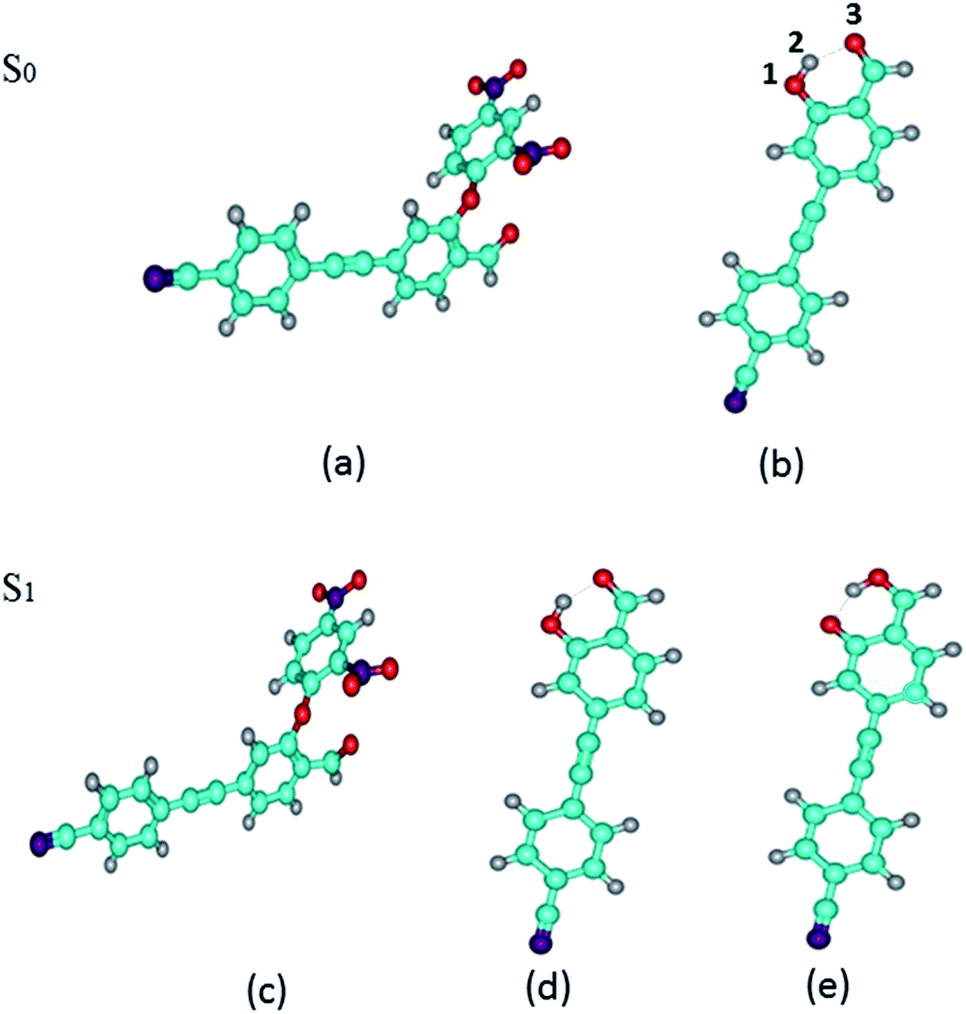
We have optimized the S0 and S1 states of DFAN and DFAH (enol and keto forms) geometries at the B3LYP/TZVP level. The optimized geometric structures are shown in Fig. 1. In Table 2, we also listed the related bond lengths and angles of enol, keto and transition state structures of DFAH. For DFAH-enol, the O1–H2 bond length is 0.987 Å in the S0 state, which increases to 0.993 Å in the S1 state, and the bond length is growing 0.006 Å; the hydrogen bond length of H2⋯O3 decreases from 1.741 Å in the S0 state to 1.693 Å in the S1 state. Moreover, the bond angle of O1–H2⋯O3 changes from 146.90° in the S0 state to 149.50° in the S1 state, which extends by 2.60°. It can be seen from the calculation results that the intramolecular hydrogen bond is strengthened in the S1 state. In particular, the DFAH-keto geometry in the S0 and S1 states are also optimized. Unexpectedly, after optimizing the S0 state, the stable structure is not obtained because of the high energy. In Table 2, we listed main bond lengths and angles of DFAH-keto in the S1 state. The change in the geometric structures of DFAH indicates that the photo excitation dominates the enhancement for the hydrogen bonds. Then, we also calculated the electron energies of DFAN and DFAH, which are listed in Table 2.
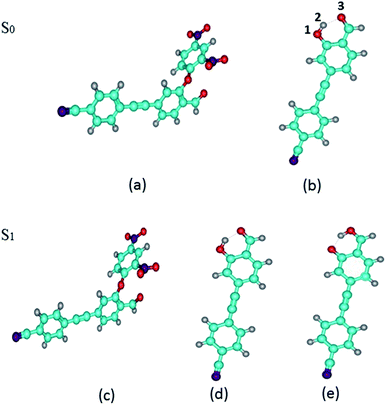 |
| | Fig. 1 Optimized geometries of DFAN and DFAH in the S0 and S1 states. (a) and (b) The S0 states of DFAN and DFAH-enol, respectively. (c)–(e) The S1 states of DFAN, DFAH-enol and DFAH-keto, respectively. The labeling of atomic color: C: blue; H: gray; O: red; N: purple. | |
Table 2 Calculated primary bond lengths (Å) and angles (°) for the enol, keto and transition-state configurations of DFAH in the S0 and S1 states
| Parameter |
Enol |
Keto (S1) |
Transition-state (S1) |
| S0 |
S1 |
| O1–H2 |
0.987 |
0.993 |
1.618 |
1.219 |
| H2–O3 |
1.741 |
1.693 |
1.016 |
1.224 |
| δ(O1–H2–O3) |
146.90 |
149.50 |
150.10 |
158.40 |
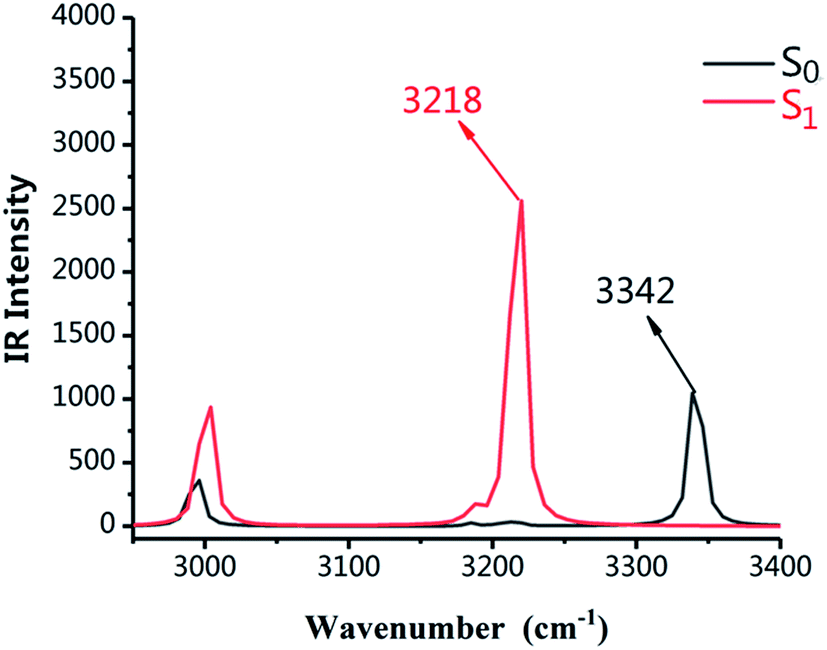
As shown in Fig. 2, the infrared vibration spectra of the main hydrogen bonds involved in the S0 and S1 states have been computed. The IR vibration frequency of H2–O3 is 3342 cm−1 and 3218 cm−1 in S0 and S1 states, respectively. In particular, from the S0 state to S1 state, the IR vibrational frequency of the H2–O3 bond of DFAH has a red-shift of 124 cm−1. Obviously, in the S1 state, the intramolecular hydrogen bond is strengthened.
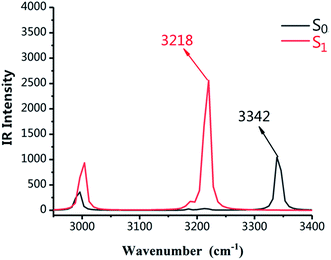 |
| | Fig. 2 Calculated IR spectra of the normal form of DFAH in the relevant spectral region of the O1–H2 stretching band in the S0 and S1 states. | |
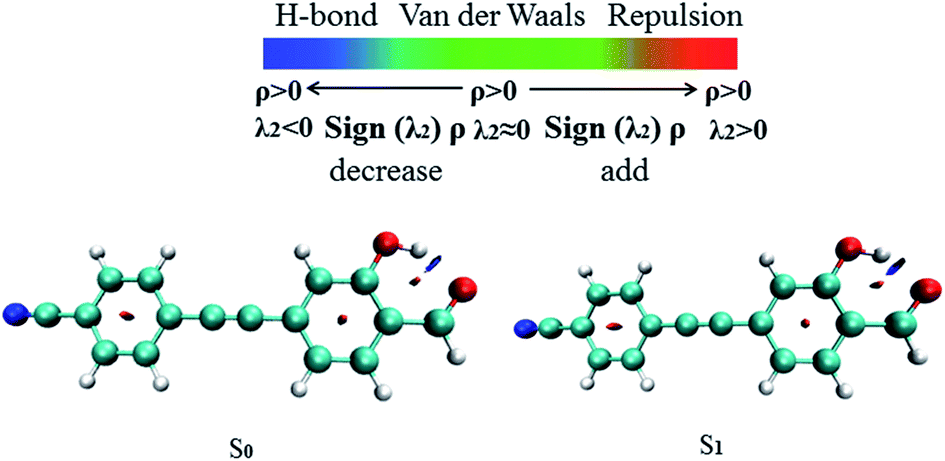
The RDG function has been used to further verify hydrogen bond properties. In Fig. 3 the color scale is blue, green and red from left to right, representing strong attraction, van der Waals force and strong mutual exclusion, respectively. The RDG isosurface range is from −0.04 to 0.02, and the isosurface of the hydrogen bond is blue. In the AIM theory, ρ(r) at the critical point of weak interactions is one of the important indexes to measure the strength of interaction, and there is a positive correlation between its value and bond strength. The larger ρ(r) of the blue region and sign(λ2) = −1, the stronger and more attractive weak interaction appears. In Fig. 3, it can be directly observed that the stronger the weak hydrogen bond interaction, the bluer the isosurface is, which proves the phenomenon of hydrogen bond enhancement in the excited state.
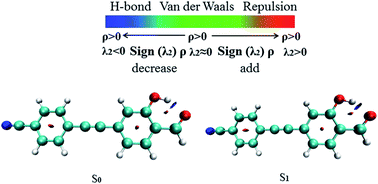 |
| | Fig. 3 The visual diagram of the RDG isosurface and color gradient axis. Blue indicates strong attractive interactions, and red indicates a strong bond-free overlap. | |
3.2 Potential energy curves of the proton transfer reactions analyses
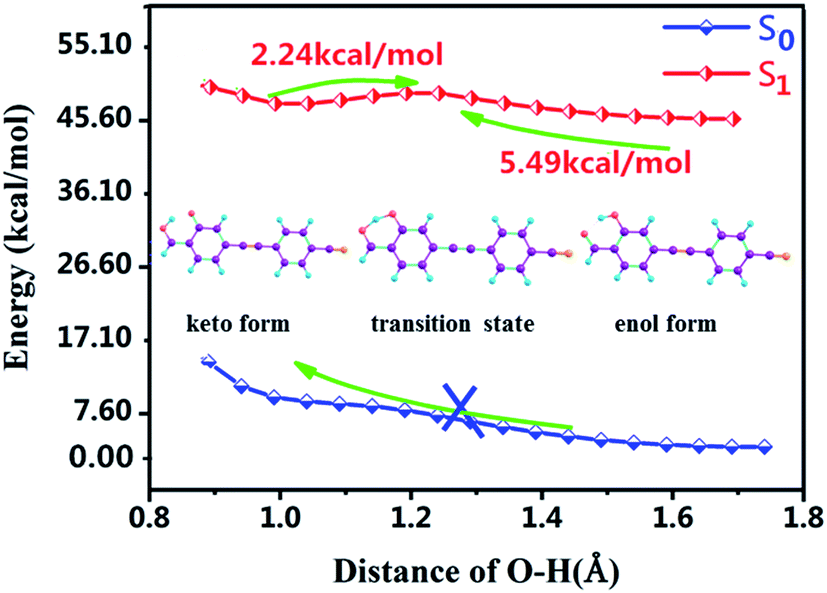
Moreover, for the lack of analyzing PT and ESIPT processes, we further plotted the potential energy curves (PECs) of the S0 and S1 states along the H2–O3 bond. As shown in Fig. 4, along the reaction pathway, the energy of DFAH system rises in the S0 state in the S0 state. Thus, the PT process is endothermic, and keto configuration cannot undergo PT. Furthermore, Fig. 4 also shows that in the S1 state, the energy barrier of DFAH-enol → DFAH-keto is 5.49 kcal mol−1, and is higher than the corresponding energy of the inverse reaction, which is only 2.24 kcal mol−1. In the S1 state, DFAH-keto is less stable than DFAH-enol, which can be easily converted into an intramolecular proton transfer enol configuration. Therefore, fluorescence can only be emitted by the enol configuration rather than the keto form, which is also consistent with the conclusion of the experiment.
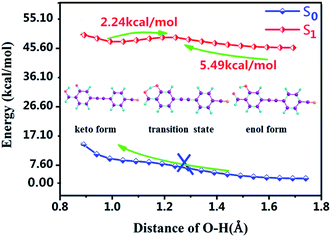 |
| | Fig. 4 Potential energy curves of the S0 and S1 states of DFAH as a function of the O3–H2 bond length. | |
3.3 Electron spectral analysis and charge population
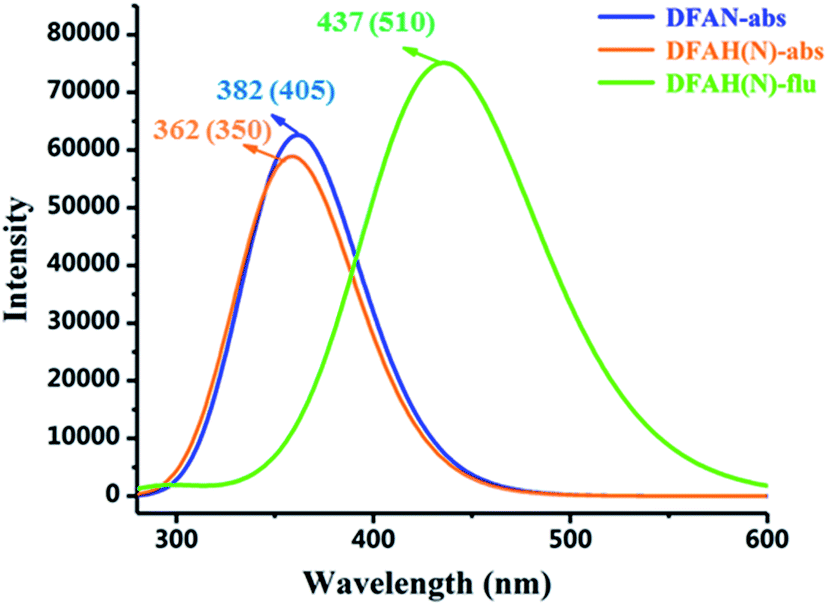
The absorption of DFAN and DFAH and the emission spectra of DFAH have been measured in the experiment.54 In order to compare with the experimental data, we calculated the absorption and emission spectra of DFAN and DFAH at the B3LYP/TZVP level (Fig. 5). In addition, the corresponding oscillator strengths for the orbital transitions and CI values involved are also listed (Tables 3 and 4). The main absorption band of DFAN is located at 382 nm, which is relatively consistent with the experimental absorption peak (350 nm). The calculated absorption peak of DFAH is located at 380 nm, which is close to the experimental value (405 nm). Kasha's rule points out that the fluorescence of the condensed matter is from S1 state. After light excitation, the calculated normal fluorescence peak of DFAH is located at 437 nm. Thus, the calculated results of the absorption spectrum and emission spectrum are in good agreement with the experiment.
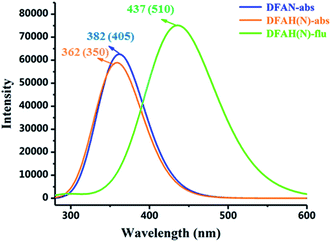 |
| | Fig. 5 The calculated absorption and fluorescence spectra of DFAN and DFAH based on the TD-DFT/B3LYP/TZVP level. The corresponding experimental values are given in the parenthesis. | |
Table 3 Comparison of experimental and calculated absorption at the TD-DFT/B3LYP/TZVP level, including energy (nm eV−1), oscillator strength (f) and orbital transition (OT) contributions to the electronic exited states (CI)
| Compound |
Electronic transition |
Energy (nm eV−1) |
Experimental absorption (nm eV−1) |
f |
Composition |
CI |
| DFAN |
S0–S1 |
382/3.25 |
350/3.54 |
0.0062 |
HOMO → LUMO |
89.6% |
| HOMO → LUMO+1 |
8.5% |
| S0–S2 |
369/3.36 |
|
0.0156 |
HOMO → LUMO+1 |
88.8% |
| HOMO → LUMO |
9.3% |
| S0–S3 |
362/3.43 |
|
1.5257 |
HOMO → LUMO+2 |
95.6% |
| DFAH(N) |
S0–S1 |
362/3.43 |
405/3.06 |
1.2227 |
HOMO → LUMO |
96.6% |
Table 4 Comparison of experimental and calculated emissions at the TD-DFT/B3LYP/TZVP level, including the energy (nm eV−1), oscillator strength (f) and orbital transition (OT) contributions to the electronic exited states (CI)
| Compound |
Electronic transition |
Energy (nm eV−1) |
Experimental emission (nm eV−1) |
f |
Composition |
CI |
| DFAN |
S1–S0 |
510/2.43 |
|
0.0072 |
LUMO → HOMO |
99.3% |
| S2–S0 |
412/3.00 |
|
0.6382 |
LUMO+1 → HOMO |
76.9% |
| LUMO+2 → HOMO |
21.2% |
| S3–S0 |
405/3.06 |
|
1.2562 |
LUMO+2 → HOMO |
76.4% |
| LUMO+1 → HOMO |
21.7% |
| S4–S0 |
397/3.12 |
|
0.0016 |
LUMO → HOMO−8 |
95.5% |
| DFAH(N) |
S1–S0 |
437/2.84 |
510/2.43 |
1.8292 |
LUMO → HOMO |
99.2% |
As shown in Fig. 6, the negative charge on O1 changed from −0.278 (S0) to −0.277 (S1), and from −0.399 (S0) to −0.408 (S1) for O3, respectively. Therefore, for DFAH-enol, the hydrogen bond is strengthened in the excited state. In order to analyze the hydrogen bonding effect on the whole electronic structure and to predict the possible electrophilic and nucleophilic reaction sites, the intramolecular electrostatic potential (MEP) was calculated. At the TD-B3LYP/TZVP level, the electrostatic potential isosurfaces on the excited state of DFAN and DFAH are plotted, as shown in Fig. 7. Different colors on the surface of the electrostatic potential mean different values. The positive electrostatic potential is represented in the red region, and the negative electrostatic potential is represented in the blue area. The H2 atom is in the hydrogen bond donor position, which represents the positive electrostatic potential. The negative potential on the O1 and O3 atoms indicate that they are the obvious hydrogen-bond acceptor sites.
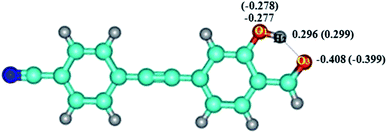 |
| | Fig. 6 Mulliken charge distribution of molecule DFAH in S0 and S1 states. The numbers in the parentheses represent the S0 state. | |
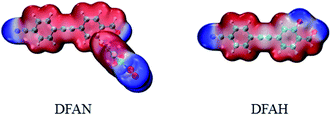 |
| | Fig. 7 The electron density isosurface mapped with the molecular electrostatic potential surface (MEPs) for DFAN and DFAH. | |
3.4 Frontier molecular orbital (FMO) analysis and sensing mechanism
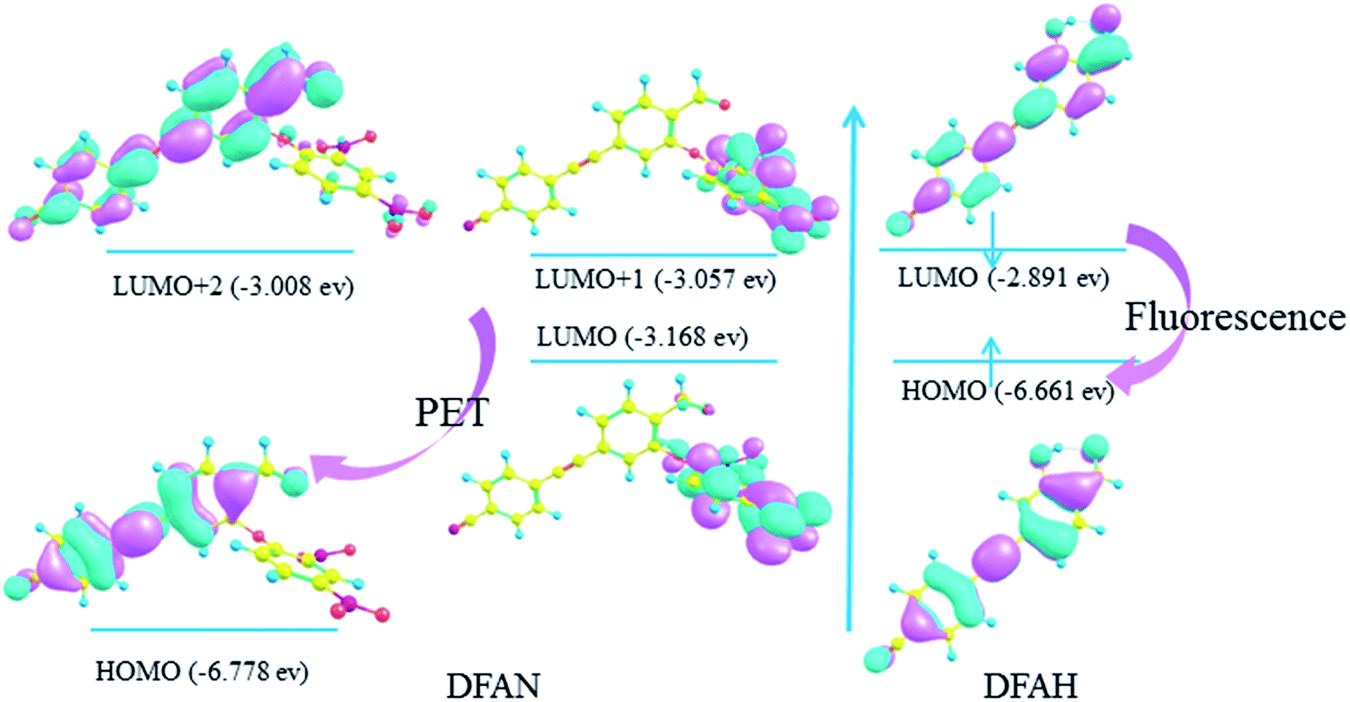
In general, the transition of electrons from one orbital to another after photo excitation accounts for induced charge redistribution.43 The calculated wavelengths, corresponding oscillator strength (f), compositions, and experimental values are listed in Tables 3 and 4, while the molecular orbitals are shown in Fig. 8. As shown in Table 3, the dominant transition of DFAN is the S0 → S1 transition, with the oscillator strength of 0.0062 lying at 350 nm, which is mainly assigned to the HOMO → LUMO (89.6%) transition with smaller components of HOMO → LUMO+1. The transition for HOMO → LUMO+2 is the local transition of ππ*-type between the cyanogroup cyan-appended benzyne unit and the carbonyl benzene unit, and the other two transitions are from the cyanogroup cyan-appended benzyne unit and the carbonyl benzene unit to the 2,4-dinitrophenyl ether group, respectively, with complete charge separation, as shown in Fig. 8. In particular, the energy of LUMO and LUMO+1 orbitals for the 2,4-dinitrophenyl ether group is located between HOMO and LUMO+2 orbitals for the cyanogroup cyan-appended benzyne unit and carbonyl benzene unit. Briefly, the complete charge separation from the cyanogroup cyan-appended benzyne unit and the carbonyl benzene unit to the 2,4-dinitrophenyl ether group in the relaxation process fits perfectly with the definition of the PET mechanism, which well-explains the fluorescence quenching effect. It can also be seen from Table 3 that the S0 → S1 transition for DFAH-enol is predicted at about 362 nm with an oscillator strength of 1.2227, and the process from HOMO to LUMO is assigned as a dominant ππ*-type transition. As shown in Fig. 8, the distribution of MO is non-localized on the DFAH conjugated system, indicating that the S1 state is the local excited state. Thus, the enol type of DFAH exhibits fluorescence emission.
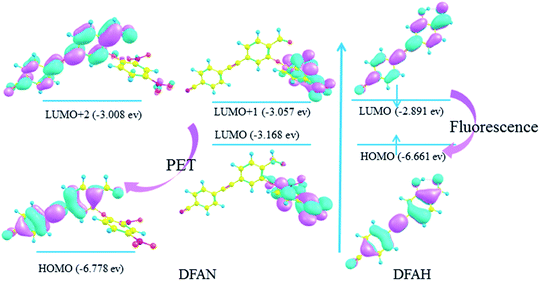 |
| | Fig. 8 Calculated frontier molecular orbitals involved in the absorption of DFAN and DFAH based on B3LYP/TZVP levels (HOMO, LUMO, LUMO+1 and LUMO+2). | |
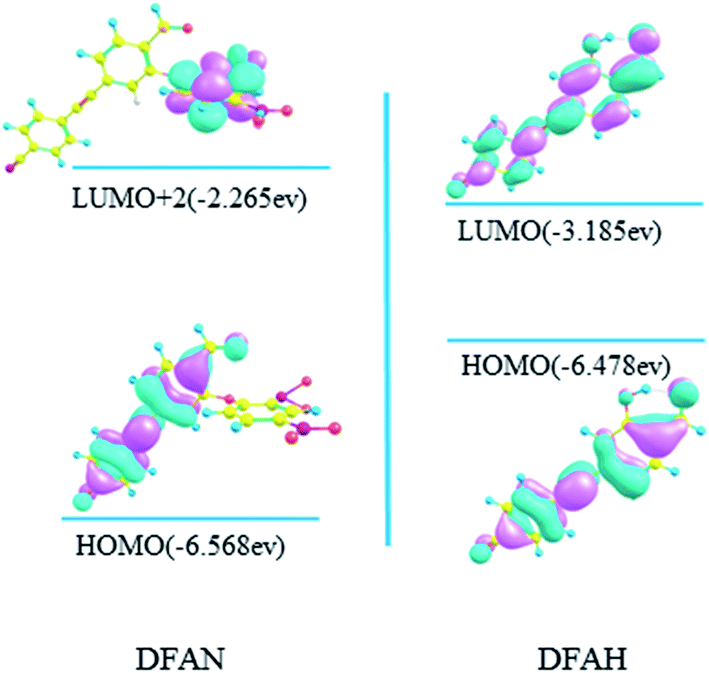
As shown in Table 4, the S1 state for DFAN is a dark state, and its oscillator strength is 0.0072. On the contrary, the S3 → S0 emission with a large oscillator strength of 1.2562 generates a ππ transition character assigned from the 2,4-dinitrophenyl ether group to the cyanogroup cyan-appended benzyne unit and the carbonyl benzene unit, as shown in Fig. 9. The fluorescence quenching effect is demonstrated by complete charge separation; moreover, the accuracy of the previous experimental and theoretical results is verified. Herein, the S1 → S0 transition of DFAH (shown in Fig. 9) corresponding to LUMO → HOMO still presents the local transition of electron density and consequently the strong fluorescence emits.
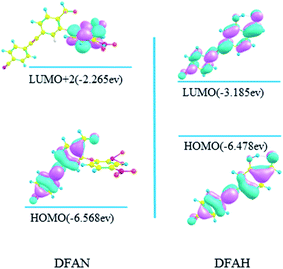 |
| | Fig. 9 Calculated frontier molecular orbitals involved in the emission of DFAN and DFAH based on B3LYP/TZVP levels (HOMO, LUMO and LUMO+2). | |
In general, the details of the sensing mechanism of this probe are as follows: the first and second excited states of DFAN are dark, and the PET process occurs with the complete charge separation between the cyanogroup cyan-appended benzyne unit, carbonyl benzene unit and 2,4-dinitrophenyl ether group, resulting in the disappearance of fluorescence. After the addition of H2S, the sulfur solution reaction of DFAN is triggered by a nucleophilic reaction, the group is eliminated, and DFAH is obtained. The S1 state of DFAH is the LE state, which produces strong fluorescence. The probe of DFAN is sensitive to discern hydrogen sulfide based on changes in the non-fluorescence signals.
4. Conclusions
In this study, the sensing mechanism of probe to H2S is studied via DFT and TDDFT methods. The structure of the S0 state and the S1 state of DFAN and DFAH were optimized, and the variation of the intramolecular hydrogen bond after light excitation was discussed by analyzing the bond length, bond angle, infrared spectrum and RDG isosurfaces, which indicate that DFAH-enol can be strengthened by the intra-HB in the excited state. The potential curve and the transition state show that both PT and ESIPT do not occur and the enol is a more stable configuration. The fluorescence change, which is the basis for the probe to detect H2S, can be explained by the PET sensing mechanism. Furthermore, the PET process caused the fluorescence quenching pathway of DFAN, and the ESIPT process of DFAH is ineffective. It is hoped that the results of our study will have some guiding significance for the design of new fluorescence probes in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Open Project of SKLMRD (Open Project of State Key Laboratory of Molecular Reaction Dynamics). The results of quantum chemical calculations described in this paper were obtained on the homemade Linux cluster of group 1101, Dalian Institute of Chemical Physics.
References
- L. Y. Chen, D. Wu, C. S. Lim, D. Kim, S. J. Nam, W. Lee, G. Kim, H. M. Kim and J. Yoon, Chem. Commun., 2017, 53, 4791–4794 RSC.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS.
- H. Kimura, Nihon Yakurigaku Zasshi, 2012, 139, 6–8 CrossRef CAS PubMed.
- R. C. O. Zanardo, V. Brancaleone, E. Distrutti, S. Fiorucci, G. Cirino and J. L. Wallace, FASEB J., 2006, 20, 2118–2120 CrossRef CAS PubMed.
- Y. Kimura, Y. I. Goto and H. Kimura, Antioxid. Redox Signaling, 2010, 12, 1–13 CrossRef CAS PubMed.
- B. Gu, N. X. Mi, Y. Y. Zhang, P. Yin, H. T. Li and S. Z. Yao, Anal. Chim. Acta, 2015, 879, 85–90 CrossRef CAS PubMed.
- S. Ulfuara, K. Min-Sik, J. N. Young and J. Y. Jung, Oxid. Med. Cell. Longevity, 2018, 1873962 Search PubMed.
- S. iorucci, E. Antonelli, A. Mencarelli, S. Orlandi, B. Renga, G. Rizzo, E. Distrutti, V. Shah and A. Morelli, Hepatology, 2005, 42, 539–548 CrossRef PubMed.
- T. Panagaki, E. B. Randi, F. Augsburger and C. Szabo, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18769–18771 CrossRef CAS PubMed.
- Y. Kaneko, Y. Kimura, H. Kimura and I. Niki, Diabetes, 2006, 55, 1391–1397 CrossRef CAS PubMed.
- S. Falconer, W. Wang, S. Gehrke, J. Cuneo, J. Britten, G. Wright and E. Brown, Chem. Biol., 2014, 21, 136–145 CrossRef CAS PubMed.
- M. Ishigami, K. Hiraki, K. Umemura, Y. k. Ogasawara, K. Ishii and H. Kimura, Antioxid. Redox Signaling, 2009, 11, 205–214 CrossRef CAS PubMed.
- N. S. Lawrence, M. Thompson, J. Davis, L. Jiang, T. G. J. Jones and R. G. Compton, Chem. Commun., 2002, 1028–1029 RSC.
- F. Chekin, F. Teodorescu, Y. Coffinier, G. H. Pan, A. Barras, R. Boukherroub and S. Szunerits, Biosens. Bioelectron., 2016, 85, 807–813 CrossRef CAS PubMed.
- J. B. Shi, C. Y. Liao, Y. W. Wang and G. B. Jiang, Guang Pu Xue Yu Guang Pu Fen Xi, 2016, 26, 336–339 Search PubMed.
- C. J. Richardson, E. A. M. Magee and J. H. Cummings, Clin. Chim. Acta, 2000, 293, 115–125 CrossRef CAS.
- D. Martin, M. Armelle and G. Erwan, ACS Sens., 2018, 3, 2138–2144 CrossRef.
- Z. Guo, G. Chen, G. Zeng, Z. Li, A. Chen, J. Wang and L. Jiang, Analyst, 2015, 140, 1772–1786 RSC.
- Y. Kimura, Y. Toyofuku, S. Koike, N. Shibuya, N. Nagahara, D. Lefer, Y. Ogasawara and H. Kimura, Sci. Rep., 2015, 5, 14774 CrossRef CAS PubMed.
- Y. Ma, C. Zhang, P. Yang, X. Li, L. Tong, F. Huang, J. Yue and B. Tang, Nanoscale, 2018, 10, 15793–15798 RSC.
- H. Zhang, Y. Xie, P. Wang, G. Chen, R. Liu, Y. W. Lam, Y. Hu, Q. Zhu and H. Sun, Talanta, 2015, 135, 149–154 CrossRef CAS PubMed.
- G. Yamamichi, W. Nakata, M. Tani, G. Tsujimura, Y. Tsujimoto, M. Nin, A. Mimura, H. Miwa and M. Tsujihata, Int. J. Clin. Oncol., 2019, 24, 1075–1080 CrossRef CAS PubMed.
- B. W. Buchan, D. A. Jobe, M. Mashock, D. Gerstbrein, M. L. Faron, N. A. Ledeboer and S. M. Callister, J. Clin. Microbiol., 2019, 57, e00513–e00519 CrossRef CAS PubMed.
- M. Gao, R. Wang, F. B. Yu and L. X. Chen, Biomaterials, 2018, 160, 1–14 CrossRef CAS PubMed.
- Y. P. Tao, L. G. Han, Y. X. Han and Z. J. Liu, Spectrochim. Acta, Part A, 2015, 137, 892–898 CrossRef CAS PubMed.
- J. Xiong, M. Zhao, X. Han, Z. Cao, X. Wei, Y. Chen, C. Duan and M. Yin, Sci. Rep., 2017, 7, 41311 CrossRef CAS PubMed.
- S. Zhuo, J. Gong, P. Zhang and C. Zhu, Talanta, 2015, 141, 21–25 CrossRef CAS PubMed.
- X. H. Wang, Z. Q. Guo, S. Q. Zhu, H. Tian and W. H. Zhu, Chem. Commun., 2014, 50, 13525–135288 RSC.
- Q. Q. Wan, Y. C. Song, Z. Li, X. H. Gao and H. M. Ma, Chem. Commun., 2013, 49, 502–504 RSC.
- H. Y. Tian, J. H. Qian, H. Y. Bai, Q. Sun, L. Y. Zhang and W. B. Zhang, Anal. Chim. Acta, 2013, 768, 136–142 CrossRef CAS PubMed.
- Y. Hao, W. Chen, L. Wang, X. Zhu, Y. Zhang, P. Qu, L. Liu, B. Zhou, Y. N. Liu and M. Xu, Talanta, 2015, 143, 307–314 CrossRef CAS.
- Y. G. Gao, K. Dan, W. J. Zhang, F. L. Liu, S. Patil, A. Qadir, A. X. Ding and A. R. Qian, Colloids Surf., B, 2020, 185, 110607 CrossRef CAS PubMed.
- K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003–18005 CrossRef CAS PubMed.
- T. S. Bailey and M. D. Pluth, J. Am. Chem. Soc., 2013, 135, 16697–16704 CrossRef CAS PubMed.
- P. W. Zhou and K. L. Han, Acc. Chem. Res., 2018, 51, 1681–1690 CrossRef CAS PubMed.
- Y. He, Y. Xu, Y. Shang, S. Zheng, W. Chen and Y. Pang, Anal. Bioanal. Chem., 2018, 410, 7007–7017 CrossRef CAS PubMed.
- X. F. Yang, Q. Huang, Y. Zhong, Z. Li, H. Li, M. Lowry, J. O. Escobedo and R. M. Strongin, Chem. Sci., 2014, 5, 2177–2183 RSC.
- Y. H. Chen, R. Sung and K. Sung, J. Phys. Chem. A, 2018, 122, 5931–5944 CrossRef CAS PubMed.
- Z. Tang, H. W. Wei and P. W. Zhou, J. Mol. Liq., 2019, 301, 112415 CrossRef.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Y. T. Qi, Z. Tang, H. B. Zhan, Y. Wang, Y. Zhao, F. Xu, T. N. Jing, Y. D. Ling and J. Y. Liu, Spectrochim. Acta, Part A, 2020, 117359 CrossRef CAS PubMed.
- Y. T. Qi, Y. Wang, Z. Tang, Q. S. Wang, Y. M. Hou, Z. Q. Gao, J. Tian and X. Fei, J. Mol. Liq., 2020, 314, 113614 CrossRef CAS.
- S. Ding, A. X. Xu, A. K. Sun, Y. Xia and Y. J. Liu, ACS Omega, 2020, 5, 19695–19701 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. M. Sanchez, J. C. Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- Y. Jiao, B. Zhu, J. H. Chen and X. H. Duan, Theranostics, 2015, 5, 173–187 CrossRef CAS PubMed.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, Phys. Chem. Chem. Phys., 2010, 12, 8914–8918 RSC.
- Y. Yang, Y. Chen, Y. Zhao, W. Shi, F. Ma and Y. Li, J. Lumin., 2019, 206, 326–334 CrossRef CAS.
- Y. Yang, Y. Ding, W. Shi, F. Ma and Y. Li, J. Lumin., 2020, 218, 116836 CrossRef CAS.
- Z. Tang and P. Zhou, J. Phys. Chem. B, 2020, 124, 3400–3407 CrossRef CAS PubMed.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- Y. Lu, B. L. Dong, W. H. Song, X. Q. Kong, A. H. Mehmood and W. Y. Lin, Anal. Methods, 2019, 11, 3301 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02511b |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Ziqing Gao
*a and
Ziqing Gao