
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTM4B180/− (TM = Hf, Ta, W, Re, Os): new structure construction with TM doped B wheel units†
Zhen Wang,
Qiuying Du and
Sung Jin Park *
*
Key Laboratory of Materials Modification by Laser, Ion and Electron Beams (Dalian University of Technology), Ministry of Education, Dalian 116024, China. E-mail: parksj@dlut.edu.cn
First published on 24th May 2021
Abstract
We report the global search for the lowest energy structures of the transition metal (TM) doped B clusters, TM4B180/− (TM = Hf, Ta, W, Re, Os) and their electronic properties. A combination of the comprehensive genetic algorithm (CGA) method with density functional theory (DFT) calculations shows that they are composed of four planar TM@B9 wheel units by sharing B atoms, except for Os4B180/−, which consists of two types of planar molecular wheels of Os@B7 and Os@B8. Among these nanoclusters, it is found that the Ta4B18 cluster has a closed-shell with a large HOMO–LUMO gap of 2.61 eV. Adaptive natural density partitioning analysis (AdNDP) reveals that the Ta4B18 cluster has σ antiaromaticity and π aromaticity, i.e., a conflicting aromaticity. The simulated photoelectron spectra (PES) of all anionic clusters are also provided for future experimental investigations.
1. Introduction
As an adjacent element of carbon (C), boron (B) has three valence electrons (2s22p1), and possesses a diverse and complex range of chemistry. Due to the characteristic of electron deficiency, B aggregates into various structures by sharing electrons and easily forms multicenter-two electron (mc–2e) bonds, which lead to various cluster structures.1–3 In the past decade, combining experimental and theoretical calculations, it was found that small and medium-sized pure B clusters could have the planar,4–6 quasi-planar,7–9 double ring,10,11 cage-like,12–15 bilayer,16,17 and core–shell18 structures. The Bn− clusters possess the planar or quasi-planar structures form up to the size of n ∼ 38, whereas the neutral counterparts from n = 20 exhibit a transition from the planar to the double-ring tubular shape.19 The discoveries of planar B360/−,9 fullerene-like B400/− (ref. 15) and bilayer B480/− (ref. 17) represent three major breakthroughs in the study of boron clusters. The planar B36 proves the viability of monolayer boron sheets with hexagonal vacancies, which leads to the concept of borophene. The cage-like B40 can be regarded as a boron analogue of C60 (ref. 20) and the bilayer B48 can be extended to a two-dimensional bilayer phase.16Doping transition-metal (TM) atoms is known as an effective approach to stabilize pure B clusters and to change their geometries and electronic properties. Up to now, the doping of B clusters with different numbers of metal atoms has led to many novel structures, e.g. (i) planar molecular wheel,21–23 (ii) half-sandwich,24 inverse sandwich25,26 and inverse triple-decker27 clusters, (iii) drum-like structures,28–30 (iv) the endohedral boron cages,31–34 and (v) metallo-borospherenes.35–38
Doping single TM atom into small-sized B clusters produces perfect TM-centered monocyclic B wheel clusters such as Co@B8−,21 Rh@B9−,22 and Ta@B10−.23 The 10 coordination number (CN) of Ta@B10− is known as the highest number among the planar species. With the increase of the number of B atoms, the structure growth pattern changes into the half-sandwich structures and the metal-centered B drum structures, such as Rh@B12−,24 Co@B16−,28 Rh@B18−,29 and Ta@B20−.30 Some highly stable endohedral B cages are also predicted by theoretical calculations, for example, Mo@B22,32 W@B24,33 and Co@B40−.34
A new class of di-metal-doped inverse sandwich complexes, including La2B7−,25 Pr2B8−,26 and La2B9−,25 have been observed by photoelectron spectra (PES) and density functional theory (DFT) calculations. Because of the unique (d–p)δ bond between metallic 5d orbitals and Bn rings, these lowest energy structures exhibit a higher level of stability than the other isomers. The first icosahedral clusters of M2B10 (M = Rh, Ir)39 were found in the theoretical investigations. Moreover, the PES results combined with DFT calculations has confirmed that the La3B14− cluster has a La–B8–La–B8–La inverse triple-decker structure, which is used to assemble 1D lanthanide B nanowires.27 More recently, the first metallo-borospherenes La3B18− and Tb3B18− (D3h)35 were observed in the experiment, and the calculations confirmed that their structures are composed of two B6 triangles linked together at their three corners with three B2 units. The core–shell spherical trihedral metallo-borospherene La3&[B2@B18]− (ref. 36) and the smallest metallo-borospherene Ta3B12− (ref. 37) were subsequently predicted. Among them, the D3h Ta3B12− compound is first metallo-borospherene with σ + π + δ triple aromaticity. The perfect core–shell La4[B@B4@B24]0/+/− clusters have been theoretically proposed to possess four equivalentinter-connected B6 triangles on the cage surface.38
In this work, we report the schematic study of the four TM atoms doped B18 clusters, TM4B180/− (TM = Hf, Ta, W, Re, Os). These clusters can be thought that they are built up with four TM doped B wheel units by sharing B atoms on their peripheral ring. Among them, the bonding pattern shows this Ta4B18 has 50 skeleton electrons on the cage surface suggesting a spherical aromatic system with filled 1s + 1p + 1d + 2s + 1f + 2p + 2d molecular orbitals. It is also found that the ground-state structure of Ta4B18 shows a conflicting aromaticity.
2. Computational methods
The optimization of the lowest energy structures of TM4B180/− (TM = Hf, Ta, W, Re, Os) were conducted using our developed comprehensive genetic algorithm (CGA) code40 incorporated with DFT calculations (CGA-DFT). The all-electron method with double-ζ numerical plus polarization d-function (DND) basis sets and the Perdew–Burke–Enzerhof (PBE) functional within the generalized gradient approximation (GGA)41 were used during each step of CGA using DMol3 package.42 Each structure was optimized without any symmetry constraint. The CGA code randomly generated sixteen initial parent configurations for each cluster system. The new structures were created by mating, perturbation, and exchange of the atom type of a pair of different types of atoms.43 In order to achieve the global minimum of potential energy surface (PES), all cluster systems had at least 3000 iterations.After the global search of CGA-DFT, the low-energy isomers were more accurately optimized by Gaussian16 program44 for TM4B180/− (TM = Hf, Ta, W, Re, Os). The previous studies proved the feasibility of the PBE0 functional45 to describe the energy differences between different isomers of TM doped B clusters.35,37,39,46–48 Moreover, our previous study of the single TM atom doped Bn (n = 7–10) clusters49 also confirmed that PBE0 functional can precisely describe the interactions between TM atom and B atom by comparing with the high-level CCSD(T)50 results. For basis set, the 6-311G* was proved enough to describe B atom in our study of pure boron clusters.51 We further calculated the equilibrium bond lengths and vibrational frequencies of TM (TM = Hf, Ta, W, Re, Os) dimer under the different basis sets and found that the def2-TZVP basis set is more suitable for TM atom (see Table S1†). Therefore, PBE0 functional combined with 6-311G* basis set for B and def2-TZVP basis set for TM atoms were chosen for our systems. Furthermore, chemical bonding analyses were performed using the adaptive natural density partitioning (AdNDP 2.0) program.52
3. Results and discussion
3.1. Lowest energy structures of TM4B180/− (TM = Hf, Ta, W, Re, Os)
The optimized lowest energy structures of neutral TM4B18 (TM = Hf, Ta, W, Re, Os) clusters and corresponding anionic species, along with their point group symmetries with 0.1 Å tolerance, are presented in Fig. 1. More information about the low-lying isomer structures is given in Fig. S1 of the ESI.† The summary of the structural and electronic properties of the ground state in both neutral and anionic series is listed in Table 1. All ground states are found to be singlet or doublet with exception of the Re4B18 cluster, which has the triplet states of spin multiplicity.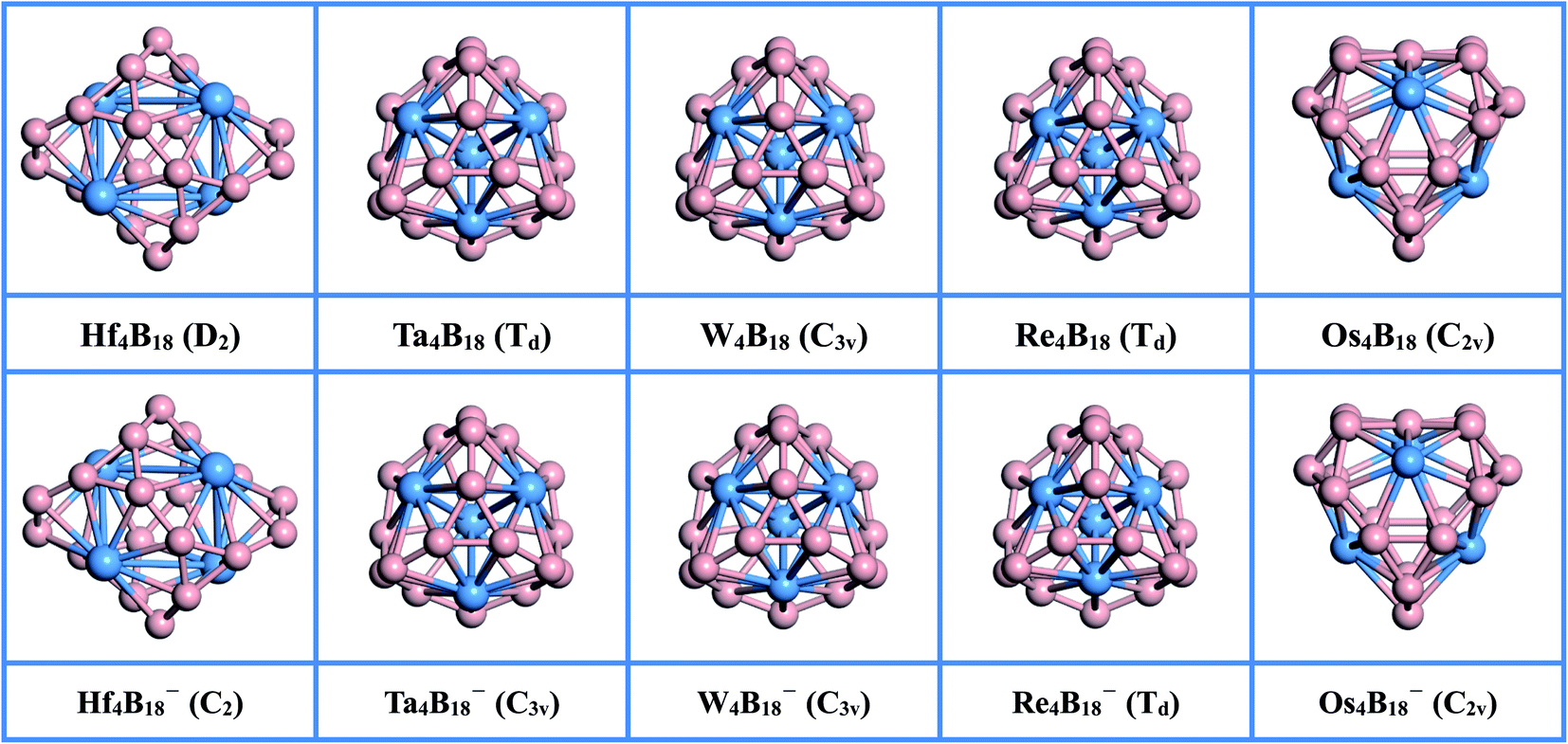 | ||
| Fig. 1 The lowest energy structures of TM4B18 (TM = Hf, Ta, W, Re, Os) clusters (upper panel) and corresponding anionic clusters (lower panel). The point group symmetry of each cluster is presented in parentheses. The blue and pink spheres are TM and B, respectively. | ||
| Min–Max (RTM–TM) | Min–Max (RTM–B) | Min–Max (RB–B) | QTM | Eb | EHL | ωmin | |
|---|---|---|---|---|---|---|---|
| Hf4B18 (D2) | 3.132–3.560 (3.346) | 2.378–2.458 (2.419) | 1.533–1.783 (1.635) | 1.148 | 5.80 | 2.60 | 120.33 |
| Hf4B18− (C2) | 3.106–3.541 (3.324) | 2.379–2.468 (2.421) | 1.537–1.792 (1.640) | 1.007 | 5.89 | 1.54 | 64.17 |
| Ta4B18 (Td) | 3.011 (3.011) | 2.351–2.358 (2.353) | 1.565–1.697 (1.631) | 0.487 | 6.02 | 2.61 | 158.80 |
| Ta4B18− (C3v) | 2.946–3.073 (3.010) | 2.332–2.378 (2.358) | 1.565–1.701 (1.634) | 0.383 | 6.13 | 1.70 | 121.59 |
| W4B18 (C3v) | 2.609–2.912 (2.761) | 2.294–2.365 (2.333) | 1.555–1.720 (1.629) | 0.068 | 6.16 | 2.41 | 136.90 |
| W4B18− (C3v) | 2.666–2.831 (2.749) | 2.315–2.352 (2.332) | 1.562–1.706 (1.629) | −0.042 | 6.30 | 1.87 | 126.07 |
| Re4B18 (Td) | 2.571–2.574 (2.573) | 2.298–2.359 (2.319) | 1.568–1.694 (1.629) | −0.166 | 6.10 | 1.85 | 216.66 |
| Re4B18− (Td) | 2.541–2.561 (2.554) | 2.289–2.391 (2.326) | 1.570–1.712 (1.639) | −0.189 | 6.26 | 1.37 | 153.41 |
| Os4B18 (C2v) | — | 2.104–2.250 (2.170) | 1.564–1.766 (1.681) | −0.089 | 5.95 | 1.69 | 102.89 |
| Os4B18− (C2v) | — | 2.108–2.278 (2.170) | 1.576–1.759 (1.687) | −0.190 | 6.09 | 1.55 | 111.05 |
As shown in the upper panel of Fig. 1, the lowest energy configuration of the neutral Hf4B18 has D2 symmetry and it is composed of four twisted umbrella-like Hf@B9 units by sharing the B atoms on their rings. The distances between the Hf atoms are in the range of 3.132 to 3.560 Å, and the average bond length between metal atoms (RTM–TM) is 3.346 Å. The bonding lengths between each Hf atom and its neighboring B atoms are in between 2.378 and 2.458 Å, and the average bond length between metal atom and B (RTM–B) is 2.419 Å. The distances between B atoms are in between 1.533 and 1.783 Å, which smallest value is slightly shorter than the B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B double bond (1.56 Å)53 and the largest value is longer than the typical B–B single bond (1.70 Å).53 Besides, the natural population analysis (NPA) shows that each Hf serves as the donor of 1.148 |e| to the B18 skeleton, implying the formation of typical charge-transfer Hf44+B184− complexes.
B double bond (1.56 Å)53 and the largest value is longer than the typical B–B single bond (1.70 Å).53 Besides, the natural population analysis (NPA) shows that each Hf serves as the donor of 1.148 |e| to the B18 skeleton, implying the formation of typical charge-transfer Hf44+B184− complexes.
The Ta4B18 forms a structure with the highly-symmetric point group of Td with the electronic state of 1A1. The Ta4B18 is composed of the previously reported the four planar molecular Ta@B9 wheels47 by sharing the B atoms on the rings. The total energy of this structure is much lower than the second low-lying isomers (C2) by 1.114 eV at the levels of PBE0/TZVP (see Fig. S1†). The interatomic distance between Ta atoms is 3.011 Å. The bonding lengths between each Ta atom and B atoms are in the range of 2.351 to 2.358 Å, which are slightly shorter than those in the freestanding Ta@B9 (2.39 Å),47 and the lengths between adjacent B atoms (1.565–1.697 Å) are longer than Ta@B9 (1.54 Å)47 at the PBE0 levels, suggesting that doping more Ta atom weakens the interaction between B atoms and strengthen the Ta and B bonding. The Ta4B18 also can be regarded as the Ta4 cluster in the middle bonded with the B atoms. We found that the lowest energy structure of Ta4 cluster has a tetrahedral structure with Td symmetry and its bonding length is 2.541 Å, which is significantly shorter than that of Ta4 moiety (3.011 Å) in Ta4B18. It shows that the strong interaction between Ta and B atoms weakens the bonding strength between Ta atoms, resulting in longer lengths. Moreover, NPA shows each Ta atom donates 0.487 |e| to the B18 skeleton forming a covalent bond due to the larger B electronegativity.
For the lowest energy structures of W4B18 and Re4B18, their geometric configurations are very similar to the Ta4B18 cluster. Doping Re atoms into the B18 framework leads to a magnetic cluster Re4B18 (Td) with the triplet states of spin multiplicity. The influence of atomic radius and magnetic moment makes it have a higher symmetry than W4B18 (C3v). The average distances of W4B18 between W atoms (RTM–TM), and between W and B atoms (RTM–B), are 2.761 and 2.333 Å, respectively. For the Re4B18 cluster, the RTM–TM and RTM–B are 2.573 Å and 2.319 Å, respectively. It can be seen that RTM–TM and RTM–B of the Re4B18 cluster are shorter than those of the W4B18 cluster, but the average bond length between B atoms (RB–B) is the same (1.629 Å). For Re4B18, compared with the previously reported planar molecular wheel Re@B9,54 we find that the bonding length of Re4B18 (1.568–1.694 Å) between adjacent B atoms are longer than the Re@B9 (1.543–1.571 Å) at the PBE0 levels.
The optimized structure of Os4B18 is a hollow cage-like structure with C2v symmetry, which is very different from the other TM4B18 (TM = Hf, Ta, W, Re) clusters. The Os4B18 is insufficient to support the large spherical B skeleton due to the further reduction of metal atomic radius. Therefore, the lowest energy structure of cage-like Os4B18 is assembled by two types of planar molecular wheels of Os@B7 and Os@B8, and the Os atoms on cage surface with the coordination numbers (CN) are 7 and 8. Moreover, the bonding length between B atoms are in the range of 1.564 to 1.766 Å, and the average (RB–B) is 1.681 Å, which is very close to the single bond value. The bonding lengths between each Os atom and its neighboring B atom are in the range of 2.104–2.250 Å, and the RTM–B is 2.170 Å. It can be seen from Table 1 that the average distance between Os and B atoms (RTM–B) becomes shorter rapidly when the size of metal atom decreases, which leads to a tighter bond between the TM and B atom. The coordinates of the lowest energy structures of TM4B18 (TM = Hf, Ta, W, Re, Os) are listed in Table S2 of the ESI.†
All corresponding global minima of anionic clusters are exhibited in the lower panel of Fig. 1. The geometric structures of TM4B18− (TM = Hf, Ta, W, Re, Os) are very similar to their corresponding neutral clusters. However, owing to the Jahn–Teller effect, the capture of one additional electron results in the low point group symmetries for Hf and Ta. The lowest energy structure of Hf4B18− has C2 symmetry, and each Hf atom transfers fewer electrons (1.007 |e|) to the B18 skeleton than the neutral. The structures of TM4B18− (TM = Ta, W, Re) are very similar like the neutral ones and consist of planar molecular wheels of the TM@B9 unit. However, compared with the corresponding neutral clusters, their RTM–TM is shorter and RB–B is slightly longer. NPA shows each TM atom donates electrons to the B18 skeleton in the range of −0.189 |e| to 0.383 |e|, which forms the typical covalent bonds. The structure of Os4B18− (C2v) is also a hollow cage-like structure, its RTM–B and RB–B are little changed. The coordinates of all anionic clusters are listed in Table S3 of the ESI.† The TM4B180/− (TM = Hf, Ta, W, Re, Os) clusters can be thought that they are constructed with four TM doped B wheel units by sharing B atoms. This approach could be a new pathway to produce various TM doped B cluster structures.
To gain a better understanding of the stability of these nanoclusters, we further examined the electronic properties. The binding energies per atom (Eb) is regarded as an effective parameter to evaluate the thermodynamic stability of a cluster, which is calculated by
| Eb = (4ETM + 18EB − ETM4B18−/0)/22 | (1) |
In the eqn (1), ETM4B18−/0, ETM and EB represent the total energy of TM4B18−/0 (TM = Hf, Ta, W, Re, Os) clusters, a TM atom, and a B atom, respectively. Here, the larger Eb value implies the more favorable thermodynamic stability of a cluster. In neutral clusters, the W4B18 has a maximum Eb value of 6.16 eV, while the Hf4B18 has a minimum Eb value (5.80 eV). The Eb values of TM4B18 (Ta, W, Re) are larger than those of others, so these clusters have a higher thermodynamic stability. The same trend is observed for the corresponding anions. As a reflection of the energy cost for an electron jumping from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), the HOMO–LUMO energy gap (EHL) can reveal the chemical stability of a cluster. In comparison with cage-like Ta3B12− which possess a EHL of 2.50 eV with three equivalent Ta@B8 octagons sharing two eclipsed B3 triangles at the top and bottom interconnected by three B2 units on the waist,37 the lowest energy structures of Hf4B18 and Ta4B18 possess a large EHL of 2.60 and 2.61 eV, respectively, being less chemically reactive than others. For all anionic clusters, due to the trapping of an electron, the EHL of these species is decreased and significantly less than the neutral clusters. Among them, the W4B18− cluster has the largest EHL value of 1.87 eV, while the EHL of the Re4B18− cluster is the smallest, only 1.37 eV. Moreover, vibrational frequency calculations confirm that there are no imaginary frequencies for all these ground-state structures, and the corresponding lowest frequencies are listed in Table 1.
3.2. Bonding analysis
The structural and electronic properties of TM4B180/−(TM = Hf, Ta, W, Re, Os) nanoclusters shows that the neutral Ta4B18 and Hf4B18 clusters are chemically more inert than the others (larger EHL)55 but relatively the Ta4B18 has larger Eb (thermodynamically more stable)56 than Hf4B18. Based on this analysis, we further investigate the bonding properties of the Ta4B18 cluster. The molecular orbital (MO) energy-level diagram and the relevant MOs of Ta4B18 derived from the Ta4 moiety and B18 skeleton, which is presented in Fig. 2. It shows the interactions between the orbitals of the four Ta atoms and the group orbitals of the B18 skeleton. Among them, LUMO, HOMO and HOMO − n represent the energy-levels of the Ta4B18 cluster, respectively. LUMO′, LUMO + n′, HOMO′ and HOMO − n′ represent the energy-levels of Ta4 moiety. LUMO′′, LUMO + n′′, HOMO′′ and HOMO − n′′ represent the energy-levels of the B18 skeleton. Since the global minimum of B18 is a planar structure, the stabilization of the 3D B18 framework is entirely due to its strong bonding with the Ta4 moiety. The 3D B18 skeleton has a small HOMO–LUMO gap of 0.85 eV. The addition of four Ta atoms results in a closed-shell Ta4B18 with a large HOMO–LUMO gap of 2.61 eV. It is noted that the Td symmetry of Ta4B18 leads to the feature level of LUMO, HOMO, HOMO − 1 and HOMO − 2 are triple degenerates. Furthermore, we find that the LUMO + 2′ of Ta4 moiety is unoccupied and mainly becomes the LUMO orbital of Ta4B18. For the occupied MOs of Ta4B18, HOMO, HOMO − 1 and HOMO − 2 are formed from LUMO + 2′′ (σ orbits), LUMO + 3′′ (σ orbits), and HOMO − 1′′ (π orbits) of B18 framework, respectively. The HOMO − 3 is a σ + π hybrid orbital, which derives from the mixing of LUMO + 1′ (π orbit) with LUMO + 1′′ (σ orbit). The HOMO − 4 of σ + σ hybrid orbital is formed from two σ orbit of HOMO − 2′ and HOMO − 4′′. Moreover, the Ta atom in Ta4B18 has total on-site Wiberg bond order (WBO) of 5.82, which well supporting the spherical coordination interactions between Ta4 moiety and B18 skeleton.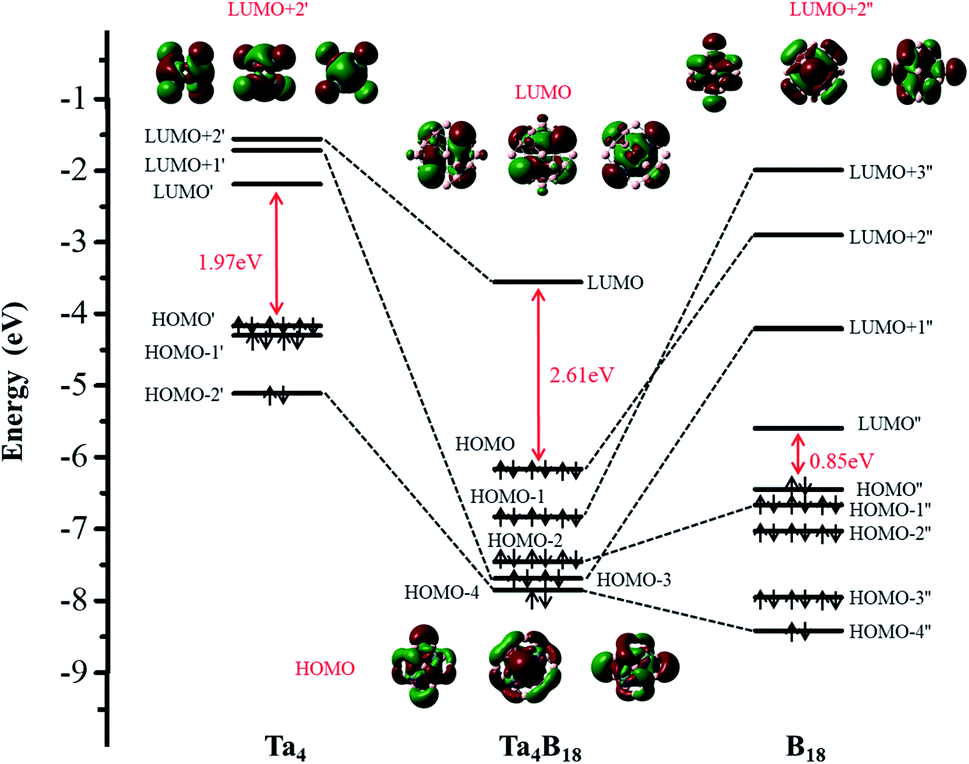 | ||
| Fig. 2 The Kohn–Sham molecular orbital correlation diagram for Ta4B18. It shows the interactions between the orbitals of the Ta4 atoms and the group orbitals of the B18 skeleton. | ||
We also performed a chemical bonding analysis of the spherical Ta4B18 using the AdNDP method. As depicted in Fig. 3, the Ta4B18 cluster contains 12 localized bonds and 25 delocalized bonds, ordered by occupation number (ON) ranging from 1.80 |e| to 2.00 |e|. The 12 localized B–B bonds are all 2c–2e σ bonds on the peripheral edge of the B18 skeleton, which are mainly composed of B 2s/2p electrons, and their ONs are 1.80 |e|. Among the 25 delocalized bonds, there are 4 equivalent 3c–2e σ bonds on the B3 triangles (ON = 1.85 |e|). The three sets of the delocalized 10c–2e Ta–B9 bonds (ON = 1.88–1.90 |e|) over the boron skeleton, which includes 4 equivalent Ta (dx2−y2)–B9 (σ) bonds, 4 equivalent Ta (dxy)–B9 (σ) bonds, and 4 equivalent Ta (dz2)–B9 (π) bonds. As shown in the bottom row of Fig. 3, there are 9 totally delocalized 22c–2e bonds distributed on the entire spherical skeleton with ON = 2.00 |e|. Among them, 6 are σ and the other 3 are π bonds. Therefore, the Ta4B18 cluster has 10 delocalized σ bonds in total (4 equivalent 3c–2e σ bonds plus 6 equivalent 22c–2e bonds), leading to the σ antiaromaticity according to 4n (n = 5) Hückel's rule. Meanwhile, the 3 totally delocalized π-bonds (22c–2e bonds) satisfies the Hückel rules of 4n + 2 (n = 1) of π aromaticity. Thus, Ta4B18 is a conflicting aromatic system with 20 σ and 6 π totally delocalized electrons. Note that 50 skeletal electrons are distributed on the cage surface is a magic number for a closed-shell three-dimensional spherical structure in which the 25 delocalized orbitals are completely filled with electron pairs leading to a closed-shell 1S21P61D102S21F142P62D10 configuration.
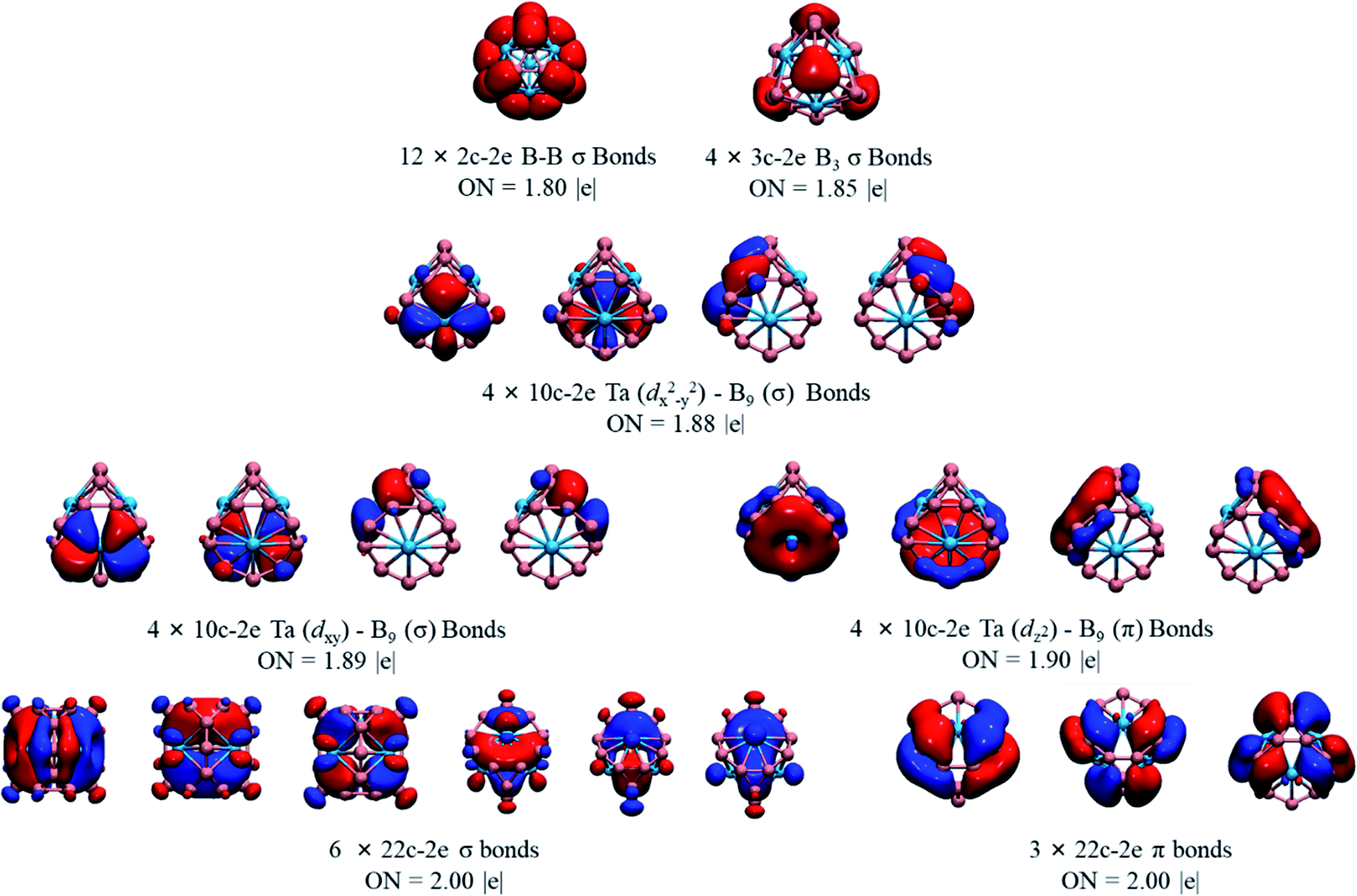 | ||
| Fig. 3 AdNDP bonding patterns of Ta4B18, with the occupation numbers (ON). | ||
3.3. Simulated photoelectron spectra of TM4B18− (TM = Hf, Ta, W, Re, Os)
Photoelectron spectra (PES) can be used as the fingerprints about the electronic structures of nanoclusters. Therefore, we simulated the PES of TM4B18− (TM = Hf, Ta, W, Re, Os) anionic clusters, hoping to help experimentally determine these lowest energy structures.First, we consider the simulated PES of Ta4B18− and Re4B18− by comparing with the experimentally reported of Ta@B9− and Re@B9−. As displayed in Fig. 4, the spectral features are labeled X, A, B, etc. In each spectrum, the X peak represents the vertical detachment energy (VDE) which denotes the transition from the anionic ground-state to the neutral ground-state, and the other (A, B, etc.) peaks indicate transitions to the excited state of the neutral complexes. The VDE of Ta4B18− is approximately 2.77 eV. After this first peak, there are five peaks between 3.5 eV and 5.5 eV. In the experimental spectrum of Ta@B9−, the first X peak is located at around 3.64 eV,47 indicating the structure of Ta4B18− assembled by Ta@B9− has the lower electron binding energy. For the simulated spectrum of Re4B18−, the first two peaks are somewhat weak and close each other. The experimental PES of Re@B9− also shows two close peaks at 4.02 eV and 4.34 eV,54 respectively. These lower binding energies of Re4B18− are similar to those of Ta4B18− by comparison with the experimental results. Presumably, this is caused by the interaction between planar molecular unit wheels (Ta@B9 and Re@B9).
 | ||
| Fig. 4 The simulated photoelectron spectra of TM4B18− (TM = Hf, Ta, W, Re, Os) clusters. | ||
The PES of Hf4B18− is simulated and shows a compact spectral pattern, and the first peak is approximately 2.16 eV, following by four consecutive peaks of the same intensity. There are four major peaks (X, A, B and C) of the simulated spectrum of W4B18−, and the VDE is approximately 3.19 eV. The simulated spectrum of Os4B18− also presents a compact spectral pattern with a VDE of 3.02 eV. To provide detailed data for the future experiment, we further calculated the vertical ionization potentials (VIP) and vertical electron affinities of neutral clusters, and adiabatic detachment energy (ADE) of anionic clusters, which are shown in Table 2. The W4B18 shows a larger VIP value (7.52 eV) and VEA value (2.82 eV) than others. The Re4B18 shows a larger ADE value of 3.07 eV than others. However, the spherical Ta4B18 has a large VIP value (7.37 eV) and a moderate VEA value (2.46 eV).
| VIPa | VEAb | VDEc | ADEd | ||
|---|---|---|---|---|---|
| a Vertical ionization energy from the ground state of the neutral to the ground state of the cation.b Vertical electronic affinity from the ground state of the neutral to the ground state of the anion.c Vertical detachment energy from the ground state of the anion to the ground state of the neutral.d Adiabatic detachment energy from the ground state of the anion to the ground state of the neutral. | |||||
| Hf4B18 (D2) | 6.65 | 1.53 | Hf4B18− (C2) | 2.16 | 1.88 |
| Ta4B18 (Td) | 7.37 | 2.46 | Ta4B18− (C3v) | 2.77 | 2.65 |
| W4B18 (C3v) | 7.52 | 2.82 | W4B18− (C3v) | 3.19 | 3.01 |
| Re4B18 (Td) | 7.25 | 2.13 | Re4B18− (Td) | 3.49 | 3.07 |
| Os4B18 (C2v) | 6.77 | 2.90 | Os4B18− (C2v) | 3.02 | 2.97 |
4. Conclusion
We carried out unbiased search for the lowest energy structures of TM4B180/− clusters (TM = Hf, Ta, W, Re, Os). The structural analysis shows that they are composed of the four planar molecular TM@B9 wheel units sharing the B atoms except for Os4B180/−, which has a hollow cage-like structure assembled by two types of planar molecular wheels of Os@B7 and Os@B8 due to the reduction of the atomic radius. According the electronic properties, spherical Ta4B18 has large Eb and EHL. The chemical bonding analyses showed that it has the σ antiaromaticity with 4n (n = 5) and π aromaticity with 4n + 2 (n = 1) from Hückel's rule, resulting in a conflicting aromatic system. Finally, the PES of all anionic clusters was simulated which provides predictive information for future experimental investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Funds for the Central Universities of China (DUT20RC(5)014), and the Supercomputing Center of Dalian University of Technology.References
- A. I. Boldyrev and L.-S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 11589–11605 RSC.
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- D. Li, J. Gao, P. Cheng, J. He, Y. Yin, Y. Hu, L. Chen, Y. Cheng and J. Zhao, Adv. Funct. Mater., 2020, 30, 1904349 CrossRef CAS.
- H.-J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS PubMed.
- A. P. Sergeeva, D. Y. Zubarev, H.-J. Zhai, A. I. Boldyrev and L.-S. Wang, J. Am. Chem. Soc., 2008, 130, 7244–7246 CrossRef CAS PubMed.
- W. Huang, A. P. Sergeeva, H.-J. Zhai, B. B. Averkiev, L.-S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed.
- W.-L. Li, Y.-F. Zhao, H.-S. Hu, J. Li and L.-S. Wang, Angew. Chem., Int. Ed., 2014, 126, 5646–5651 CrossRef.
- W.-L. Li, Q. Chen, W.-J. Tian, H. Bai, Y.-F. Zhao, H.-S. Hu, J. Li, H.-J. Zhai, S.-D. Li and L.-S. Wang, J. Am. Chem. Soc., 2014, 136, 12257–12260 CrossRef CAS PubMed.
- Z. A. Piazza, H.-S. Hu, W.-L. Li, Y.-F. Zhao, J. Li and L.-S. Wang, Nat. Commun., 2014, 5, 3113 CrossRef PubMed.
- B. Kiran, S. Bulusu, H.-J. Zhai, S. Yoo, X. C. Zeng and L.-S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 961–964 CrossRef CAS PubMed.
- X. Wu, L. Sai, S. Zhou, P. Zhou, M. Chen, M. Springborg and J. Zhao, Phys. Chem. Chem. Phys., 2020, 22, 12959–12966 RSC.
- J. Zhao, X. Huang, R. Shi, H. Liu, Y. Su and R. B. King, Nanoscale, 2015, 7, 15086–15090 RSC.
- Y.-J. Wang, Y.-F. Zhao, W.-L. Li, T. Jian, Q. Chen, X.-R. You, T. Ou, X.-Y. Zhao, H.-J. Zhai, S.-D. Li, J. Li and L.-S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Nanoscale, 2014, 6, 11692–11696 RSC.
- H.-J. Zhai, Y.-F. Zhao, W.-L. Li, Q. Chen, H. Bai, H.-S. Hu, Z. A. Piazza, W.-J. Tian, H.-G. Lu, Y.-B. Wu, Y.-W. Mu, G.-F. Wei, Z.-P. Liu, J. Li, S.-D. Li and L.-S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed.
- L. Sai, X. Wu, N. Gao, J. Zhao and R. B. King, Nanoscale, 2017, 9, 13905–13909 RSC.
- W.-J. Chen, Y.-Y. Ma, T.-T. Chen, M.-Z. Ao, D.-F. Yuan, Q. Chen, X.-X. Tian, Y.-W. Mu, S.-D. Li and L.-S. Wang, Nanoscale, 2021, 13, 3868–3876 RSC.
- J. Zhao, L. Wang, F. Li and Z. Chen, J. Phys. Chem. A, 2010, 114, 9969–9972 CrossRef CAS PubMed.
- T. Jian, X. Chen, S.-D. Li, A. I. Boldyrev, J. Li and L.-S. Wang, Chem. Soc. Rev., 2019, 48, 3550–3591 RSC.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- C. Romanescu, T. R. Galeev, W.-L. Li, A. I. Boldyrev and L.-S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334–9337 CrossRef CAS PubMed.
- W.-L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L.-S. Wang, J. Am. Chem. Soc., 2012, 134, 165–168 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W.-L. Li, A. I. Boldyrev and L.-S. Wang, Acc. Chem. Res., 2013, 46, 350–358 CrossRef CAS PubMed.
- I. A. Popov, W.-L. Li, Z. A. Piazza, A. I. Boldyrev and L.-S. Wang, J. Phys. Chem. A, 2014, 118, 8098–8105 CrossRef CAS PubMed.
- T.-T. Chen, W.-L. Li, J. Li and L.-S. Wang, Chem. Sci., 2019, 10, 2534–2542 RSC.
- W.-L. Li, T.-T. Chen, D.-H. Xing, X. Chen, J. Li and L.-S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6972–E6977 CrossRef CAS PubMed.
- T.-T. Chen, W.-L. Li, W.-J. Chen, J. Li and L.-S. Wang, Chem. Commun., 2019, 55, 7864–7867 RSC.
- I. A. Popov, T. Jian, G. V. Lopez, A. I. Boldyrev and L.-S. Wang, Nat. Commun., 2015, 6, 8654 CrossRef CAS PubMed.
- T. Jian, W.-L. Li, X. Chen, T.-T. Chen, G. V. Lopez, J. Li and L.-S. Wang, Chem. Sci., 2016, 7, 7020–7027 RSC.
- W.-L. Li, T. Jian, X. Chen, H.-R. Li, T.-T. Chen, X.-M. Luo, S.-D. Li, J. Li and L.-S. Wang, Chem. Commun., 2017, 53, 1587–1590 RSC.
- P. Jin, Q. Hou, C. Tang and Z. Chen, Theor. Chem. Acc., 2015, 134, 13 Search PubMed.
- Y. Wang, X. Wu and J. Zhao, J. Cluster Sci., 2018, 29, 847–852 CrossRef CAS.
- J. Lv, Y. Wang, L. Zhang, H. Lin, J. Zhao and Y. Ma, Nanoscale, 2015, 7, 10482–10489 RSC.
- J. Liu, Y. Zhang, C. Li, W. Jin, G. Lefkidis and W. Hübner, Phys. Rev. B, 2020, 102, 024416 CrossRef CAS.
- T.-T. Chen, W.-L. Li, W.-J. Chen, X.-H. Yu, X.-R. Dong, J. Li and L.-S. Wang, Nat. Commun., 2020, 11, 2766 CrossRef CAS PubMed.
- X.-Y. Zhao, M. Yan, Z. Wei and S.-D. Li, RSC Adv., 2020, 10, 34225–34230 RSC.
- Y. Zhang, X.-Y. Zhao, M. Yan and S.-D. Li, RSC Adv., 2020, 10, 29320–29325 RSC.
- X.-Q. Lu, C.-Y. Gao, Z. Wei and S.-D. Li, J. Mol. Model., 2021, 27, 130 CrossRef PubMed.
- W.-y. Liang, J. Barroso, S. Jalife, M. a. Orozco-Ic, X. Zarate, X. Dong, Z.-h. Cui and G. Merino, Chem. Commun., 2019, 55, 7490–7493 RSC.
- J. Zhao, R. Shi, L. Sai, X. Huang and Y. Su, Mol. Simul., 2016, 42, 809–819 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- L. Sai, L. Tang, J. Zhao, J. Wang and V. Kumar, J. Chem. Phys., 2011, 135, 184305 CrossRef PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Revision A. 03, Gaussian. Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- W.-L. Li, A. S. Ivanov, J. Federic, C. Romanescu, I. Cernušák, A. I. Boldyrev and L.-S. Wang, J. Chem. Phys., 2013, 139, 104312 CrossRef PubMed.
- C. Romanescu, T. R. Galeev, W.-L. Li, A. I. Boldyrev and L.-S. Wang, J. Chem. Phys., 2013, 138, 134315 CrossRef PubMed.
- B. L. Chen, W. G. Sun, X. Y. Kuang, C. Lu, X. X. Xia, H. X. Shi and G. Maroulis, Inorg. Chem., 2018, 57, 343–350 CrossRef PubMed.
- X. Wu, Y. Wang, X. Zhao, S. Zhou, S. Li, M. Chen and J. Zhao, Eur. Phys. J. Plus, 2021, 136, 328 CrossRef CAS.
- G. D. Purvis and R. J. Bartlet, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS.
- F. Li, P. Jin, D.-e. Jiang, L. Wang, S. B. Zhang, J. Zhao and Z. Chen, J. Chem. Phys., 2012, 136, 074302 CrossRef PubMed.
- N. V. Tkachenko and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2019, 21, 9590–9596 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- T.-T. Chen, W.-L. Li, H. Bai, W.-J. Chen, X.-R. Dong, J. Li and L.-S. Wang, J. Phys. Chem. A, 2019, 123, 5317–5324 CrossRef CAS PubMed.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- M. Rahm, T. Zeng and R. Hoffmann, J. Am. Chem. Soc., 2019, 141, 342–351 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02525b |
| This journal is © The Royal Society of Chemistry 2021 |