
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free multicomponent synthesis of novel macrocyclic tetrathiadienes with cyano and amino groups†
Nail S. Akhmadiev,
Ekaterina S. Mescheryakova,
Vnira R. Akhmetova * and
Askhat G. Ibragimov
* and
Askhat G. Ibragimov
Institute of Petrochemistry and Catalysis, Russian Academy of Science, 141 Prospekt Octyabrya, 450075 Ufa, Russian Federation. E-mail: vnirara@mail.ru; Fax: +7 3472 842750; Tel: +7 3472 842750
First published on 25th May 2021
Abstract
The first synthesis of 5,12-diamino-7,14-bis(aryl)-1,4,8,11-tetrathiacyclotetradeca-5,12-diene-6,13-dicarbonitriles was performed as a multicomponent macroheterocyclization of malononitrile, aryl aldehydes, and 1,2-ethanedithiol in the presence of a catalytic amount of triethylamine in ethanol. The structures of the obtained macroheterocycles were confirmed by spectral methods, X-ray diffraction, and MALDI TOF mass spectrometry.
1 Introduction
Organic sulfides form an abundant cluster of biologically active molecules, providing a specific physiological effect. For example, the methionine amino acid serves as a donor of methyl groups in the body; allicin is formed upon mechanical destruction of garlic cells and exhibits bactericidal properties; biotin (vitamin) occurs as a part of enzymes and regulates the protein and fat balance; umifenovir is an antiviral agent, and so on.1–3 Unlike the traditional Ullmann and Chan Lam coupling reactions, a simple strategy of a non-halide pathway to sulfur compounds is the use of diaryl sulfides as starting substrates.4–6 Another approach is based on the formation of a C–S bond by transition metal-catalyzed thiomethylation via condensation of thioacetals with CH-acids.7–9Meanwhile, the growing demand for new materials, increasing complexity of molecular drug targets, and chemotherapeutic drug and antibiotic resistance account for the increasing relevance of new thiamacrocyclic compounds.10,11 An example is provided by the synthesis of a macroheterocyclic product containing a disulfide linker, octreotide, used for the treatment of tumors overexpressing growth hormone (somatostatin).12
In this connection, a relevant task is to search for conceptually new approaches to the synthesis of sulfur-containing macroheterocycles.13 One such approach does not require the use of metal ions as template agents.14 Another original method is multicomponent self-assembly of macroheterocyclic compounds by (2 + 4 + 2)-cyclocondensation of amino alcohols, formaldehyde, and α,ω-dithiols.15 It is noteworthy that saturated and unsaturated crown ethers of this type possess high complexing capacity towards soft transition and alkali metals and small organic molecules.16–20 Moreover, unsaturated thiacrown ethers can be used for the creation of hybrid materials in form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complexes with electron transfer to endo-lanthanofullerene [La@C82-A(C2v)].21
:1 host–guest complexes with electron transfer to endo-lanthanofullerene [La@C82-A(C2v)].21
Conformational features contribute to the unique nature of thia macrocycles. For unsaturated thiacrown ethers oxidative transformations and Z/E isomerization of double bonds upon heating have been described.22 It was shown that tetrathiadiene macrocyclic rings (–S–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–S–)2 preferably exist in a stepped configuration, forming close crystal packing (column structures) provided by tight (zigzag-like) non-covalent contacts between the sulfur atoms of the neighboring rings, which are similar for hydrogen and π–π stacking bonds.23 These patterns of intermolecular contacts determine the properties of conducting materials and organic electrodes.24,25
CH–S–)2 preferably exist in a stepped configuration, forming close crystal packing (column structures) provided by tight (zigzag-like) non-covalent contacts between the sulfur atoms of the neighboring rings, which are similar for hydrogen and π–π stacking bonds.23 These patterns of intermolecular contacts determine the properties of conducting materials and organic electrodes.24,25
The single examples of unsaturated S-containing macroheterocycles and their high practical value stimulated us to design new substituted 1,4,8,11-tetrathiacyclotetradeca-5,12-dienes. The introduction of sulfur atoms with a lone pair of electrons into the macrocycle and the presence of cyano and amino groups can endow macromolecules with complexing and antioxidant properties.
2 Results and discussion
Previously, it was shown that a multicomponent condensation of malononitrile with aldehydes and S-mono- or S,S-binucleophilic reagents in the presence of a basic catalyst gives pyridine skeleton I or 6,7-dihydro-1,4-dithiepine structure II (Scheme 1).26,27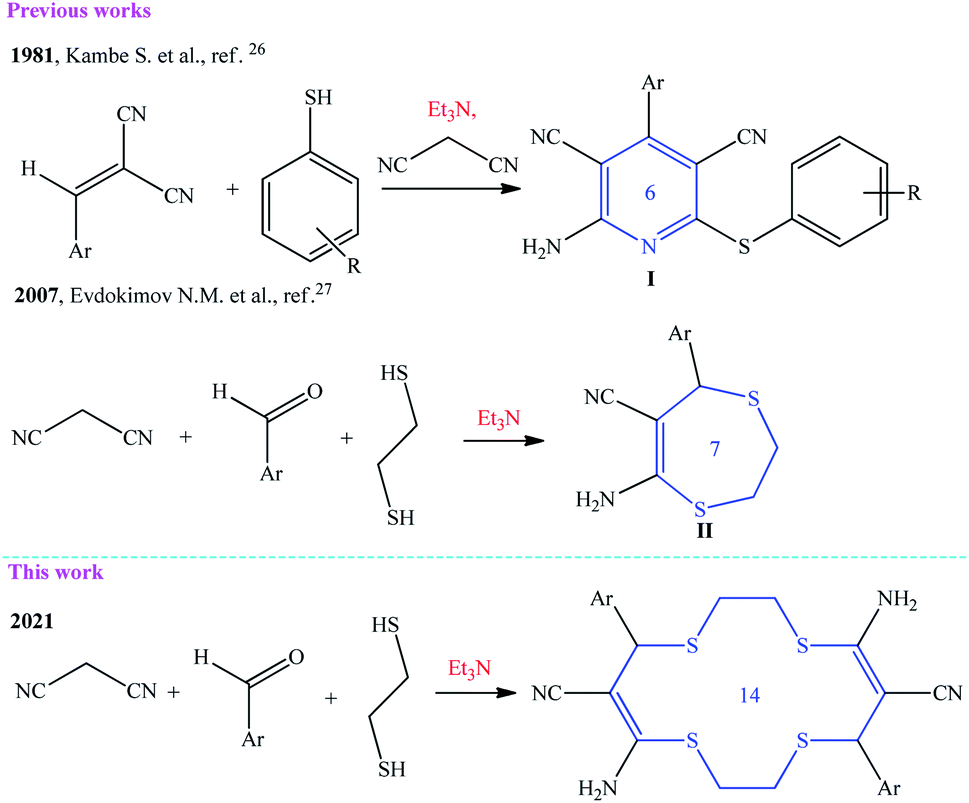 | ||
| Scheme 1 Evolution of the multicomponent reaction of malononitrile with aldehydes and SH acids in the synthesis of six- to fourteen-membered hetero(macro)cycles. | ||
In this study, we demonstrated for the first time a new potential of malononitrile macroheterocyclization with aromatic aldehydes and 1,2-ethanedithiol to give previously unknown macroheterocyclic 5,12-diamino-7,14-bis(aryl)-1,4,8,11-tetrathiacyclotetradeca-5,12-diene-6,13-dicarbonitriles (Scheme 1).
The base-catalyzed condensation of malononitrile with nucleophilic reagents is, most often, highly selective and gives heterocyclic products in high yields.28 The model reaction involving malononitrile, 1,2-ethanedithiol, and aryl aldehyde containing electron-withdrawing fluorine atom in the para-position was utilized to study the effect of the catalyst type on the yield and selectivity of the reaction. This reaction proceeded successfully when the reactant molar ratio was 1:1:1, triethylamine (5 mol%) was present as the catalyst, and the reaction mixture was heated to 70 °C in ethanol or water. In this case, the target macrocycle was formed in 67% or 61% yield (entries 1 and 3, Table 1).
| No | Catalyst | Solvent | Product | Yield, % |
|---|---|---|---|---|
| a Reaction conditions: malononitrile 1 (2.5 mmol), 4-fluorobenzaldehyde 3a (2.5 mmol), 1,2-ethanedithiol 2 (2.5 mmol), 5 mol% of the catalyst, 8 mL of solvent, 70 °C, stirring for 5 h.b DBU – diazabicycloundecene. | ||||
| 1 | Et3N | C2H5OH | 4a | 67 |
| 2 | — | C2H5OH | 4a | 17 |
| 3 | Et3N | H2O | 4a | 61 |
| 4 | Et3N | CH2Cl2 | 4a | 34 |
| 5 | Piperidine | C2H5OH | 4a | 59 |
| 6 | DBUb | C2H5OH | 4a | 63 |
| 7 | Morpholine | C2H5OH | 4a | 29 |
| 8 | K2CO3 | C2H5OH | 4a | 51 |
| 9 | L-Lysine·H2O | C2H5OH | 4a | — |
| 10 | H3BO3 | C2H5OH | 4a | — |
| 11 | BF3·OEt2 | C2H5OH | 4a | — |
| 12 | NiCl2·6H2O | C2H5OH | 4a | — |
Ethanol was chosen as the solvent because malononitrile is readily soluble in ethanol, while the resulting macrocycles are poorly soluble (Scheme 2). The reaction was also efficiently implemented with piperidine (59% yield of 4a) and DBU (63%, entries 5 and 6, Table 1) used as organocatalysts. In the absence of a catalyst, the yield of product 4a decreased to 17%. Product 4a was not formed when Lewis acids (H3BO3, BF3·OEt2, and NiCl2·6H2O) were used as catalysts (5 mol%) (entries 10–12, Table 1).
 | ||
| Scheme 2 Model synthesis of amino- and cyano-substituted 1,4,8,11-tetrathiacyclotetradeca-5,12-diene 4a. | ||
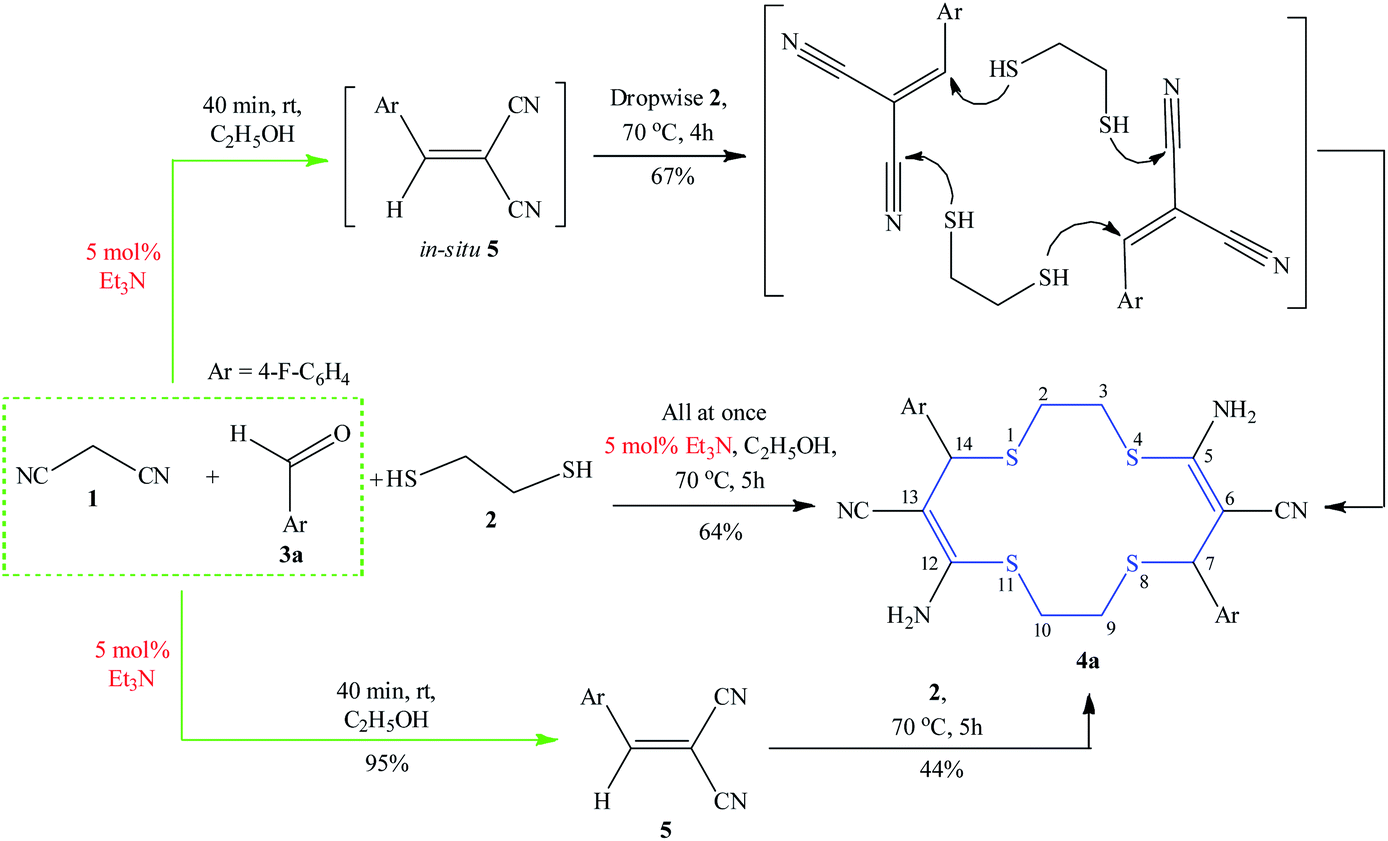
Fast addition of 1,2-ethanedithiol to the intermediate 5 resulted in complete gumming of the reaction mixture, evidently due to the competing polymerization. A similar outcome was obtained when long-chain α,ω-dithiols (1,3-propane-, 1,4-butanedithiols) were used as S-nucleophiles.
The use of 2-furaldehyde, formylferrocene, and acetaldehyde under the developed conditions resulted in the formation of powdered products insoluble in organic solvents such as CHCl3, DMSO, or DMF, which precluded the spectroscopic characterization of the reaction mixture.
Another approach that can be successfully implemented is the three-component reaction when reagents have added all at once to the reaction mixture to give thiacrown ether 4a in 64% yield (Scheme 2). Two-component procedure consisting in preliminary mixing of malononitrile with aldehyde results in the in situ intermediate formation of the Knoevenagel product, 2-(4-fluorobenzylydene)malononitrile 5, within 40 min. Moreover, product 5 was isolated in a pure state in 95% yield and described by X-ray and mass spectral method (Fig. 1). The use of ready compound 5 as the reactant under similar conditions gave macrocycle 4a in 44% yield (Scheme 2).
 | ||
| Fig. 1 Molecular structure of the compound 5. | ||
The crystals of 5 were obtained by slow evaporation of a DMSO-d6 solution. The molecules of 5 crystallize in the triclinic system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; the crystal lattice parameters are close to those described previously.29 The C
; the crystal lattice parameters are close to those described previously.29 The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond length is in line with established parameters,30 being 1.1384(18) and 1.1380(16) Å for the C6–N11 and C5–N8 bonds, respectively. The molecule has a nearly planar geometry, the root-mean-square deviation of atoms is 0.055 Å.
N bond length is in line with established parameters,30 being 1.1384(18) and 1.1380(16) Å for the C6–N11 and C5–N8 bonds, respectively. The molecule has a nearly planar geometry, the root-mean-square deviation of atoms is 0.055 Å.
Thus, as shown in Scheme 2, three reactions follow the same route: first, the Knoevenagel product 5 is formed and then the self-organization of dithiol molecules occurs by location between two molecules of product 5 to realize the (2 + 2)-cycloaddition. Considering the yield of product 4a, two macroheterocyclization reactions either with simultaneous mixing of all three reactants or via in situ synthesis of intermediate 2-(4-fluorobenzylidene)malononitrile 5 are of preparative value.
The 1H NMR spectrum of macroheterocycle 4a exhibited a triple set of high-field signals at δH 2.74–2.84, 3.02–3.05 ppm, and 3.46–3.51 ppm with integrated intensity ratio of 1:1:2 (Fig. 2). According to HSQC 1H–13C NMR experiments, this group of signals was assigned to the ethylene protons (CH2(2,3,9,10)) between sulfur atoms. Splitting of the geminal proton signals of the (SCH2CH2S)2 moieties was due to the magnetic non-equivalence (anisochronous nuclei) of the hydrogen atoms on the NMR time scale and the lack of structural symmetry at the sulfur atoms. It is noteworthy that hydrogen atoms of the CH2 groups in the SCH2CH2S chains are diastereotopic, obviously due to the rigid conformation of the macroheterocycle 4a.31
 | ||
| Fig. 2 Splitting of proton signals of the ethylene group (SCH2CH2S)2 in the 1H NMR spectrum of macroheterocycle 4a in DMSO-d6 (400 MHz) at room temperature (δH ppm). | ||
As a result of the symmetry and conformational rigidity of the macrocycles, the 13C NMR spectra exhibit a moderate number of signals. The 13C NMR spectrum of compound 4a shows two high-field signals at δC 29.2 ppm (C(3)) and 31.5 (C(2)) ppm corresponding to the ethylene moiety between the sulfur atoms. The characteristic signal of the methine carbon atom occurs at δC 45.4 ppm. The signal at δC 85.6 ppm corresponds to the quaternary carbon atom at the CN-substituted double bond. The enamine carbon atom resonates in a low field at 163.0 ppm. The 13C NMR spectrum of compound 4a shows splitting of the aromatic carbon signals, due to the presence of fluorine in the para-position of the ring, with spin–spin coupling constants corresponding to published data: δC 161.8 (1JCF 242.8 Hz), 152.4, 136.0 (4JCF 2.4 Hz), 129.5 (3JCF 8.1 Hz), 120.3, and 115.9 (2JCF 21.4 Hz) ppm.32 The MALDI TOF mass spectrum shows intense molecular ion peaks with m/z 555.0307 and 570.9909 corresponding to [M + Na]+ and [M + K]+ ions of the structure ascribed to macroheterocycle 4a.
The heteronuclear 1H–13C HMBC 2D NMR spectrum of compound 4a showed a correlation between the C(2)H2 protons and C(14) methine carbon atom; however, there were no clear-cut cross-peaks with the C(5) quaternary carbon atom at the NH2 group, probably, due to slow conformational exchange (Fig. 3).
 | ||
| Fig. 3 Fragment of molecule 4a with indicated long-range interactions according to 1H–13C HMBC data (δC ppm). | ||
Under the developed conditions, the reaction was carried out for the para- and meta-substituted aromatic aldehydes 3a–i with electron-donating (CH3, OCH3, N(CH3)2) and withdrawing (F, Cl, CF3) substituents (Scheme 3). The yields of the macroheterocycles 4a–i varied in the 44–73% range. The presence of para-substituents in the aromatic ring of aldehydes was favorable for increasing the yield of the target macrocycles (Table 2). With participation of para- and meta-substituted 1,3-benzodioxol-5-ylmethanal (piperonal) the thiacrown ether 4i was efficiently synthesized in 73% yield. However, there is information in the literature that in the presence of ortho-,ortho′-substituents in aromatic aldehydes, this reaction gave substituted 6,7-dihydro-1,4-dithiepines II.27 Obviously, the meta- and/or para-substituents in the benzene ring in the Knoevenagel product 5 complicate the intramolecular nucleophilic addition of 1,2-dithiol 2 giving 6,7-dihydro-1,4-dithiepines, but promote intermolecular (2 + 2) cycloaddition of 2 to 5, resulting in the formation of macrocyclic tetrathiadienes with cyano and amino groups.
 | ||
| Scheme 3 Catalytic macroheterocyclization of malononitrile with aryl aldehydes and 1,2-ethanedithiol. | ||
| Compounds | Substituent Ar | Yield 4a, % | Compounds | Substituent Ar | Yield 4a, % |
|---|---|---|---|---|---|
| a The given yields for compounds 4a–i were obtained by method B. | |||||
| 4a | 4-F-C6H4 | 67 | 4f | 3,4-(CH3O)2–C6H3 | 53 |
| 4b | 4-Cl-C6H4 | 61 | 4g | 4-CH3-C6H4 | 63 |
| 4c | 3-F-C6H4 | 58 | 4h | 4-(CH3)2N–C6H4 | 44 |
| 4d | 4-CF3-C6H4 | 68 | 4i | 1,3-Benzodioxol-5-yl | 73 |
| 4e | 4-(CH3O)–C6H4 | 59 | |||
Unsaturated thiacrown ethers 4a and 4b form crystal solvates (Fig. 5) in which the molecules of the major substance are linked by N–H⋯O hydrogen bonds to DMSO molecules. Note that the solvent molecules are disordered. 14-Membered cyclic products trans-4a and trans-4b have E-geometry of the olefinic moieties. The C(sp2)–S bond lengths are 1.771(2) and 1.752(6) Å, which agrees with the data for previously studied compounds.33 There exist endo- and exo-conformations of macrocyclic thioethers, depending on whether the sulfur lone electron pairs point inside or outside the macrocyclic cavity, respectively. Like other relatively simple thioethers,33 structures 4a and 4b have the exo-conformation. The para-fluoro- or para-chloroaryl moieties are arranged equatorially relative to the macrocycle plane. The obtained trans-thiacrown ether molecules have a symmetry center, with the distances between the opposite sulfur atoms, S1–S1′ and S2–S2′, in 4a and 4b differing insignificantly (Fig. 4).
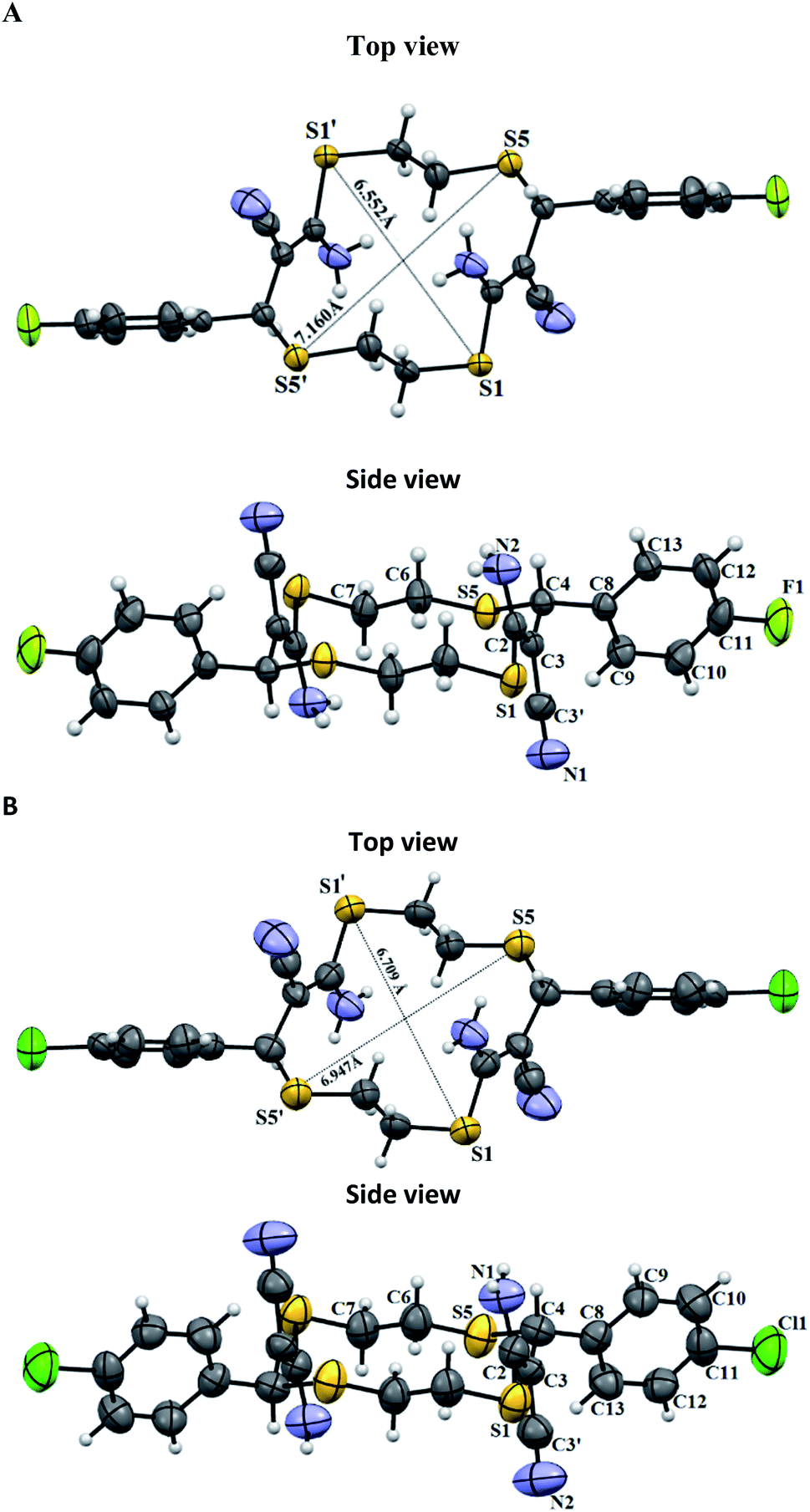 | ||
| Fig. 4 Molecular structure of unsaturated thiacrown ethers trans-4a and trans-4b. Non-hydrogen atoms are shown by thermal ellipsoids (p = 50%). | ||
The molecules of compound 4a form crystals with a triclinic crystal lattice (P), while molecules of 4b crystallize in the orthorhombic system (space group Pbca). In the crystals of 4a, thiacrown ether molecules form stacks along the a axis via C–H⋯N hydrogen bonds, whereas molecules of 4b are connected into chains along the b axis via DMSO molecules (Fig. 5). According to published data,34 molecules in the crystals of unsaturated thiacrown ethers are linked by S⋯S contacts; the distances between the sulfur atoms are equal to or are shorter than the sum of the van der Waals radii (S⋯S = 3.7 Å). In the crystals of 4a and 4b, the distances between the sulfur atoms of the tetrathiadiene rings are markedly longer than the sum of the van der Waals radii of sulfur atoms (Fig. 5).
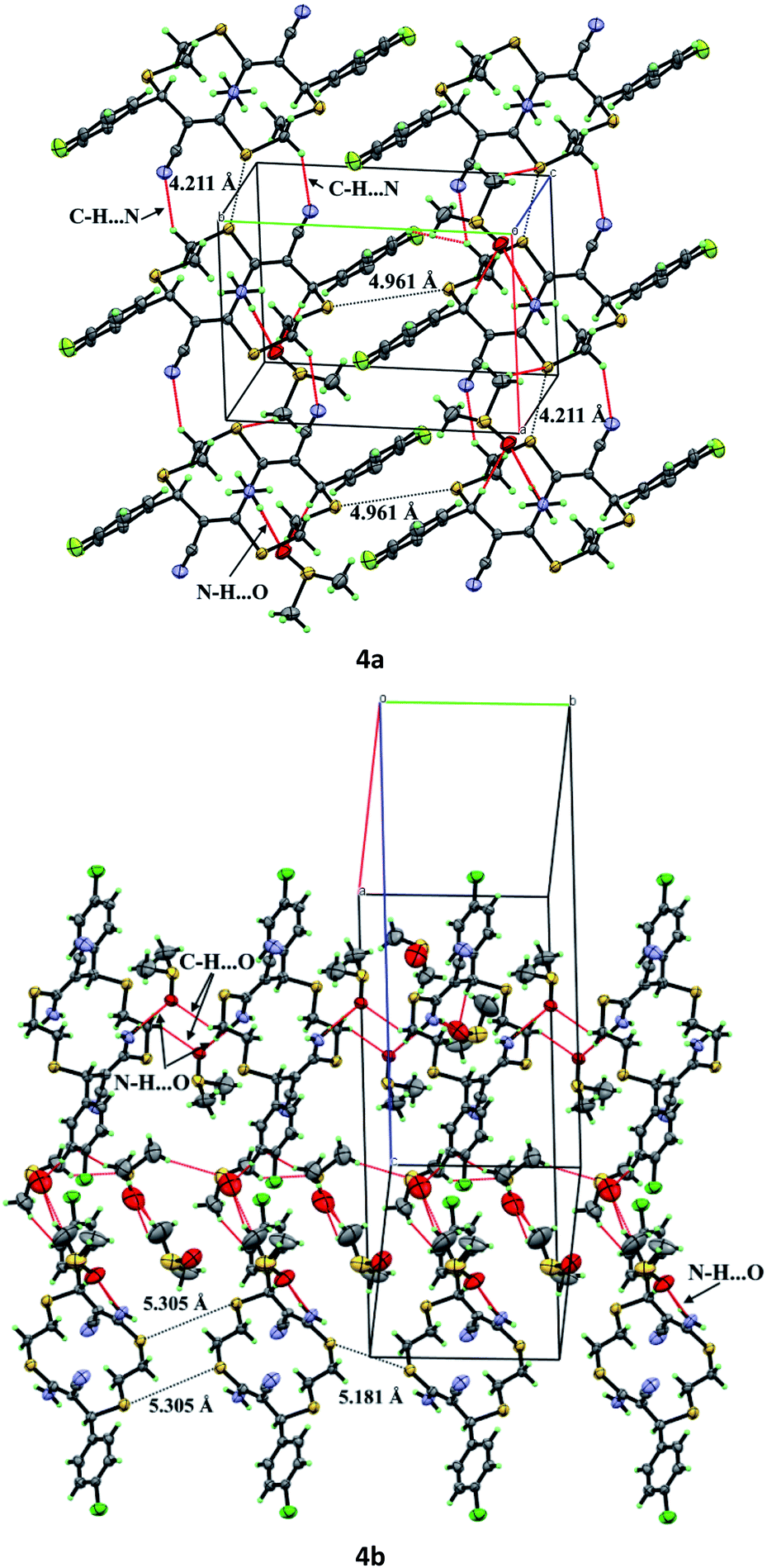 | ||
| Fig. 5 Crystal packing of compounds 4a and 4b with indicated intermolecular interactions (red dashed line) and S⋯S distances (black dashed line). | ||
3 Conclusions
Thus, here we report the first efficient method for constructing macroheterocyclic structures from available reactants within the framework of the metal-free concept. This method is based on multicomponent macroheterocyclization of malononitrile, meta- or para-substituted arylaldehydes, and 1,2-ethanedithiol in the presence of triethylamine as a base catalyst. Due to the presence of bifunctional groups in the reactants pseudo-six-component cycloaddition occurs with the participation of SH-groups of dithiol, CC- and CN-bonds of the Knoevenagel adduct. The resulting 14-membered macroheterocycles represent a conceptually novel type of unsaturated systems containing cyano and amino groups. According to X-ray diffraction data 1,4,8,11-tetrathiacyclotetradeca-5,12-dienes have a E-geometry of the olefinic moieties and the exo-conformation of the macrocyclic thioethers. Owingh to the presence of several functional groups, these tetrathiamacrocycles (thiacrown ethers) would be of interest for chemists specializing in the coordination, supramolecular, and medicinal chemistry.
4 Experimental section
4.1. Materials and instruments
The reaction products were characterized by 1H and 13C NMR spectra that were recorded on spectrometers Bruker Avance 400 NMR (400.13 MHz and 100.62 MHz) and Bruker Ascend III HD 500 (500.17 MHz and 125.78 MHz). Also 2D homo- (COSY) and hetero- (HSQC, HMBC) nuclear spectra were obtained on a Bruker Avance 500 in DMSO-d6 by Bruker standard procedures, internal reference standard TMS. IR spectra were obtained on a Bruker Vertex-70V FT-IR spectrometer for samples prepared as a Nujol mull. UV spectra were recorded on a Perkin Elmer Lambda 750 UV/VIS-spectrometer for DMSO solutions in the wavelength range of 200–1000 nm using a 1 cm thick cuvette. Matrix-assisted laser desorption/ionization (MALDI) mass spectrum was recorded on a Bruker's device MALDI TOF Autoflex III with sinapinic acid as a matrixes. GC-MS analysis of compound 5 was performed on a Shimadzu GC 2010 chromatograph equipped with a Shimadzu GCMS-QP2010 Ultra mass selective detector and a Supelco 5 ms capillary column (60 m × 0.25 m × 0.25 μm), with helium as carrier gas. Elemental analysis was performed on a Carlo Erba Model 1106 elemental analyzer. Melting points were determined on a Kofler hot-stage microscope (RNMK 80/2617) apparatus. The reaction progress was monitored by TLC method on Sorbfil plates (PTSKh-AF-A), eluent cyclohexane–CH2Cl2–EtOAc, 1:2:10, visualization with iodine vapor.
Method A. 4-Fluorobenzaldehyde (0.27 mL, 2.5 mmol) and triethylamine (0.02 mL, 0.0125 mmol) are added to a solution of malononitrile (0.17 g, 2.5 mmol) in ethanol (8 mL), and then 1,2-ethanedithiol (0.21 mL, 2.5 mmol) is added dropwise under argon. The reaction mixture is stirred at 70 °C for 4 h, filtered, washed with ethanol (2 × 10 mL), and dried in air to give 5,12-diamino-7,14-bis(4-fluorophenyl)-1,4,8,11-tetrasulfanylcyclotetradeca-5,12-diene-6,13-dicarbonitrile 4a in 67% yield.
Method B. A mixture of malononitrile (0.17 g, 2.5 mmol), 4-fluorobenzaldehyde (0.27 mL, 2.5 mmol) and triethylamine (0.02 mL, 0.0125 mmol) in 8 mL of ethanol was stirred under argon at room temperature for 40 min. Then 1,2-ethanedithiol (0.21 mL 2.5 mmol) is added dropwise. The reaction mixture is stirred at 70 °C for 4 h, filtered, washed with ethanol (2 × 10 mL), and dried in air to give macroheterocycle 4a is obtained with a yield of 64%.
Method C. To a suspension of 2-(4-fluorobenzylidene)malononitrile (0.43 g, 2.5 mmol) in 8 mL of ethanol, 1,2-ethanedithiol (0.21 mL, 2.5 mmol) is added dropwise in an argon atmosphere. The reaction mixture is stirred at 70 °C for 4 h, filtered, washed with ethanol (2 × 10 mL) and dried in air to give macroheterocycle 4a is obtained with a yield of 44%.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2). 13C-NMR (ppm): δ = 153.1, 144.3, 128.5 (2JCF 32 Hz), 128.4 (4JCF 1 Hz), 126.1 (3JCF 3.7 Hz), 124.6 (1JCF 271 Hz), 120.1, 84.7, 45.8, 31.4, 29.2. MS (MALDI TOF) 655.0454 [M + Na]+, 670.9974 [M + K]+.3). 13C-NMR (ppm): δ = 152.0, 137.2, 136.8, 129.5, 127.4, 120.3 (C-16,25), 86.1 (C-6,13), 45.9, 31.4, 29.2, 21.1 (C-33,34). MS (MALDI TOF) 547.0865, [M + Na]+, 563.0848 [M + K]+.C, 55%), 95 (M − FC6H5, 15%).
2). 13C-NMR (ppm): δ = 153.1, 144.3, 128.5 (2JCF 32 Hz), 128.4 (4JCF 1 Hz), 126.1 (3JCF 3.7 Hz), 124.6 (1JCF 271 Hz), 120.1, 84.7, 45.8, 31.4, 29.2. MS (MALDI TOF) 655.0454 [M + Na]+, 670.9974 [M + K]+.3). 13C-NMR (ppm): δ = 152.0, 137.2, 136.8, 129.5, 127.4, 120.3 (C-16,25), 86.1 (C-6,13), 45.9, 31.4, 29.2, 21.1 (C-33,34). MS (MALDI TOF) 547.0865, [M + Na]+, 563.0848 [M + K]+.C, 55%), 95 (M − FC6H5, 15%).4.2. X-ray diffraction experiments
The X-ray diffraction measurements for compounds 4a, 4b and 5 were performed on an Agilent XCalibur (Eos, Gemini) automated four-circle diffractometer (graphite monochromator, MoKα radiation, λ = 0.71073 Å, ω-scan mode, 2θmax = 62°). Data collection, cell refinement, data reduction were carried out using the CrysAlisPro.36 The structures 4a and 5 were solved by direct methods with the SHELXS program37 and structure 4b was solved by with the SHELXT program.38 Positional parameters of non-hydrogen atoms were refined by the full-matrix least-squares method in the anisotropic approximation using the SHELXL 2018/3 program.39 All hydrogen atoms in compounds 4a and 4b were generated using the proper HFIX command and refined isotropically using the riding model. The hydrogen atoms in compound 5 were located in the Difference Fourier map and refined isotropically. Crystal data and structure refinement parameters are shown in Table S1 (see ESI).† The crystallographic data, coordinates of atoms, and geometric parameters for compounds 4a, 4b and 5 were deposited at the Cambridge Crystallographic Data Centre (entry no. CCDC 2064004 4a, CCDC 2064005 4b, and CCDC 2064003 5).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially partly supported by the Russian Science Foundation (project no. 19-73-00070). The structural data were obtained with the financial support of the Russian Ministry of Education and Science (project no. 2019-05-595-000-058) on unique equipment at the “Agidel” Collective Usage Center (Ufa Federal Research Center, Russian Academy of Sciences) and within the framework of the State Assignment AAAA-A19-119022290010-9.References
- S. Shekh, K. K. Amarnath Reddy and K. H. Gowd, J. Sulfur Chem., 2021, 42, 109, DOI:10.1080/17415993.2020.1817457.
- C. Jacob, Nat. Prod. Rep., 2006, 23, 851, 10.1039/B609523M.
- Z. V. F. Wright, N. C. Wu, R. U. Kadam, I. A. Wilson and D. W. Wolan, Bioorg. Med. Chem. Lett., 2017, 27, 3744, DOI:10.1016/j.bmcl.2017.06.074.
- R. Zhang, H. Ding, X. Pu, Z. Qian and Y. Xiao, Catalysts, 2020, 10, 1339, DOI:10.3390/catal10111339.
- P. Mampuys, C. R. McElroy, J. H. Clark, R. V. A. Orru and B. U. W. Maes, Adv. Synth. Catal., 2020, 362, 3, DOI:10.1002/adsc.201900864.
- Y. Qin, Y. Han, Y. Tang, J. Wei and M. Yang, Chem. Sci., 2020, 11, 1276, 10.1039/c9sc04169a.
- V. R. Akhmetova, N. S. Akhmadiev, Z. A. Starikova, A. R. Tulyabaev, E. S. Mescheryakova and A. G. Ibragimov, Tetrahedron, 2015, 71, 7722, DOI:10.1016/j.tet.2015.07.055.
- N. S. Akhmadiev, E. S. Mescheryakova, V. R. Khairullina, V. R. Akhmetova, L. M. Khalilov and A. G. Ibragimov, J. Mol. Struct., 2021, 1228, 129734, DOI:10.1016/j.molstruc.2020.129734.
- V. R. Akhmetova, N. S. Akhmadiev, R. A. Zainullin, V. R. Khayrullina, E. S. Mescheryakova and N. A. Glushkova, Can. J. Chem., 2020, 98, 725, DOI:10.1139/cjc-2019-0186.
- A. K. Yudin, Chem. Sci., 2015, 6, 30, 10.1039/C4SC03089C.
- X. Yu and D. Sun, Molecules, 2013, 18, 6230, DOI:10.3390/molecules18066230.
- A. Iarov, I. Avrutov, N. Lazarowych and S. Houldsworth, WO Pat., 2005087794A1, 2005 Search PubMed.
- A. V. Chuchuryukin, P. A. Chase, H. P. Dijkstra, B. M. J. M. Suijkerbuijk, A. M. Mills, A. L. Spek, G. P. M. van Klink and G. van Koten, Adv. Synth. Catal., 2005, 347, 447, DOI:10.1002/adsc.200404282.
- N. E. Borisova, M. D. Reshetova and Yu. A. Ustynyuk, Russ. Chem. Rev., 2007, 76, 785, DOI:10.1070/RC2007v076n09ABEH003705.
- G. R. Khabibullina, V. R. Akhmetova, M. F. Abdullin, T. V. Tyumkina, L. M. Khalilov, A. G. Ibragimov and U. M. Dzhemilev, Tetrahedron, 2014, 70, 3502, DOI:10.1016/j.tet.2014.03.053.
- Y. Suzaki, H. Nagai and K. Osakada, Chem. Lett., 2014, 43, 714, DOI:10.1246/cl.131213.
- Y.-F. Han, H. Lia and G.-X. Jin, Chem. Commun., 2010, 46, 6879, 10.1039/C0CC00770F.
- T. Tsuchiya, T. Shimizu, K. Hirabayashi and N. Kamigata, J. Org. Chem., 2002, 67, 6632, DOI:10.1021/jo020269w.
- M. Ashram, G. M. Al-Mazaideh, W. Al-Zereini, A. Al-Mustafa and S. Mizyed, J. Sulfur Chem., 2019, 40, 277, DOI:10.1080/17415993.2019.1579816.
- A. Holzberger and E. Kleinpeter, Magn. Reson. Chem., 2004, 42, 589, DOI:10.1002/mrc.1378.
- T. Takahiro, K. Hiroki, S. Kumiko, W. Takatsugu, A. Takeshi, S. Toshio, K. Nobumasa, M. Naomi and N. Shigeru, Chem. Commun., 2006, 3585, 10.1039/b606183d.
- T. Shimizu, S. Komatsuzaki and K. Hirabayashi, Heteroat. Chem., 2011, 22, 287, DOI:10.1002/hc.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010, DOI:10.1021/acs.chemrev.7b00449.
- J. Heiska, M. Nisula and M. Karppinen, J. Mater. Chem. A, 2019, 7, 18735, 10.1039/C9TA04328D.
- A. I. Konovalova, I. S. Antipina and V. A. Burilov, et al., Russ. J. Org. Chem., 2018, 54, 157, DOI:10.1134/S107042801802001X.
- S. Kambe, K. Saito, A. Sakurai and H. Midorikawa, Synthesis, 1981, 531, DOI:10.1055/s-1981-29513.
- N. M. Evdokimov, A. S. Kireev, A. A. Yakovenko, M. Yu. Antipin, I. V. Magedov and A. Kornienko, J. Org. Chem., 2007, 72, 3443, DOI:10.1021/jo070114u.
- V. V. Dotsenko, S. G. Krivokolysko and A. M. Semenova, Chem. Heterocycl. Compd., 2018, 54, 989, DOI:10.1007/s10593-018-2383-y.
- M. Yu. Antipin, V. N. Nesterov, S. Jiang, O. Y. Borbulevych, D. M. Sammeth, E. V. Sevostianova and T. V. Timofeeva, J. Mol. Struct., 2003, 650, 1, DOI:10.1016/S0022-2860(02)00468-4.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1, 10.1039/P298700000S1.
- A. de Leon, J. Pons, J. García-Antón, X. Solans, M. Font-Bardia and J. Ros, Polyhedron, 2007, 26, 2498, DOI:10.1016/j.poly.2006.12.034.
- N. S. Akhmadiev, E. S. Mescheryakova, V. R. Khairullina, V. R. Akhmetova and A. G. Ibragimov, Chem. Heterocycl. Compd., 2020, 56, 473, DOI:10.1007/s10593-020-02683-8.
- S. J. Lange, J. W. Sibert, C. L. Stern, A. G. M. Barrett and B. M. Hoffman, Tetrahedron, 1995, 51, 8175, DOI:10.1016/0040-4020(95)00439-F.
- T. H. Staeb, R. Gleiter and F. Rominger, Eur. J. Org. Chem., 2002, 2815–2822, DOI:10.1002/1099-0690(200208)2002:16<2815::AID-EJOC2815>3.0.CO;2-9.
- M. Trilla, R. Pleixats, M. W. Chi Man and C. Bied, Green Chem., 2009, 11, 1815, 10.1039/b916767f.
- Agilent A. T. Ltd, CrysAlis PRO/2012, Yarnton, Oxfordshire, England Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112, DOI:10.1107/S0108767307043930.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3, DOI:10.1107/S2053229614024218.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2064003–2064005. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02616j |
| This journal is © The Royal Society of Chemistry 2021 |