
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinguishing chemically induced NADPH- and NADH-related metabolic responses using phasor analysis of UV-excited autofluorescence
Audrey H. Short ,
Nazar Al Aayedi,
Madhu Gaire,
Max Kreider,
Chong Kai Wong and
Paul Urayama*
,
Nazar Al Aayedi,
Madhu Gaire,
Max Kreider,
Chong Kai Wong and
Paul Urayama*
Department of Physics, Miami University, Oxford, OH 45056, USA. E-mail: urayampk@MiamiOH.edu; Fax: +1-513-529-5629; Tel: +1-513-529-9274
First published on 24th May 2021
Abstract
NADPH and NADH are well known for their role in antioxidant defense and energy metabolism, respectively, however distinguishing their cellular autofluorescence signals is a challenge due to their nearly identical optical properties. Recent studies applying spectral phasor analysis to autofluorescence emission during chemically induced metabolic responses showed that two-component spectral behavior, i.e., spectral change acting as a superposition of two spectra, depended on whether one or multiple metabolic pathways were affected. Here, we use this property of spectral behavior to show that metabolic responses primarily involving NADPH or NADH can be distinguished. We start by observing that the cyanide-induced response at micro- and millimolar concentrations does not follow mutual two-component spectral behavior, suggesting their response mechanisms differ. While respiratory inhibition at millimolar cyanide concentration is well known and associated with the NADH pool, we find the autofluorescence response at micromolar cyanide concentration exhibits two-component spectral behavior with NADPH-linked EGCG- and peroxide-induced responses, suggesting an association with the NADPH pool. What emerges is a spectral phasor map useful for distinguishing cellular autofluorescence responses related to oxidative stress versus cellular respiration.
Introduction
Reduced pyridine nucleotides (e.g., nicotinamide adenine dinucleotide phosphate (NADPH) and nicotinamide adenine dinucleotide (NADH)) are ubiquitous metabolic cofactors known for their role not only in antioxidant defense and energy metabolism, respectively, but also calcium homeostasis, gene expression, immunological functions, aging, and cell death.1,2 Studies of cellular autofluorescence have revealed the existence of multiple conformations for these molecules, significant because the change in conformation correlates with metabolic condition.3–5 The ability to sense conformation indicates that detailed information regarding metabolism exists in the autofluorescence signal, leading to a renewed interest in developing conformation as a metabolic indicator and endogenous biomarker6 for monitoring disease,3–5,7–12 for cytotoxicity,13,14 and for investigating NADPH/NADH balance.1,15Because protein binding affects the excited-state emission spectrum,16 spectrum shape is a source for contrast when monitoring cellular reduced pyridine nucleotide conformation.17 NAD(P)H denotes the uncertain origin of the autofluorescence signal15—a challenge in fluorescence-based sensing due to the nearly identical optical properties of NADPH and NADH. The ability to distinguish their signals would allow for more precise identification of metabolic state and response because the metabolic roles of NADPH and NADH are distinct. Approaches for separating NADPH and NADH signals include the development of genetically encoded sensors18 and differentiation based on excited-state lifetimes.15 Here we report an approach for distinguishing emission signals associated with NADPH- and NADH-related autofluorescence response based on an analysis of spectral behavior.
Previously, spectral phasor analysis on autofluorescence signals was used to assess two-component spectral behavior (i.e., spectra that can be modeled as a superposition of two spectra) during chemically induced metabolic change involving a range of NAD(P)H-linked metabolic pathways.17,19 Because autofluorescence emission is an ensemble average signal of NAD(P)H conformations present in the system, two-component behavior suggests the signal arises from a superposition of two ensembles, e.g., conformational ensembles associated with the activated and inactivated states of a pathway. Non-two-component behavior represents situations where more than two ensembles are needed to describe the observed emission behavior.
A recent study tested this hypothesis and demonstrated a correlation between two-component spectral behavior in the autofluorescence and metabolic function.20 In this metabolic interpretation, responses having the same mechanism follow two-component behavior, while responses having different mechanisms may not follow two-component behavior. For example, the autofluorescence response to various alcohols were observed to follow mutual two-component behavior20 because a general mechanism accounted for alcohol-induced respiratory inhibition.21 By comparison, respiratory inhibition using ethanol and cyanide did not follow two-component behavior,20 consistent with their different inhibitory mechanisms.21,22 Here, “mechanism” refers to the effects of a metabolic response on the NADH(P)H conformational ensemble, not necessarily to a biochemical pathway. This metabolic interpretation for two-component behavior in the autofluorescence response provides a functionally significant way of monitoring cellular metabolism.
Here we use spectral phasor analysis on UV-excited autofluorescence to show that metabolic responses primarily involving NADPH or NADH can be distinguished. We start by noting a concentration dependent autofluorescence response to cyanide, observing that responses induced at micromolar and millimolar cyanide concentrations do not follow mutual two-component spectral behavior. While the millimolar response is associated with the inhibition of the mitochondrial electron transport chain (ETC)23 and so is predominantly linked to NADH, we find the micromolar response follows two-component behavior with responses induced by epigallocatechin gallate (EGCG) and oxidative stress, which are linked to the NADPH pool.1 We hypothesize that the micromolar cyanide response is related to reactive oxygen species (ROS) production by the ETC. Overall, we find that NADPH- and NADH-related responses follow distinct phasor behavior which can be represented on a response map useful for distinguishing oxidative stress from changes in cellular respiration.
Experimental
Instrumentation
The spectrofluorometric system is described previously17,19 and consists of a nitrogen-gas discharge laser (model GL-3300, Photon Technology International) operating at a 3 Hz repetition rate and having a 1 ns nominal pulse width at 337 nm wavelength. Spectra are acquired using a spectrograph (model MS 125, Spectra-Physics/Newport) coupled to a nanosecond-gated intensified CCD (ICCD) (model iStar 734, Andor) with a picosecond-resolution digital delay generator (DDG) for controlling gate delay. An optical constant fraction discriminator (model OCF-401, Becker & Hickl) serves as a low temporal-jitter reference for gate-delay timing. So that the sample's emission falls within the range of gate delays accessible by the ICCD/DDG, the excitation pulse is delayed using a 25 m multimode optical fiber. Emission is filtered through a 385 nm long-pass filter and collected at 90-degrees from the excitation using a separate optical fiber.The spectrograph utilizes a 400 l mm−1 grating, and the system is calibrated for wavelength using a mercury–argon lamp (cat. no. 6035, Newport). The 1024 × 1024-pixel CCD chip is fully binned along one axis during readout. The measured spectral width of atomic lines is 2 nm with the 1024-channel output spanning a 250 nm wavelength range (400–650 nm wavelength). The ICCD gate opens 5 ns prior to signal arrival and integrates for 80 ns, sufficiently long to collect time-integrated emission. A full spectrum is collected for each excitation pulse, although spectra are averaged over multiple pulses to increase peak-signal-to-noise ratio. The CCD is temperature regulated and the CCD dark current is subtracted from measured spectra prior to analysis.
Sample preparation and data acquisition
The protocols for preparing cellular suspensions and for real-time autofluorescence monitoring are found elsewhere.19 Briefly, Saccharomyces cerevisiae is grown on YPD agar medium (cat. no. Y1000, TekNova) rather than in liquid medium to minimize background fluorescence. Cells are grown for two or three days, triple washed in phosphate-buffered saline (PBS, cat. no. 20012, Life Technologies) prior to measurement. Cells are then resuspended in PBS for a final volume of 3 ml and absorbance of 1 (approximately 107 cells per ml, calibrated using a hemocytometer). Measurements are made every 30 s, on a sample housed in a UV-transparent plastic cuvette open to air. Samples are continuously stirred during all measurements to facilitate mixing, to minimize photobleaching, and to minimize any UV-induced physiological response. The lack of measurable photobleaching was confirmed on a representative sample by varying the time interval between measurements and by varying the excitation intensity. Samples were not temperature regulated; room temperature was measured at 22 ± 2 °C. An autofluorescence response is induced once the emission intensity is observed to be steady for at least 10 min, about 30 min after resuspension in PBS. To induce a response, the chemical is added to the cuvette using a pipette. Samples grown on agar and suspended in PBS in this manner are confirmed to have a UV-excited autofluorescence intensity that responds to oxygenation and to additions of cyanide, ethanol, and glucose19 in manners similar to starved yeast cultures maintained in a batch reactor.24Unless otherwise stated, chemicals are added from a concentrated stock solution to the final concentration indicated such that each addition is limited to a 2% volume increase. Chemicals used to induce an autofluorescence response are potassium cyanide (cat. no. 60178, Sigma-Aldrich) in PBS, EGCG (cat. no. E4143, Sigma-Aldrich) in PBS, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, cat. no. C2920, Sigma-Aldrich) in dimethyl sulfoxide (DMSO, cat. no. 41639, Sigma-Aldrich), and hydrogen peroxide (cat. no. 88597, Sigma-Aldrich) in deionized water. Cyanide is a respiratory inhibitor that binds to complex IV of the ETC;22 EGCG is an inhibitor of glucose-6-phosphate dehydrogenase (G6PD), an enzyme utilizing NADP+ as a coenzyme,25 and known to not effect NADH binding;15 FCCP is a protonophore known to uncouple oxidative phosphorylation and increase cellular respiration;26 and hydrogen peroxide is a physiological oxidant capable of inducing oxidative stress and limiting NADH availability via inhibition of α-ketoglutarate dehydrogenase in the citric acid cycle (CAC).27,28 No more than 1 vol% DMSO is present in the sample; no autofluorescence response is observed for a DMSO-only control.
Spectral phasor analysis
Initially developed for the rapid identification of regions within hyperspectral images,29 a spectral phasor is a mathematical “projection” (inner product) of a spectrum onto a complex exponential function over one cycle. Phasor analysis is a non-iterative, computationally rapid approach for graphically representing spectrum shape and for analyzing two- and three-component mixtures.For N spectral channels, the spectral phasor A is
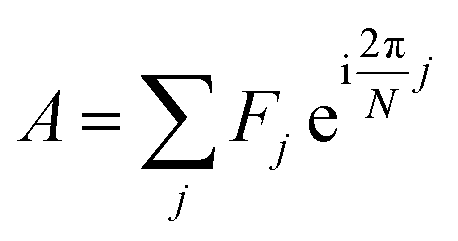 | (1) |
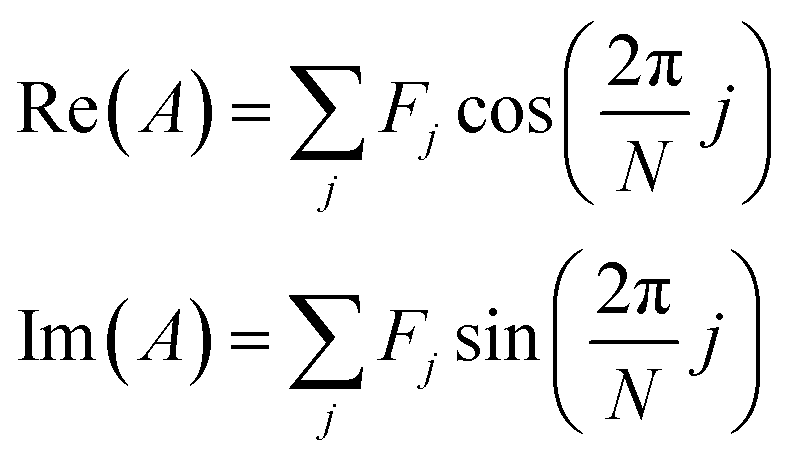 | (2) |
Fig. 1a shows phasors calculated for simulated Gaussian spectra as width and peak-intensity wavelength are varied. Phasors for spectra of a given width form circle-like trajectories, traveling counterclockwise as the peak-intensity wavelength varies from the long to short wavelength ends of the spectral interval. Phasors for spectra of a given peak-intensity wavelength diverge out from the origin as the width decreases and lie on a unit circle in the limit of an infinitely narrow spectrum. Trajectories of constant width and of constant wavelength are orthogonal along the negative real axis, corresponding to the spectral interval being centered on the Gaussian spectrum.
 | ||
Fig. 1 Visualizing spectral phasors. (a) A phasor plot calculated from simulated Gaussian spectra. Wavelength is expressed as a unitless quantity between 0–100 over the spectral interval. Red lines connect spectra of constant width; from the outer to inner loops, the width is 5–20, in uniform increments. Blue lines connect spectra of constant peak-intensity wavelength; proceeding counterclockwise, the peak-intensity wavelength is from 0–100, in uniform increments. The green region provides a sense for scale by showing data from Fig. 3a. (b) A three-component interpretation. The phasor A calculated using emission from a three-component system lies within the triangle formed by the pure-component phasors A1, A2, and A3. The fractional contribution αi is proportional to the opposing area. (c) Effects of background on the phasor shift direction. Assuming the fractional contribution of the background A2 is small,  remain near A1 and A3, and so the direction of shifts (A3 − A1) and remain near A1 and A3, and so the direction of shifts (A3 − A1) and 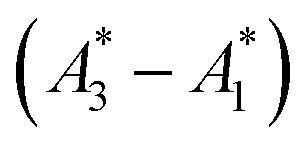 is nearly unchanged. is nearly unchanged. | ||
Phasors calculated from a three-components system have a graphical interpretation (Fig. 1b).29 Phasor A lies somewhere within a triangle formed by the pure-component phasors A1, A2, and A3. The fractional contribution αi is proportional to the opposing area. Phasors for a two-component system are collinear because one of the areas is zero.
In practice, phasor values calculated from the autofluorescence of independently prepared samples show a small variation. Therefore, when assessing collinearity of spectral phasors across multiple samples, it is helpful to shift the axes so that the time-averaged initial phasor is at the origin. Such phasor plots will have axes labeled ΔRe(A) and ΔIm(A) as opposed to Re(A) and Im(A) for plots showing results of eqn (2). In theory, the direction of a phasor response depends on this variation. Nonetheless, if the sample-to-sample variation is small, the phasor-shift direction is not significantly affected as illustrated in Fig. 1c. Let A1 and A3 represent the “before” and “after” pure-component phasors with  being the observed phasors which are shifted due to an arbitrary component A2. Assuming the fractional contribution of A2 is small,
being the observed phasors which are shifted due to an arbitrary component A2. Assuming the fractional contribution of A2 is small,  remain near A1 and A3. Thus, the directions associated with shifts (A3 − A1) and
remain near A1 and A3. Thus, the directions associated with shifts (A3 − A1) and 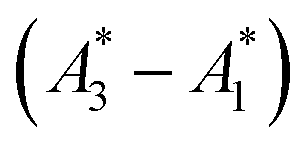 are nearly unchanged.
are nearly unchanged.
The collinearity of spectral phasors indicates two-component spectral behavior in the autofluorescence response. We hypothesize a metabolic interpretation17,20 in which two-component behavior occurs when metabolic change involves the same response mechanism and non-two-component behavior is possible when metabolic change involves different response mechanisms. This behavior has been shown for a range of chemically induced autofluorescence responses and could not be explained as an artifact of the chemical addition.20 For example, sequential chemical additions of ethanol and cyanide into a sample gave non-two-component responses while sequential additions of D-glucose and deoxyglucose gave two-component responses. Further, L-glucose (a metabolically inactive enantiomer) gave no response with subsequent D-glucose and deoxyglucose additions once again giving a two-component response. Other controls demonstrated how the autofluorescence response was not an artifact of the chemical addition, e.g., a given chemical (i.e., cyanide) gave different, non-collinear responses depending on sample incubation in glucose and, conversely, that different chemicals (i.e., various alcohols) gave collinear responses of varying magnitude correlating to the degree of respiratory inhibition by each alcohol.
Results
Fig. 2a shows that the autofluorescence response to cyanide added to a cellular sample is concentration dependent with the change in intensity showing a good empirical fit to an exponential. A comparison of the autofluorescence responses at micromolar and millimolar cyanide concentrations within a given cellular sample is possible by sequentially inducing autofluorescence responses (Fig. 2b and c), first at 10 or 50 μM concentration then at 5 mM concentration.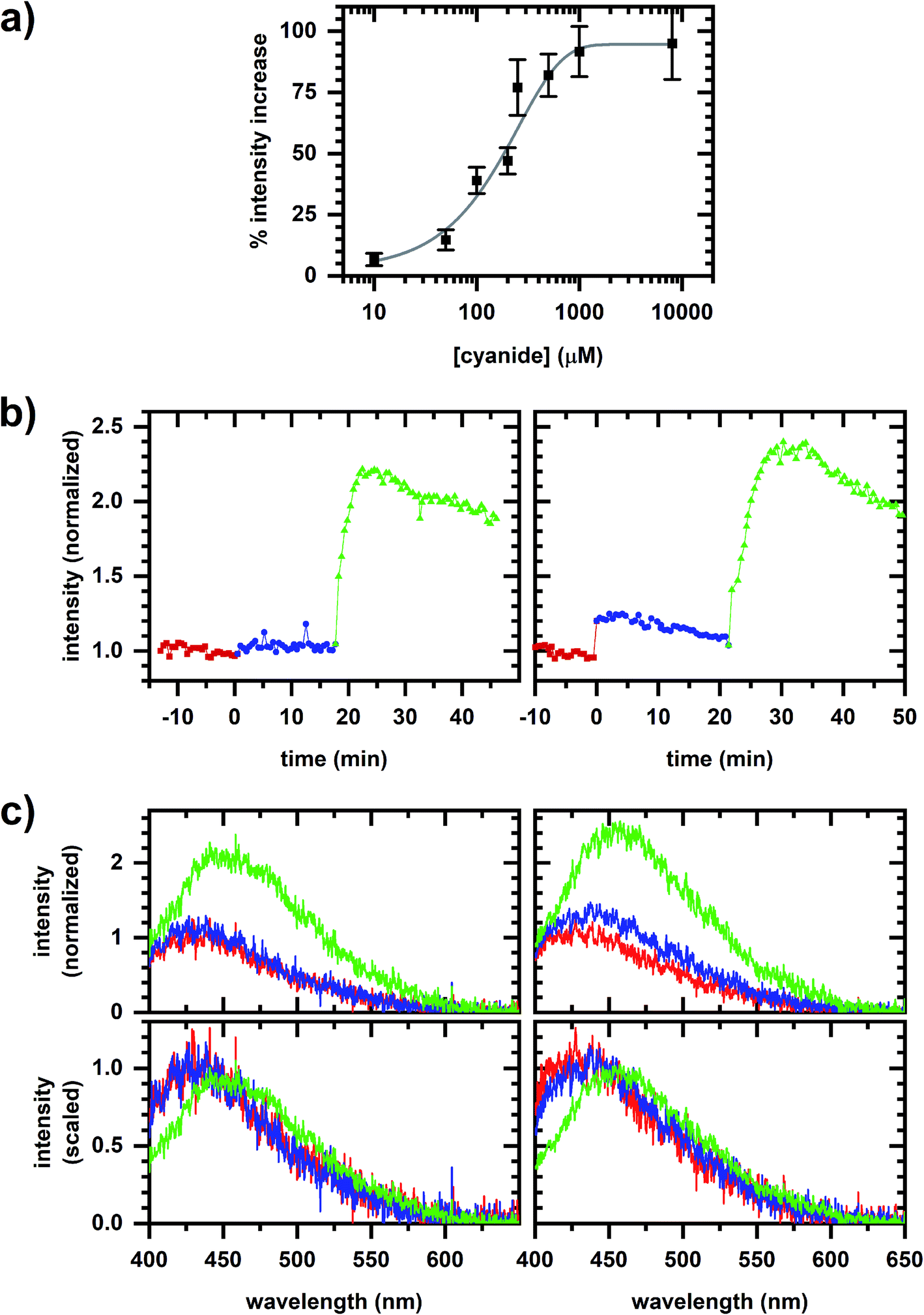 | ||
| Fig. 2 Autofluorescence response to cyanide. (a) Change in emission intensity versus cyanide concentration. The spectrally integrated intensity is averaged over 10 min intervals prior to and after cyanide addition to the cellular sample. The fit is to a one-phase exponential association equation. Error bars are the standard deviation of measurements from independently prepared samples. (b) Spectrally integrated emission intensity. Red/square is prior to any cyanide addition to the cellular sample. Left plot shows the sequential additions of 10 μM (blue/circle) and 5 mM (green/triangle) cyanide. Right plot shows the sequential additions of 50 μM (blue/circle) and 5 mM (green/triangle) cyanide. Intensities are normalized to its value averaged over a 10 min interval prior to cyanide addition. (c) Representative emission spectra for the dataset shown in (b). Plot color matches the sequence in (b). Spectra in the top row are normalized to the fitted peak intensity prior to chemical addition. Spectra in the bottom row are scaled to minimize least-square difference. | ||
A spectral phasor analysis on emission spectra (Fig. 3a) shows that sequentially induced phasor shifts are not collinear, indicating that a two-component model cannot account for the overall spectral response. When sequential micromolar and millimolar cyanide additions are repeated in independently prepared samples, spectral phasors calculated from emission after the micromolar cyanide response and after the millimolar cyanide response are separately collinear (Fig. 3b). Thus, emission spectra over multiple samples follow two-component behavior when cyanide concentrations are low and high separately, but not over the entire range in concentration, evidencing that the cyanide response for the two concentration regimes utilizes different metabolic mechanisms.
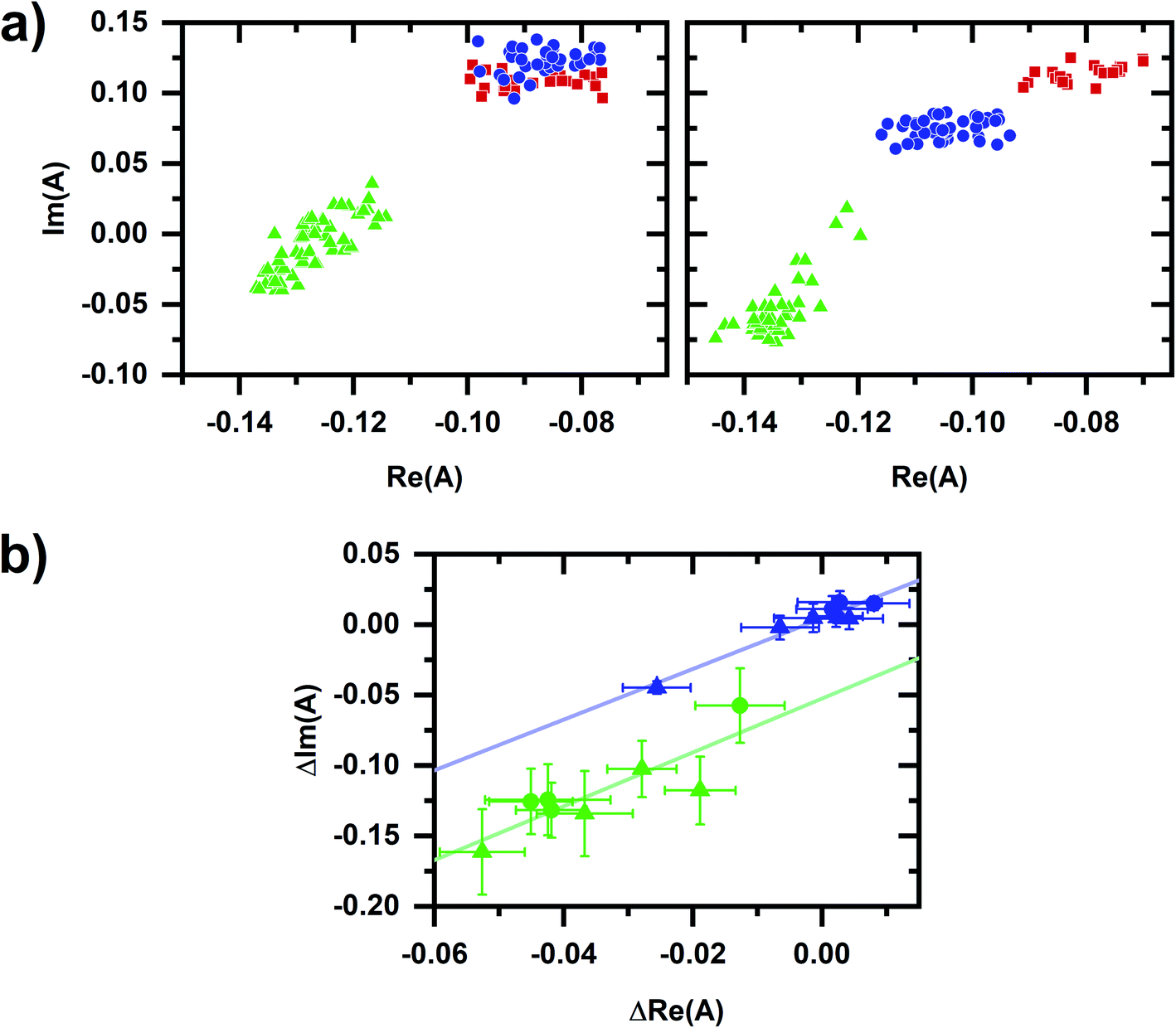 | ||
| Fig. 3 Phasor plots for the autofluorescence response to cyanide. (a) Phasor plots for datasets shown in Fig. 2b. Red/square is prior to any cyanide addition to the cellular sample. Left plot shows the sequential additions of 10 μM (blue/circle) and 5 mM (green/triangle) cyanide. Right plot shows the sequential additions of 50 μM (blue/circle) and 5 mM (green/triangle) cyanide. (b) Comparison of spectral phasor values for independently prepared samples. Phasor values are shifted so that the phasor value averaged over 10 min prior to the first chemical addition to the cellular sample is at the origin. Blue and green symbols are phasor values after the micromolar- and millimolar-cyanide addition, respectively. Circles are for the 10 μM, 5 mM cyanide sequential additions. Triangles are for the 50 μM, 5 mM cyanide sequential additions. The symbol and error bars are the average and standard deviation of phasor values measured during 10 min after chemical addition. Blue and green lines are linear least-squares fits to the blue and green symbols, respectively. For the entire figure, phasor analysis is performed over the first 400 pixels (400–500 nm wavelength range). | ||
It is known that the millimolar response is associated with the inhibition of the mitochondrial ETC23 and so predominantly linked to NADH emission. Thus we seek an interpretation for the response at micromolar concentration.
A comparison of emission spectra from 5 μM NADH in MOPS buffer without cyanide and with 10 mM cyanide added shows that NADH solvatochromism does not account for the micromolar cyanide response. In considering metabolic interpretations for the micromolar cyanide response, we consider the possibility of NADPH-linked pathways. EGCG competitively inhibits NADPH-binding enzymes25 and so it can be used to induce a metabolic response affecting cellular NADPH emission only.15 Fig. 4a shows the cellular autofluorescence intensity in response to sequential additions of EGCG (20–60 μM), micromolar cyanide (10 or 50 μM), then millimolar cyanide (5 mM) to the cellular sample. The addition of EGCG results in a concentration-dependent increase in autofluorescence intensity. Subsequent micromolar and millimolar cyanide responses show a fractional increase in intensity that is similar to the no-EGCG case (Fig. 2b). Emission spectra for a representative case (30 μM EGCG, 10 μM cyanide, 5 mM cyanide) are shown in Fig. 4b with the phasor plot shown in Fig. 5a. The EGCG-induced phasor shift is large and in roughly the opposite direction as the shift induced by millimolar cyanide. Additionally, phasors for the millimolar cyanide response follow an altered dynamic (Fig. 5a, arrow) as compared with the same response but without EGCG (Fig. 3a). Fig. 5b and c show phasor plots for multiple samples using the same range of chemical additions found in Fig. 4a.
 | ||
| Fig. 4 Autofluorescence response to sequential EGCG, micromolar cyanide, millimolar cyanide additions. (a) Spectrally integrated emission intensity. Color/symbol indicates different sequences of addition to the cellular sample: red/square – 30 μM EGCG, 10 μM cyanide, 5 mM cyanide; blue/circle – 20 μM EGCG, 50 μM cyanide, 5 mM cyanide; green/triangle – 60 μM EGCG, 10 μM cyanide, 5 mM cyanide. EGCG added at time 0 min. Micromolar and millimolar cyanide are added at the first and second arrows, respectively. Intensities are normalized to its value averaged over a 10 min interval prior to the first chemical addition. (b) Representative emission spectra. Dataset is the same as for the red/square plot in (a). Color indicates the chemical addition to the cellular sample – red (prior to addition), blue (30 μM EGCG), green (10 μM cyanide), and orange (5 mM cyanide). Spectra in the top plot are normalized to the fitted peak intensity prior to chemical addition. Spectra in the bottom plot are scaled to minimize least-square difference. | ||
 | ||
| Fig. 5 Phasor plots for sequential EGCG, micromolar cyanide, millimolar cyanide additions. (a) Representative phasor plot showing the same dataset as in Fig. 4b. Red/square is prior to any chemical addition to the cellular sample. Subsequent chemical additions are 30 μM EGCG (blue/circle), 10 μM cyanide (green/upright triangle), and 5 mM cyanide (orange/inverted triangle). The millimolar cyanide response shows significant variation over time, shifting the length of the arrow in the first 5 min after chemical addition. (b) Phasor plots showing values for multiple samples. Symbols and colors indicate different sequences of addition to the cellular sample, which are the same as for Fig. 4a. Red/square – 30 μM EGCG, 10 μM cyanide, 5 mM cyanide; blue/circle – 20 μM EGCG, 50 μM cyanide, 5 mM cyanide; green/triangle – 60 μM EGCG, 10 μM cyanide, 5 mM cyanide. Two datasets are shown for each condition, indicated by filled or open symbols. The dataset from (a) is in open red/square symbols. (c) Phasor plots showing data for multiple samples, shifted so that the phasor value averaged over 10 min prior to the first chemical addition to the cellular sample is at the origin. The datasets, colors, and symbols are the same as for (b). The symbol and error bars are the average and standard deviation of phasor values measured during 10 min after chemical addition. The larger error bars for the millimolar-cyanide phasor values are due to large variations, e.g., as seen in (a). For the entire figure, phasor analysis is performed over the first 400 pixels (400–500 nm wavelength range). | ||
Least-squares fits (Fig. 6) show a collinear relationship for phasors after the EGCG and micromolar cyanide additions to the cellular sample. Note that these phasors also share a collinearity with phasors after the micromolar cyanide addition without EGCG, i.e., the best-fit line from the no-EGCG case (Fig. 3b) is nearly identical with best-fit lines from the EGCG-containing data (Fig. 6). Because the autofluorescence after EGCG and micromolar cyanide additions follow two-component behavior and because EGCG action specifically affects the cellular NADPH pool, we hypothesize that the micromolar-cyanide autofluorescence response is associated with metabolic processes predominantly affecting the NADPH pool. These results suggest two mechanisms for the cyanide-induced autofluorescence response – one that primarily affects the NADPH pool at micromolar cyanide concentration and another that primarily affects the NADH pool at millimolar cyanide concentration. Metabolic pathways which account for the two mechanisms are considered in the Discussion section.
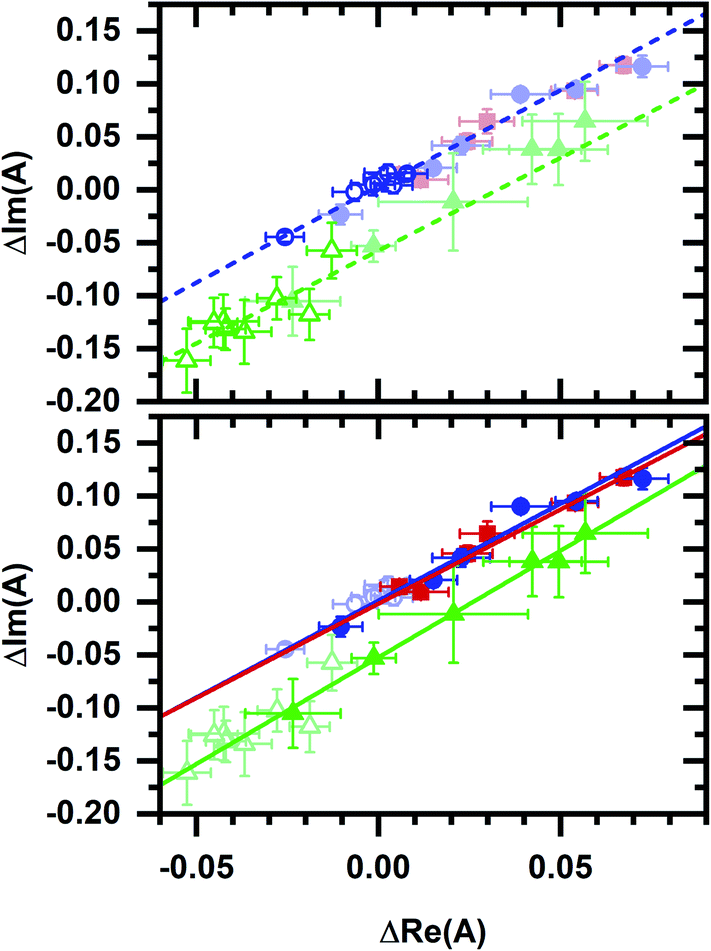 | ||
| Fig. 6 Phasor plots comparing data from Fig. 3b and 5c. Phasors and fit lines are color coded by condition: red – after EGCG addition to the cellular sample; blue – after micromolar cyanide addition; green – after millimolar cyanide addition. Open symbols are data from Fig. 3b. Closed symbols are data from Fig. 5c. Top plot, fit lines using data from Fig. 3b only. Lines are linear least-squares fits. Data from Fig. 5c (closed symbols, dimmed) appear consistent with fit lines based on data from Fig. 3b. Bottom plot, fit lines using data from Fig. 5c only. Lines are linear least-squares fits. Data from Fig. 3b (open symbols, dimmed) appear consistent with fit lines based on data from Fig. 5c. | ||
Phasor values also retain a collinear relationship after the millimolar cyanide addition to the cellular sample (Fig. 6). Here again, datasets with and without the initial EGCG addition are collinear, i.e., the best-fit line after the millimolar cyanide response without EGCG (Fig. 3b) is nearly identical with the best-fit line after the millimolar cyanide response with EGCG (Fig. 6). The millimolar-cyanide response does not change the sample ordering along the best-fit lines in Fig. 6, suggesting that cumulative changes to the cellular NAD(P)H conformational ensemble are additive.
With these results, a map of NADPH–NADH phasor space emerges such that the location along the collinearity line represents cellular NADPH status and shifting between collinearity lines represents a change in cellular respiration. In this way, we demonstrate that NADPH-related (i.e., EGCG and micromolar-cyanide induced) and NADH-related (i.e., millimolar-cyanide induced) responses are distinguishable.
As examples of navigating this NADPH–NADH “map” and as demonstration that cellular micro- and millimolar cyanide responses remain distinguishable even when the initial metabolic condition is altered, we consider two additional sequences of chemical additions. The first sequence of FCCP, micromolar cyanide, then millimolar cyanide added to the cellular sample is used to illustrate spectral behavior at an increased respiration rate. A second sequence of hydrogen peroxide, micromolar cyanide, then millimolar cyanide added to the cellular sample is used to directly compare a response due to oxidative stress with the micromolar-cyanide response.
Fig. 7a shows a representative response for the FCCP and hydrogen peroxide sequences. There is a decrease in cellular autofluorescence intensity after FCCP addition due an increased ETC flux, an NADH oxidizing pathway. There is a slight decrease in intensity after hydrogen peroxide addition, a physiological oxidant. The micromolar- and millimolar-cyanide responses for both sequences appear similar to the response in Fig. 2b. Fig. 7b and c also show phasor plots for three samples each using sequences of FCCP (20 μM), micromolar cyanide (10 μM), millimolar cyanide (5 mM); and hydrogen peroxide (0.02%/vol), micromolar cyanide (10 μM), millimolar cyanide (5 mM) being added to the cellular sample.
 | ||
| Fig. 7 Autofluorescence response to sequential additions involving FCCP and hydrogen peroxide. (a) Spectrally integrated emission intensity. Color/symbol indicates different sequences of addition to the cellular sample: red/square – 20 μM FCCP, 10 μM cyanide, 5 mM cyanide; blue/circle – 0.02% hydrogen peroxide, 10 μM cyanide, 5 mM cyanide. FCCP or hydrogen peroxide added at time 0 min. Micromolar and millimolar cyanide are added at the first and second arrows, respectively. Intensities are normalized to its value averaged over a 10 min interval prior to the first chemical addition. (b) Phasor plots showing data for multiple samples. Color (red or blue) indicates different sequences of addition to the cellular sample, which is the same as for Fig. 7a. There are three independently prepared samples for each sequence, distinguished by symbol (square, circle, triangle) and color intensity. (c) Phasor plots showing data for multiple samples, shifted so that the phasor value averaged over 10 min prior to the first chemical addition to the cellular sample is at the origin. Shown are the same datasets as from (b). Symbols, color, and color intensity are the same as for (b). The symbol and error bars are the average and standard deviation of phasor values measured during 10 min after chemical addition. For the entire figure, phasor analysis is performed over the first 400 pixels (400–500 nm wavelength range). | ||
Fig. 8 compares results from Fig. 7 with the proposed NADPH–NADH map (Fig. 6), confirming that phasor values for the FCCP and hydrogen-peroxide sequences show a relationship with the two collinearity lines. For the FCCP sequence (Fig. 8a, top), the phasors after the FCCP-induced response are above the upper collinearity line, consistent with an increased respiratory rate. Interestingly, the FCCP-induced shift is oblique to the millimolar-cyanide-induced shift, consistent with the two chemicals not following the same mechanism. NAD(P)H conformations involved in one of the autofluorescence responses are not involved in the other response or the proportions of conformational involvement differ.
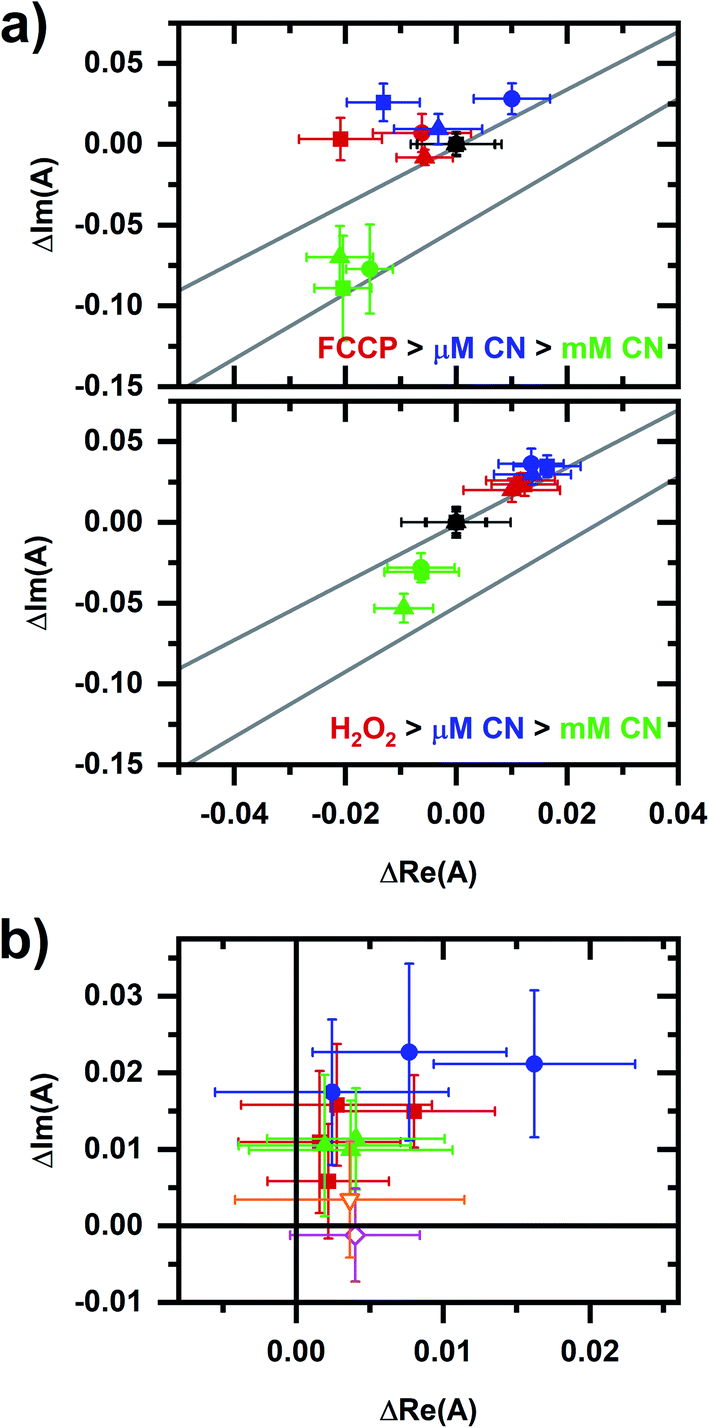 | ||
| Fig. 8 A comparison of data from Fig. 7c with the collinearity lines from Fig. 6. (a) Details of the autofluorescence response to sequential additions to the cellular sample involving FCCP (top plot) and peroxide (bottom plot). Symbol color indicates the chemical addition: black (prior to the addition, starting phasor value has been shifted to the origin), red (after FCCP or peroxide addition), blue (after micromolar-cyanide addition), and green (after millimolar-cyanide addition). Three independently prepared samples are shown for each plot, corresponding to the square, circle, triangle symbols. The upper and lower lines are the red and green fit lines from Fig. 6, bottom. (b) Comparison of micromolar-cyanide shifts. Only the phasor shift associated with the 10 μM cyanide response is plotted. Color/symbol indicates conditions: red/square – without prior chemical addition to the cellular sample (from Fig. 3); blue/circle – after addition of FCCP; green/upright triangle – after addition of hydrogen peroxide. Two additional responses are shown: orange/open-inverted triangle – 144 mM methanol; and magenta/open diamond – 2 mM diamide. Methanol and diamide responses are from ref. 20. For the entire figure, phasor analysis is performed over the first 400 pixels (400–500 nm wavelength range). | ||
For the hydrogen-peroxide sequence (Fig. 8a, bottom), the peroxide- and micromolar-cyanide-induced responses are in the same direction, further supporting an interpretation that the micromolar-cyanide response is associated with oxidative stress and so primarily affecting the cellular NADPH pool.
The micromolar-cyanide responses for both sequences appear collinear (Fig. 8b) with the micromolar-cyanide responses from Fig. 3b, suggesting the same response mechanism for each. Further comparisons of the micromolar-cyanide response to two chemicals associated with oxidative stress, methanol30 and diamide,31 are made using previously published results.20 The methanol-induced response is in the same general direction as the micromolar-cyanide responses while the diamide-induced response is primarily in the positive real phasor-axis direction. Note that diamide quenches NADH emission in solution,20 which may account the altered shift direction.
Finally, returning to Fig. 8a, phasors after the millimolar-cyanide response for both the FCCP and peroxide sequences lie below the upper collinearity line consistent with a decrease in cellular respiration.
Discussion and conclusions
Fig. 3 shows the cellular autofluorescence response to micromolar and millimolar cyanide concentrations does not follow two-component spectral behavior, evidencing their different origins. The micromolar-cyanide response follows two-component behavior with the EGCG response (Fig. 5 and 6), suggesting involvement of the cellular NADPH pool. As the primary catechin in green tea, EGCG is known to have a range of biological effects.32,33 Nonetheless, it is an effective inhibitor of NADPH binding25 and has been shown to preferentially decrease the amount of protein-bound NADPH but not protein-bound NADH during live-cell microscopy.15 By comparison, the millimolar-cyanide response is known to primarily involve NADH-linked pathways.23 Thus, Fig. 6 also demonstrates how autofluorescence responses linked to NADPH and NADH are distinguishable. Fig. 6 and 8 describe a proposed NADPH–NADH phasor map for which position along a collinearity line indicates NADPH status while a response going between collinearity lines indicates a change in cellular respiration.Next, we consider whether such a phasor map and spectral behavior are consistent with the metabolic effects of the various chemicals used. FCCP, a protonophore and mitochondrial uncoupler, increases cellular respiration,26 while cyanide inhibits cellular respiration at complex IV of the ETC;22 these chemicals produce spectral phasor shifts above and below the initial collinearity line consistent with cellular respiration being activated and inhibited, respectively. The non-two-component behavior between FCCP and millimolar-cyanide responses (Fig. 8) suggests their different mechanisms. Because only reduced pyridine nucleotides are fluorescent,34 there is also an associated decrease or increase in emission intensity due to the activation or inhibition of the electron transport chain (an oxidizing pathway) by FCCP or cyanide, respectively.
EGCG and hydrogen peroxide both have a metabolic response associated with the NADPH pool. EGCG competitively inhibits NADP+ binding to G6PD.25 Hydrogen peroxide oxidizes NADPH as part of antioxidant defense pathways involving glutathione peroxidase or peroxiredoxin antioxidant systems via the glutathione and thioredoxin reductase enzymes.1 Hydrogen peroxide concentrations used here (0.02 vol% H2O2) are above the physiological regime and so are associated with oxidative stress rather than signaling.28 Notably, the hydrogen-peroxide-induced phasor response is similar to methanol- and diamide-induced responses reported previously,20 suggesting this may be a spectral phasor signature for oxidative stress.
A metabolic link between the micromolar-cyanide response and NADPH also exists. Superoxide production occurs with electron leakage from the mitochondrial ETC35,36 and there is only partial inhibition of complex IV at micromolar cyanide concentrations.37,38 Because cyanide inhibition (at complex IV) occurs downstream of ROS production (at complex III), partial inhibition results in greater electron transport leakage,39 resulting in increased superoxide production. Subsequent conversion to hydrogen peroxide via superoxide dismutase results is a rapid peroxide accumulation in response to cyanide.40 We hypothesize that this rapid peroxide accumulation is the reason for the micromolar-cyanide autofluorescence response. Because there is only a partial inhibition of respiration37,38 and because hydrogen peroxide inhibits the CAC,27 the NADH-related response may not be significant at micromolar cyanide concentrations, leaving the NADPH-related response as the predominant effect. As evidence, FCCP treated samples exhibit a larger micromolar-cyanide response suggesting a larger electron leakage due to the partial inhibition of a higher ETC flux (Fig. 8b).
Note that although EGCG, hydrogen peroxide, and micromolar-cyanide responses share two-component spectral behavior, the autofluorescence intensity increases for EGCG and decreases for hydrogen peroxide and micromolar cyanide (Fig. 4 and 7). The intensity increase may be due to the inhibition by EGCG of the associated NADPH oxidizing reactions and the intensity decrease may be due to the peroxide-induced oxidation of NADPH. The lack of correspondence between intensity and spectral response indicates information contained in the two signals are complementary; intensity is generally sensitive to NAD(P)H concentration while spectrum is sensitive to NAD(P)H conformation.
Thus, the proposed NADPH–NADH phasor map is consistent with the metabolic effects of the various chemicals. We have utilized a metabolic interpretation for the two-component spectral behavior in a cellular autofluorescence response,17,20 that two-component behavior occurs when metabolic change involves the same mechanism and non-two-component behavior can occur when metabolic change involves different mechanisms. Note that a “mechanism” does not refer to a biophysical or biochemical pathway, but rather to the effect a metabolic response has on the cellular NAD(P)H conformational ensemble. In this sense, the two-component spectral behavior between EGCG, hydrogen peroxide, and micromolar-cyanide responses indicate that they effect the same NAD(P)H conformations which is not inconsistent with their effects occurring via different biochemical pathways.
Since this idea is central to the interpretations here, it is worth considering the physical origins of two-component spectral behavior. Because cellular autofluorescence emission is an ensemble average of NAD(P)H conformations present, two-component behavior occurs when there is a superposition of two ensembles, e.g., the conformations associated with the activated and inactivated states of a pathway. Non-two-component behavior means more than two conformational ensembles are needed to describe the observed emission behavior.
Note that two-component behavior is not equivalent to saying the emission is comprised of two spectral components. In fact, the emission may be comprised of many spectral components, perhaps identifiable using an approach such as singular value decomposition. Two-component behavior can result if spectral components change in a correlated manner, e.g., because the components are due to conformations associated with the same metabolic response.
The cellular autofluorescence response may also include solvatochromic effects, e.g., due to changes in the intracellular environment, as opposed to a change in the NAD(P)H conformational ensemble. If present here, solvatochromic effects are either correlated with the metabolic response, e.g., a change in solvent condition like pH that is part of the metabolic response, or are small compared with the change in emission associated with conformation.
We assess two-component behavior by observing the collinearity of spectral phasors. While linearity cannot be proven formally (mathematically) from real observations, the collinear relationships reported here can be extrapolated well beyond the fit range and have agreement among fits using various data sets (Fig. 6). Nonetheless, further analysis may reveal additional features of the autofluorescence response and so the two-component determination here is meant to provide a zeroth-order description.
Spectral phasors are a straightforward, real time analysis, providing information on NAD(P)H conformation for a more precise description of the metabolic response than the commonly used “free vs. bound” description of NAD(P)H conformation. In addition to a two-component interpretation, there is also a three-component interpretation for spectral phasors29 where a phasor from such a mixture lies within the triangle formed by the three pure-component phasors (Fig. 1b) and so it may be that conformations associated with multiple NAD(P)H pools can be distinguished simultaneously.
In addition to spectroscopy, cellular NAD(P)H conformation can be sensed from excited-state, energy-transfer, and anisotropy-decay measurements of autofluorescence signals.3,41,42 Phasor analysis for fluorescence lifetime signals is well established,34,43–46 initially applied to imaging microscopy for multicomponent calculations on fluorescence lifetime imaging microscopy (FLIM) data47 and for simplifying FLIM/fluorescence resonance energy transfer (FRET) analysis.48 With regards to autofluorescence signals, it can be used to distinguish different metabolic states during label-free FLIM imaging49 with a path in phasor space representing a “metabolic trajectory” of changing cellular behavior.9,50,51 Phasor FLIM can also identify pharmacological disruptions to mitochondrial function in cerebral tissue.52
“Phasors” generally describe the (graphical) representation of a complex quantity with a phasor analysis existing for both spectral and lifetime signals. Despite the similar nomenclature however, the physical origins for conformational contrast differ and so spectral and lifetime approaches do not contain identical information, i.e., one cannot be calculated from the other. Spectral change occurs when there is a change in the electronic energy level structure, e.g., due to coupling with the environment, whereas lifetime change is associated with a change in relaxation rates, e.g., due to dynamical quenching. Spectral and lifetime phasor approaches may therefore be complementary in providing metabolic information.
In conclusion, a phasor approach was used to show that chemically induced cellular autofluorescence responses linked to NADPH and NADH pathways can be distinguished. Considering the NADPH- and NADH-linked spectral responses together allows for a more precise identification of metabolic state because the metabolic roles of NADPH and NADH are distinct. A phasor map of NADPH/NADH status is proposed and tested using sequences of chemically induced metabolic responses. This approach may be useful for distinguishing cellular autofluorescence response related to oxidative stress versus cellular respiration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by funds from Miami University's College of Arts and Science's Dean's Scholar program and the Office of Research for Undergraduate's Undergraduate Summer Scholars Program. We thank Zhifan Cai, Kevin Karney, and Carolina Vissers for assistance on this project.References
- T. S. Blacker and M. R. Duchen, Free Radicals Biol. Med., 2016, 100, 53–65 CrossRef CAS PubMed
.
- W. H. Ying, Antioxid. Redox Signaling, 2008, 10, 179–206 CrossRef CAS PubMed
- H. D. Vishwasrao, A. A. Heikal, K. A. Kasischke and W. W. Webb, J. Biol. Chem., 2005, 280, 25119–25126 CrossRef CAS
- J. Vergen, C. Hecht, L. V. Zholudeva, M. M. Marquardt, R. Hallworth and M. G. Nichols, Microsc. Microanal., 2012, 18, 761–770 CrossRef CAS PubMed
- M. A. Yaseen, S. Sakadzic, W. Wu, W. Becker, K. A. Kasischke and D. A. Boas, Biomed. Opt. Express, 2013, 4, 307–321 CrossRef CAS PubMed
- A. C. Croce and G. Bottiroli, Eur. J. Histochem., 2014, 58, 2461 CAS
- J. M. Salmon, E. Kohen, P. Viallet, J. G. Hirschberg, A. W. Wouters, C. Kohen and B. Thorell, Photochem. Photobiol., 1982, 36, 585–593 CrossRef CAS
- J. A. Palero, A. N. Bader, H. S. de Bruijn, A. v. d. P. van den Heuvel, H. J. C. M. Sterenborg and H. C. Gerritsen, Biomed. Opt. Express, 2011, 2, 1030–1039 CrossRef CAS
- C. Stringari, J. L. Nourse, L. A. Flanagan and E. Gratton, PLoS One, 2012, 7, e48014 CrossRef CAS PubMed
- K. Drozdowicz-Tomsia, A. G. Anwer, M. A. Cahill, K. N. Madlum, A. M. Maki, M. S. Baker and E. M. Goldys, J. Biomed. Opt., 2014, 19, 086016 CrossRef PubMed
- A. Rueck, C. Hauser, S. Mosch and S. Kalinina, J. Biomed. Opt., 2014, 19, 096005 CrossRef PubMed
- C. W. Shuttleworth, Neurochem. Int., 2010, 56, 379–386 CrossRef CAS PubMed
- R. M. Rodrigues, P. Macko, T. Palosaari and M. P. Whelan, Toxicol. Lett., 2011, 206, 281–288 CrossRef CAS PubMed
- A. Bednarkiewicz, R. M. Rodrigues and M. P. Whelan, Toxicol. in Vitro, 2011, 25, 2088–2094 CrossRef CAS PubMed
- T. S. Blacker, Z. F. Mann, J. E. Gale, M. Ziegler, A. J. Bain, G. Szabadkai and M. R. Duchen, Nat. Commun., 2014, 5, 3936 CrossRef CAS PubMed
- T. G. Scott, R. D. Spencer, N. J. Leonard and G. Weber, J. Am. Chem. Soc., 1970, 92, 687–695 CrossRef CAS
- J. Maltas, L. Amer, Z. Long, D. Palo, A. Oliva, J. Folz and P. Urayama, Anal. Chem., 2015, 87, 5117–5124 CrossRef CAS PubMed
- R. K. Tao, Y. Z. Zhao, H. Y. Chu, A. X. Wang, J. H. Zhu, X. J. Chen, Y. J. Zou, M. Shi, R. M. Liu, N. Su, J. L. Du, H. M. Zhou, L. Y. Zhu, X. H. Qian, H. Y. Liu, J. Loscalzo and Y. Yang, Nat. Methods, 2017, 14, 720–728 CrossRef CAS PubMed
- Z. Long, J. Maltas, M. C. Zatt, J. Cheng, E. J. Alquist, A. Brest and P. Urayama, J. Biophotonics, 2015, 8, 247–257 CrossRef CAS PubMed
- J. Maltas, D. Palo, C. K. Wong, S. Stefan, J. O'Connor, N. Al Aayedi, M. Gaire, D. Kinn and P. Urayama, RSC Adv., 2018, 8, 41526–41535 RSC
- H. N. Carlsen, H. Degn and D. Lloyd, J. Gen. Microbiol., 1991, 137, 2879–2883 CrossRef CAS PubMed
- D. E. Griffiths and D. C. Wharton, J. Biol. Chem., 1961, 236, 1850–1856 CrossRef CAS
- M. R. Duchen and T. J. Biscoe, J. Physiol., 1992, 450, 13–31 CrossRef CAS PubMed
- S. A. Siano and R. Mutharasan, Biotechnol. Bioeng., 1989, 34, 660–670 CrossRef CAS PubMed
- E. S. Shin, J. Park, J. M. Shin, D. Cho, S. Y. Cho, D. W. Shin, M. Ham, J. B. Kim and T. R. Lee, Bioorg. Med. Chem., 2008, 16, 3580–3586 CrossRef CAS PubMed
- P. G. Heytler and W. W. Prichard, Biochem. Biophys. Res. Commun., 1962, 7, 272–275 CrossRef CAS
- L. Tretter and V. Adam-Vizi, J. Neurosci., 2000, 20, 8972–8979 CrossRef CAS
- H. Sies, Redox Biol., 2017, 11, 613–619 CrossRef CAS PubMed
- F. Fereidouni, A. N. Bader and H. C. Gerritsen, Opt. Express, 2012, 20, 12729–12741 CrossRef CAS PubMed
- D. Yasokawa, S. Murata, Y. Iwahashi, E. Kitagawa, R. Nakagawa, T. Hashido and H. Iwahashi, Appl. Biochem. Biotechnol., 2010, 160, 1685–1698 CrossRef CAS PubMed
- N. S. Kosower, E. M. Kosower, B. Wertheim and W. S. Correa, Biochem. Biophys. Res. Commun., 1969, 37, 593–596 CrossRef CAS
- H.-S. Kim, M. J. Quon and J.-a. Kim, Redox Biol., 2014, 2, 187–195 CrossRef CAS PubMed
- L. Bartosikova and J. Necas, Vet. Med., 2018, 63, 443–467 CrossRef CAS
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed
- Q. Chen, E. J. Vazquez, S. Moghaddas, C. L. Hoppel and E. J. Lesnefsky, J. Biol. Chem., 2003, 278, 36027–36031 CrossRef CAS PubMed
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed
- H. B. Leavesley, L. Li, K. Prabhakaran, J. L. Borowitz and G. E. Isom, Toxicol. Sci., 2008, 101, 101–111 CrossRef CAS PubMed
- H. Nůsková, M. Vrbacký, Z. Drahota and J. Houštěk, J. Bioenerg. Biomembr., 2010, 42, 395–403 CrossRef PubMed
- G. E. Isom and J. L. Borowitz, in Toxicology of cyanides and cyanogens, ed. A. H. Hall, G. E. Isom and G. A. Rockwood, John Wiley and Sons, West Sussex, England, 2015, ch. 5, pp. 70–81, DOI:10.1002/9781118628966.ch5
- A. G. Kanthasamy, B. Ardelt, A. Malave, E. M. Mills, T. L. Powley, J. L. Borowitz and G. E. Isom, Toxicol. Lett., 1997, 93, 47–54 CrossRef CAS PubMed
- A. Gafni and L. Brand, Biochemistry, 1976, 15, 3165–3171 CrossRef CAS PubMed
- R. V. Hull, P. S. Conger and R. J. Hoobler, Biophys. Chem., 2001, 90, 9–16 CrossRef CAS
- G. Weber, J. Phys. Chem., 1981, 85, 949–953 CrossRef CAS
- E. Gratton, D. M. Jameson and R. D. Hall, Annu. Rev. Biophys. Bioeng., 1984, 13, 105–124 CrossRef CAS PubMed
- K. M. Hirshfield, D. Toptygin, B. S. Packard and L. Brand, Anal. Biochem., 1993, 209, 209–218 CrossRef CAS PubMed
- L. Malacrida, S. Ranjit, D. M. Jameson and E. Gratton, Annu. Rev. Biophys., 2021, 50, 575–593 CrossRef CAS PubMed
- A. H. A. Clayton, Q. S. Hanley and P. J. Verveer, J. Microsc., 2004, 213, 1–5 CrossRef CAS PubMed
- M. A. Digman, V. R. Caiolfa, M. Zamai and E. Gratton, Biophys. J., 2008, 94, L14–L16 CrossRef CAS PubMed
- C. Stringari, A. Cinquin, O. Cinquin, M. A. Digman, P. J. Donovan and E. Gratton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13582–13587 CrossRef CAS PubMed
- C. Stringari, R. A. Edwards, K. T. Pate, M. L. Waterman, P. J. Donovan and E. Gratton, Sci. Rep., 2012, 2, 568 CrossRef PubMed
- S. Ranjit, L. Malacrida, M. Stakic and E. Gratton, J. Biophotonics, 2019, 12, e201900156 CrossRef PubMed
- C. A. Gomez, J. Sutin, W. C. Wu, B. Y. Fu, H. Uhlirova, A. Devor, D. A. Boas, S. Sakadzic and M. A. Yaseen, PLoS One, 2018, 13, e0194578 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2021 |