
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic–inorganic hybrid perovskite for low-cost and high-performance xerographic photoreceptors†
Wei-Min Guab,
Chuanxi Wangc,
Cai-Yan Gao a,
Xin-Heng Fana,
Lian-Ming Yang*a and
Ke-Jian Jiang*a
a,
Xin-Heng Fana,
Lian-Ming Yang*a and
Ke-Jian Jiang*a
aKey Laboratory of Green Printing, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: kjjiang@iccas.ac.cn; yanglm@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100190, P. R. China
cBeijing National Laboratory for Molecular Sciences (BNLMS), State Key Lab for Structural Chemistry of Unstable and Stable Species, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100190, P. R. China
First published on 21st June 2021
Abstract
Solution-processable organic–inorganic hybrid perovskites are being widely investigated for many applications, including solar cells, light-emitting diodes, photodetectors, and lasers. Herein, we report, for the first time, successful fabrication of xerographic photoreceptors using methylammonium lead iodide (CH3NH3PbI3) perovskite as a light-absorbing material. With the incorporation of polyethylene glycol (PEG) into the perovskite film, the ion migration inherent to the perovskite material can be effectively suppressed, and the resulting photoreceptor exhibits a high and panchromatic photosensitivity, large surface potential, low dark decay, and high environmental resistance and electrical cycling stability. Specifically, the energies required to photodischarge one half of the initial surface potential (E0.5) are 0.074 μJ cm−2 at 550 nm and 0.14 μJ cm−2 at 780 nm, respectively. The photosensitivites outmatch those of the conventionally used organic pigments having narrow spectral responses. Our findings inform a new generation of highly efficient and low-cost xerographic photoreceptors based on perovskite materials.
1. Introduction
Xerographic technology was invented in the 1930s by Chester Carlson, followed by the introduction of the first xerographic copying machine in 1949.1,2 This technology has caused a new revolution in modern office copying/printing. In xerography, the photoreceptor is a key element to accept and retain charges in the dark and undergo photo-discharge upon light exposure for forming an electrostatic latent image. In principle, it is a good insulator in the dark and becomes conductive when exposed to light. A typical photoreceptor, along with the xerographic process, is shown in Scheme 1. The photoreceptor has a multilayer structure composed of a charge generation layer (CGL) and a charge transport layer (CTL) on a metallic substrate. The photoreceptor is first charged negatively in the dark by a corotron, forming a high density of charge on its upper surface with a surface potential (V0), which is followed by a slow decay from dark conductivity. Upon light illumination, electron–hole (e–h) pairs are generated and separated in the CGL with the aid of the electrical field. Subsequently, the holes inject to the CTL and drift across the CTL to discharge the device while the electrons move to neutralize the positive countercharge residing on the substrate, leading to rapid photodischarge in the exposed areas as shown in Scheme 1b.3,4 For the photoreceptor, E0.5, the energy required to photo-discharge one half of the initial potential (Vi), is usually used to assess the photosensitivity of the device. The E0.5 is the product of I and t, where I (μJ cm−2 s−1) is the intensity of a certain monochromatic light, and t (s) is the corresponding time. The lower E0.5 value, the higher photosensitivity. | ||
| Scheme 1 (a) Schematic of the configuration and the photo-discharge process of a dual-layer photoreceptor: CGL, charge generation layer; CTL, charge transport layer. (b) Schematic of a photo-induced discharge curve. | ||
In the early stage, inorganic semiconductor, such as amorphous selenium and amorphous silicon (a-Si), were predominantly used as photoconductive materials for the photoreceptors, which were incorporated by vacuum deposition method.5,6 Currently, organic pigments have been extensively employed due to their low cost.7,8 However, these organic photoconductive materials are insoluble in common organic solvents, and must be elaborately dispersed in a solvent for the prevention of their aggregation and precipitation. On the other hand, organic-based photoconductors generally hold narrow absorption bands, and thus exhibit photosensitivity only to a specific light source.1,2 Thus, it is of high value and considerable importance to find out other CGM candidates for simplifying the fabrication process, reducing the cost, and elevating the device performance.
Organic–inorganic hybrid perovskite materials with a chemical structure of ABX3 (A: methylammonium and formamidinium; B: Pb, and Sn; and X: I and Br) have attracted tremendous attention due to their optoelectronic properties (such as a direct and tunable band gap, a high absorption coefficient, a long charge carrier diffusion length) as well as solution processability.9–12 The fabrication of the perovskite film based on a solution process enables a variety of relatively easy deposition techniques, such as inkjet printing, screen printing, and roller coating. In the recent decade, the perovskite materials have been intensely investigated for their applications in solar cells, light-emitting diodes, photodetectors, and laser.13–15 These research activities inspired us to explore their application in xerographic photoreceptors.
In this study, CH3NH3PbI3 (MAPbI3) perovskite was employed as a charge generation material for the fabrication of xerographic devices. It was well known that organic–inorganic hybrid perovskites are a class of ionic conductors with loose crystal structures, and thus ion migration is an intrinsic property of perovskite materials.16–18 This phenomenon may be problematic for the photoreceptors in a xerographic process, where a high electric field (ca. 105 V cm−1) is created across the device.19 Here we found that the ion migration can be greatly suppressed with the incorporation of polyethylene glycol (PEG) into the perovskite film. As a result, the as-prepared perovskite photoreceptor exhibits a high photosensitivity and a broad spectral response with high electrical and environmental stability.
2. Results and discussion
The perovskite photoreceptor was constructed in a dual-layer configuration consisting of a perovskite charge generation layer (CGL) and a triaryl amine charge transport layer (CTL). Both the layers were sequentially coated on an aluminum substrate by a doctor blading method. For the CGL, a mixture of perovskite precursor and polyethylene glycol (PEG) with a certain ratio was dissolved in dimethylformamide (DMF). The use of PEG was expected to control the perovskite crystallization process, and suppress its ion migration. In addition, the PEG could improve the adhesion between the perovskite layer and the aluminum substrate; for the CTL, a mixture of N,N,N,N-tetrakis(4-methylphenyl)-benzidine (TPD) as a hole charge transport material and Makrolon as a binder resin, was dissolved in dichloromethane. The fabrication procedure is described in detail in the Experimental section.The morphology of the perovskite CGL was investigated by scanning electron microscope (SEM), as shown in Fig. 1 and in Fig. S1 (ESI†). The pristine film without PEG comprises dendrite-like perovskite domains on the several micrometers long scale with large voids between them, which is consistent with the literature reports on the similar films.20 The low surface coverage can cause light penetrating through the surface layer, reducing the light absorption in the CGL. Besides, the uncovered areas lead to direct contact between the CTL and the substrate, affecting the charge acceptance and photo-discharge of the devices prepared. With the addition of PEG at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of PEG monomer (C2H4O) to MAPbI3, the large domains seemed fused, and the surface coverage was remarkably improved with few pinholes in the film. Increasing the ratio to 1.5:1, the domain size was reduced significantly, and a uniform and full coverage perovskite film was attained. No obvious change in morphology was observed upon further increasing the ratio up to 2:1 (see ESI, Fig. S1†). The results suggested that the addition of PEG greatly improved the morphology of the perovskite film, in accordance with the previous reports.20,21 The PEG used here as a scaffold may enable the production of more nucleation sites and smaller perovskite grains in the film formation. The UV-vis absorption and X-ray diffraction spectra for the above-mentioned films are shown in Fig. S2 and S3 (ESI†).
:1 molar ratio of PEG monomer (C2H4O) to MAPbI3, the large domains seemed fused, and the surface coverage was remarkably improved with few pinholes in the film. Increasing the ratio to 1.5:1, the domain size was reduced significantly, and a uniform and full coverage perovskite film was attained. No obvious change in morphology was observed upon further increasing the ratio up to 2:1 (see ESI, Fig. S1†). The results suggested that the addition of PEG greatly improved the morphology of the perovskite film, in accordance with the previous reports.20,21 The PEG used here as a scaffold may enable the production of more nucleation sites and smaller perovskite grains in the film formation. The UV-vis absorption and X-ray diffraction spectra for the above-mentioned films are shown in Fig. S2 and S3 (ESI†).
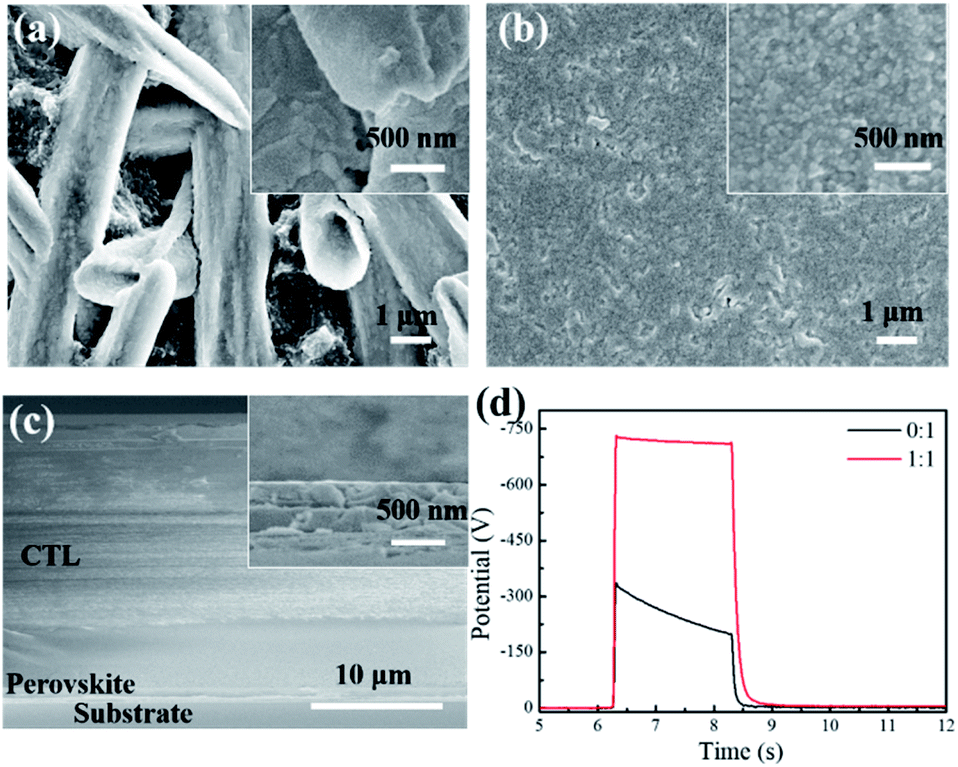 | ||
| Fig. 1 Top SEM images of the perovskite films without (a) and with PEG (b) (perovskite:PEG = 1:1 mol/mol). (c) Cross-sectional SEM image of a typical perovskite photoreceptor (inset: higher magnification SEM image). (d) PIDCs of the perovskite photoreceptors without and with PEG (perovskite:PEG = 1:1, mol/mol). | ||
Fig. 1c shows the cross sectional SEM image of a typical perovskite photoreceptor, where the thicknesses of the CGL and the CTL are ∼700 nm and ∼22 μm, respectively. The xerographic performance of the perovskite photoreceptors was analyzed by measuring their photo-induced discharge curves (PIDC) under illumination at 550 nm, including the initial surface potential (V0), the dark decay (Rd) of the surface potential, photosensitivity (E0.5), and residual potential (Vr). For the perovskite devices with different amounts of PEG, the parameters were listed in Table 1. It was found that all the perovskite devices presented a high photosensitivity with E0.5 < 0.1 μJ cm−2. However, the device based on the pristine perovskite CGL provided only a poor set of xerographic parameters: a very low V0 of −329 V and a considerably high Rd value of 65 V s−1 albeit with the highest photosensitivity. It should be pointed out that a xerographic process depends mainly on the difference in surface potentials between the exposed and unexposed areas to create an electrostatic latent image. Thus, a high contrast potential between the two areas is essential for the image formation. A working photoreceptor requires a sufficiently high surface potential (>600 V), lower dark decay (<40 V s−1), high photosensitivity (E0.5 < 2 μJ cm−2), and as low residual potential as possible.1,2 The high dark conductivity of the pristine perovskite device is related to a high dark decay and a lower surface potential, and supposed to arise from the perovskite charge generation layer. Previous studies indicated that the exciton binding energy of MAPbI3 perovskite is on the order of kBT (25 meV) at room temperature.22,23 Such, free carriers may prevail and flow directly under the strong electric field even in the dark. And also, I− and MA+ ions in the perovskite layer would migrate across the device under an electric field.22–25 Therefore, both the charge carriers and the mobile ions may be responsible for the high dark conductivity in the pristine perovskite device.
| Device (PEG:MAPbI3) |
V0 [V] | Rd [V s−1] | E0.5 [μJ cm−2] | Vr [V] |
|---|---|---|---|---|
| a All photoconductive data listed in the table were measured with a corona voltage of −5 kV and a monochromatic light of 1.0 μW (λ: 550 nm). | ||||
| 0:1 |
−329 ± 5 | 65 ± 1.3 | 0.049 ± 0.001 | −2 ± 0.6 |
| 1:1 |
−721 ± 3.5 | 19 ± 0.5 | 0.074 ± 0.003 | −8 ± 1 |
| 1.5:1 |
−761 ± 13 | 18 ± 2.6 | 0.091 ± 0.002 | −8 ± 2 |
| 2:1 |
−780 ± 14 | 11 ± 3.5 | 0.092 ± 0.009 | −5 ± 1 |
As shown in Fig. 1d, with the PEG addition (PEG:MAPbI3, 1:1 mol/mol), the surface potential of the device greatly increased from −329 V to −721 V, followed by a decrease of the dark decay from 65 V s−1 to 19 V s−1. With increasing the PEG content of (1.5:1 and then 2:1), the surface potentials slightly increased, accompanied by slow decrease of the photosensitivity (the E0.5 is increased from 0.074 to 0.091 and 0.092 μJ cm−2), probably ascribed to the reduced conductivity of the device due to the insulating nature of the PEG employed. Even so, the photosensitivities for all the PEG-contained perovskite photoreceptors are several times higher than those for the conventional organic-based devices.26–30 Accordingly, it is safe to say that the perovskite photoreceptor has the possibility to be applied in high-speed, high-resolution copying machines. Furthermore, the photosensitivity of the devices was investigated under the illumination at 780 nm, and the xerographic parameters were listed in Table S1 (ESI†). The optimized device (PEG:MAPbI3, 1:1) exhibits a high charge acceptance (V0: −705 V), low decay (Rd: 12 V s−1), and good photosensitivity (E0.5: 0.138 μJ cm−2). The E0.5 value is comparable to those of phthalocyanine-based photoreceptors for printer.31,32 Considering the fact that the absorption coefficient of MAPbI3 is relatively low at 780 nm, such a photosensitivity is encouraging as well. Further compositional engineering of perovskite materials may improve the photosensitivity in near infrared region.11 To the best of our knowledge, very rare are the organic pigments highly photosensitive to both the visible and near-infrared region in xerography.1,2 This implies that the perovskite photoreceptors would be used in both copier and printer.
As mentioned above, the dark conductivity may arise from both charge carriers and mobile ions of the MAPbI3 film. To make a distinction between them, the direct current (DC) temperature-dependent conductivity was measured in the dark for the vertical devices with structure Au/perovskite/Au (Fig. 2a). For both the devices with and without PEG, two slopes can be identified from each dataset, as shown in Fig. 2b. They may correspond to two conduction mechanisms: electronic conduction in the low-temperature region and ionic conduction in the high-temperature region.33 The activation energy of the electronic and ion migration can be extracted from the temperature-dependent electrical conductivity by the Nernst–Einstein relation:
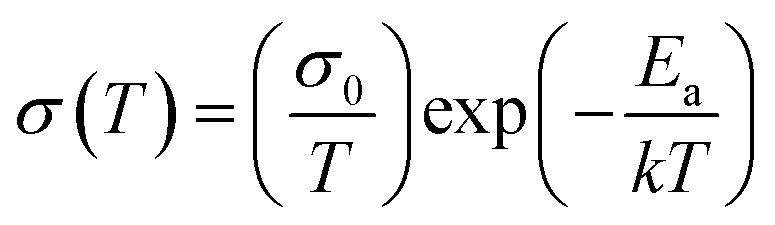 | (1) |
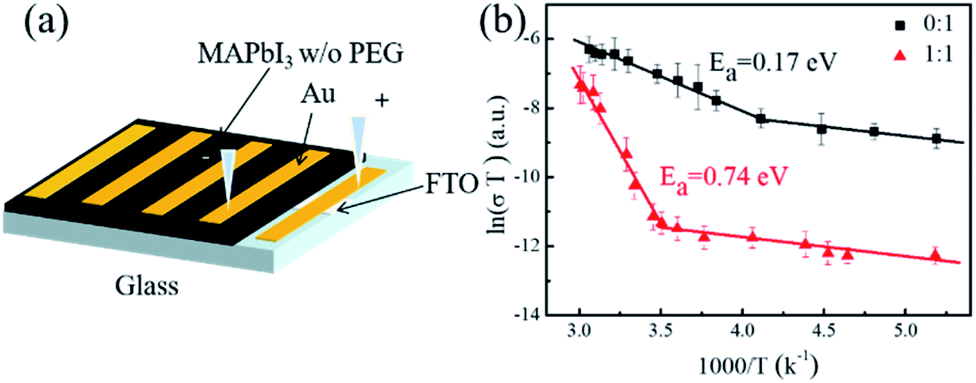 | ||
| Fig. 2 (a) Temperature-dependent conductivity measurement geometry. (b) The temperature-dependent conductivity result of MAPbI3 without and with PEG (PEG:MAPbI3 = 1:1, mol/mol). | ||
For the PEG-containing perovskite film, a similar activation energy of ∼43 meV was recorded for the electron conduction, indicating that the incorporation of PEG do not affect the exciton dissociation significantly. But for the film with PEG, a higher transition temperature of 300 K from the electronic to ionic migration-dominated conduction was found with a higher ionic conduction Ea of 0.74 eV, which is about four times larger than that for the pristine perovskite film. The results indicate that the incorporated PEG may locate at the grain boundaries and suppress the ion dissociation and migration via chemical interaction and/or physical barrier. In addition, it can be found that over the entire temperature region from 190 to 330 K, the conductivity for the PEG-containing device is about 1 to 2 orders of magnitude lower than that for the pristine perovskite device, suggesting that the incorporation of PEG not only inhibits the ion migration, but also reduces the charge carrier transfer.
To verify the presence of intermolecular interactions between MAPbI3 and PEG, Fourier-transform infrared spectroscopy (FTIR) was measured. As shown in Fig. S4 (ESI†), the stretching vibration of C–O bond appears at 1050–1060 cm−1 in the pure PEG, and shifts to 1100–1130 cm−1 in the PEG–perovskite system. The shift may be ascribed to the formation of a coordination bond between PEG and the perovskite by sharing the lone pair electrons on the C–O of PEG with the empty 6p orbital of Pb2+ in the perovskite.38 Besides, hydrogen bonding could form between MA+ and O in the PEG chain.20 To examine the change in the chemical bonding states of Pb and I after the incorporation of the PEG, X-ray photoelectron spectroscopy (XPS) was recorded. Fig. S5a and 5b (ESI†) reveal the shifts of I 3d and Pb 4f XPS peaks of the MAPbI3 after the incorporation of PEG, further indicating the chemical interaction between PEG and the perovskite.39
The stability of the perovskite photoreceptors was preliminarily evaluated. Fig. 3a shows the photo-induced discharging curves (PIDCs) of the PEG-based perovskite photoreceptor (PEG:MAPbI3 = 1:1, mol/mol). The measurement was performed with a repeatedly charging and photo-discharging process for nine cycles, where the photoreceptor was charged with a corona voltage of −5 kV, and then photo-discharged under a 550 nm monochromatic light with an intensity of 1.0 μW cm−2. These PIDCs and the corresponding key parameters are almost the same, suggesting their good electrical cycling stability. We also examined the thermal and humidity stability of the device which stayed for 30 days at a temperature of 50 °C in an atmosphere of 40–50 RH%. It was found that its PIDC curves (shown in Fig. 3b) and xerographic parameters (Table S2, ESI†) remain almost unchanged.
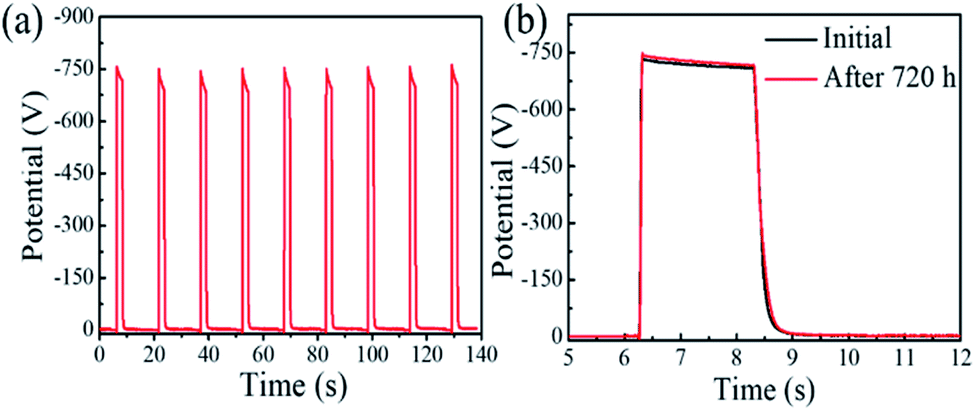 | ||
| Fig. 3 (a) Photo-induced discharge curves of the perovskite photoreceptor (PEG:MAPbI3 = 1:1) with a repetitive charge–photodischarge process for nine cycles, where the photoreceptor is charged with a corona voltage of −5 kV, and photo-discharged using 550 nm monochromatic light with intensity of 1.0 μW cm−2. (b) PIDCs before and after aging for 30 days at 50 °C with humidity of 40–50 RH%. | ||
3. Conclusions
In summary, MAPbI3 perovskite was for the first time utilized as a light-absorbing charge generation material (CGM) for xerographic photoreceptor. It was observed that the incorporation of PEG could effectively inhibited the ion migration in the perovskite film, and improved the charge acceptance of the device. The resultant perovskite photoreceptor exhibits an excellent set of xerographic parameters: good and panchromatic photosensitivity, high surface potential, low dark decay, and low residual potential. In addition, the perovskite photoreceptor presents high electrical cycling and environmental stability. The perovskite photoreceptor may be applicable to copier and printer. Further studies, particularly aimed at practical use, are ongoing in our group.4. Experimental section
4.1. Materials
Lead(II) iodide (99.999%) and methylammonium iodide (>99.5%) were purchased from Xi'an Polymer Light Technology in China. Y-type phthalocyanin (Y–TiOPC), N,N,N,N-tetrakis(4-methylphenyl)-benzidine (S100) was purchased from Nippon Shizai Co., Ltd in Japan. Polycarbonate Makrolon was purchased from Idemitsu Kosan Co., Ltd in Japan. Polymer polyethylene glycol (PEG Mn = 20000) was purchased from Alfa Aesar in America. N,N-dimethylformamide (anhydrous, 99.8%), chlorobenzene (anhydrous, 99.8%) and dichloromethane (AR, 99.5%) were purchased from Sigma-Aldrich. Aluminum substrates were purchased from HG Technologies Co., Ltd in China.
4.2. Fabrication of photoreceptors
All chemicals were directly used without any further treatment. Aluminum substrates were cleaned with ultrasonic wave in basic water, deionized water and ethanol successively. The cleaned aluminum plates (5 × 5 cm) were dried under N2 atmosphere and then treated with oxygen plasma for 30 min before use. CH3NH3I and PbI2 (1:1, mol/mol) for 227 and 659 mg mL−1 solution was mixed with PEG which was dissolved in N,N-dimethylformamide earlier (20% concentration) and stirred in N,N-dimethylformamide at 60 °C for 3 h. The molar ratio of PEG monomers (C2H4O) to ultimate product MAPbI3 is from 0:1 to 2:1. The resulting solution was then coated onto the aluminum substrate by doctor-blade coating method. The thickness of perovskite film is around 700 nm. Then the perovskite-coated plate was dried on a hot plate at 60 °C for 45 min and at 105 °C for 70 min, respectively, in a nitrogen filled glove box. The CTL solution was prepared by mixing 6.7 g of S100, 8.3 g of Makrolon and 85 g dichloromethane, which was stirred for at least 12 hours. After the plate was cooled to room temperature, the CTL solution was coated upon the perovskite layer using doctor-blade coating method in ambient air. Then the sample was dried on a hot plate at 95 °C for 45 min. The thickness of CTL layer is around 22 μm.
4.3. Characterization
Photo-induced discharge curves (PIDCs) of the perovskite photoreceptors were measured by an EPA-8200 electrostatic paper analyzer. The scan area was 3.14 cm2 (Φ 2 cm). For each charge–discharge cycling, the sample undergoes five processes: pre-exposure (4 s), charging (2 s), dark holding (2 s), exposure (2 s), and rest (5 s). The corona voltage for charging and monochromatic light for exposure was fixed to −5 kV and 1.0 μW (light: 780 nm and 550 nm), respectively. From these PIDCs, the values of the initial surface potential (V0, charged by the corona), the surface potential decay in dark (Rd), and the real surface potential just before exposure (Vi = V0 − Rd), the exposure energy for surface potential decrease to a half of Vi (E0.5), and residual potential after exposure (Vr) can be derived.The direct current (DC) temperature-dependent conductivity of the perovskite films were measured using a variable-temperature probe station equipped with a Keithley model 2400 digital source meter. The perovskite devices were measured in the dark for the vertical devices with structure Au/perovskite/Au to better consistency with normal working condition. The electrodes area and thickness were 0.04 cm2 and 700 nm, respectively. The perovskite devices were first cooled down to 193 K, and their resistances were then measured after being heated to each target temperature (5 K min−1 heating rate) and stabilized for 5 min.
The surface and cross-section morphology of the perovskite thin films were examined by scanning electron microscopy (SEM, S-4800). The X-ray diffraction (XRD) measurements were using an X-ray diffractometer (Rigaku, D/MAX RINT-2500) with Cu Kα radiation (λ = 1.54 Å) at a speed of 2° min−1. The absorption spectra were collected using a UV-vis spectrometer (SHIMADZU, UV-1800 UV-vis Spectrophotometer) in the wavelength range of 300–850 nm. The Fourier-transform infrared spectroscopy (FTIR) spectra were measured using BRUKER TENSOR 27 infrared spectrometer. X-ray photoelectron spectroscopy (XPS) were performed on the Thermo Scientific ESCALAB 250Xi using 200 W monochromated Al Kα radiation, and 500 μm X-ray spot was used for XPS analysis.
Author contributions
K. J. and L. M. developed the basic concept, and coordinated the project. W. M. fabricated and characterized the photoreceptors, and optimized device performance. C. X. contributed to the characterization of PIDCs. C. Y. and X. H. contributed to the SEM measurement and analyzed the data. K. J. and L. M. contributed to the supervision of the project. W. M, K. J. and L. M. co-wrote the manuscript. All authors reviewed the paper.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Nature Science Foundation of China (Grant No. 61874123, 21572235). L.-M. Yang and K.-J. Jiang thank Prof. Yuan Wang from College of Chemistry and Molecular Engineering, Peking University (China) for helping in xerographic measurements and inspired discussion in the manuscript formation.References
- K.-Y. Law, Chem. Rev., 1993, 93, 449–486 CrossRef CAS.
- D. S. Weiss and M. Abkowitz, Chem. Rev., 2010, 110, 479–526 CrossRef CAS PubMed.
- K.-Y. Law, Chem. Mater., 1992, 4, 605–611 CrossRef CAS.
- Y. Wang and D. J. Liang, Adv. Mater., 2010, 22, 1521–1525 CrossRef CAS PubMed.
- G. Pfister, Contemp. Phys., 1979, 20, 449–479 CrossRef CAS.
- W. E. Spear and P. G. Lecomber, Philos. Mag. Lett., 1976, 33, 935–949 CrossRef CAS.
- K.-J. Jiang, H. Chen, M. Wang and S. Yang, Mater. Sci. Eng., B, 1999, 57, 87–91 CrossRef.
- Y. W. X. Zhang, Y. Ma, Y. Ye, Y. Wang and K. Wu, Langmuir, 2006, 22, 344–348 CrossRef PubMed.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y.-B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 9898–9903 CrossRef CAS PubMed.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Hunphry-Baker, J. H. Yum, J. E. Moser, M. Grätzel and N. G. Park, Sci. Rep., 2012, 2, 591–597 CrossRef PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Gratzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- F. Li, Y. Zhang, K.-J. Jiang, C. Zhang, J.-H. Huang, H. Wang, H. Fan, P. Wang, Y. Chen, W. Zhao, X. Li, L.-M. Yang, Y. Song and Y. Li, Adv. Mater., 2018, 30, 1804454 CrossRef PubMed.
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S.-H. Jang, J. Zhang, X. Lu, D. Lei, C.-S. Lee, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- H. Z. Lu, A. Krishna, S. M. Zakeeruddin, M. Gratzel and A. Hagfeldt, iScience, 2020, 23, 101359 CrossRef CAS PubMed.
- Z. Xiao, Y. Yuan, Y. Shao, Q. Wang, Q. Dong, C. Bi, P. Sharma, A. Gruverman and J. Huang, Nat. Mater., 2015, 14, 193–198 CrossRef CAS PubMed.
- J. Mort, M. Machonkin, R. Ziolo and I. Chen, Appl. Phys. Lett., 1992, 61, 1829–1831 CrossRef CAS.
- Y. Zhao, J. Wei, H. Li, Y. Yan, W. Zhou, D. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- Y. Zong, Y. Zhou, Y. Zhang, Z. Li, L. Zhang, M.-G. Ju, M. Chen, S. Pang, X. C. Zeng and N. P. Padture, Chem, 2018, 4, 1404–1415 CAS.
- Y. Lin, Y. Bai, Y. Fang, Q. Wang, Y. Deng and J. Huang, ACS Energy Lett., 2017, 2, 1571 CrossRef CAS.
- S. Tan, I. Yavuz, N. D. Marco, T. Huang, S.-J. Lee, C. S. Choi, M. Wang, S. Nuryyeva, R. Wang, Y. Zhao, H.-C. Wang, T.-H. Han, B. Dunn, Y. Huang, J.-W. Lee and Y. Yang, Adv. Mater., 2020, 32, 1906995 CrossRef CAS PubMed.
- X. Li, X. Wang, W. Zhang, Y. Wu, F. Gao and J. Fang, Org. Electron., 2015, 18, 107–112 CrossRef CAS.
- Y. Zhao, W. Zhou, Z. Han, D. Yuac and Q. Zhao, Phys. Chem. Chem. Phys., 2021, 23, 94–106 RSC.
- K.-Y. Law and I. W. Tarnawskyj, US Pat.4797337, 1989 Search PubMed.
- R. O. Loutfy, A. M. Hor, P. Kazmaler and M. Tam, J. Imaging Sci., 1989, 33, 151–159 CAS.
- K.-Y. Law and F. Court Balley, J. Imaging Sci., 1987, 31, 172–177 CAS.
- K. Y. Law and F. C. Bailey, Dyes Pigm., 1988, 9, 85–107 CrossRef CAS.
- R. O. Loutfy, A. M. Hor, C. K. Hsiao, G. DiPaola-Baranyi and P. M. Kazmaier, Pure Appl. Chem., 1988, 60, 1047–1054 CAS.
- R. O. Loutfy, A. M. Hor and A. Rucklldge, J. Imaging Sci., 1987, 31, 31–37 CAS.
- T. Enokida, R. Hirohashi and T. Nakamura, J. Imaging Sci., 1990, 34, 234–242 CAS.
- T. Y. Yang, G. Gregori, N. Pellet, M. Gratzel and J. Maier, Angew. Chem., Int. Ed., 2015, 54, 7905–7910 CrossRef CAS PubMed.
- Y. Yuan, J. Chae, Y. Shao, Q. Wang, Z. Xiao, A. Centrone and J. Huang, Adv. Energy Mater., 2015, 5, 1500615 CrossRef.
- X. Li, W. Zhang, Y. C. Wang, W. Zhang, H. Q. Wang and J. Fang, Nat. Commun., 2018, 9, 3806 CrossRef PubMed.
- J. Xing, Q. Wang, Q. Dong, Y. Yuan, Y. Fang and J. Huang, Phys. Chem. Chem. Phys., 2016, 18, 30484–38490 RSC.
- Y. Zhou, Y. Yin, X. Zuo, L. Wang, T.-D. Li, Y. Zhou, N. P. Padture, Z. Yang, Y. Guo, Y. Xue, K. Kisslinger, M. Cotlet, C.-Y. Nam and M. H. Rafailovich, Chem. Mater., 2020, 32, 5104–5117 CrossRef CAS.
- M. Kim, S. G. Motti, R. Sorrentino and A. Petrozza, Energy Environ. Sci., 2018, 11, 2609–2619 RSC.
- S. Fu, X. Li, L. Wan, Y. Wu, W. Zhang, Y. Wang, Q. Bao and J. Fang, Adv. Energy Mater., 2019, 9, 1901852 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02717d |
| This journal is © The Royal Society of Chemistry 2021 |