
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceJejucarbazoles A–C, carbazole glycosides with indoleamine 2,3-dioxygenase 1 inhibitory activity from Streptomyces sp. KCB15JA151†
Gil Soo Kim‡
ab,
Jun-Pil Jang‡a,
Mincheol Kwonab,
Tae Hoon Ohac,
Kyung Taek Heoab,
Byeongsan Leeac,
Jung-Sook Leed,
Sung-Kyun Ko a,
Young-Soo Hongab,
Jong Seog Ahn*ab and
Jae-Hyuk Jang*ab
a,
Young-Soo Hongab,
Jong Seog Ahn*ab and
Jae-Hyuk Jang*ab
aAnticancer Agent Research Center, Korea Research Institute of Bioscience and Biotechnology, Cheongju 28116, Korea. E-mail: jsahn@kribb.re.kr; jangjh@kribb.re.kr
bDepartment of Biomolecular Science, KRIBB School of Bioscience, University of Science and Technology, Daejeon 34113, Korea
cCollege of Pharmacy, Chungbuk National University, Cheongju, 28160, Korea
dKorean Collection for Type Cultures, Korea Research Institute of Bioscience and Biotechnology, Jeongeup 56212, Korea
First published on 1st June 2021
Abstract
A bioassay-guided investigation led to the isolation of three new carbazole glycosides, jejucarbazoles A–C (1–3), from Streptomyces sp. KCB15JA151. Their planar structures were elucidated by detailed NMR and MS spectroscopic analysis with a literature study. Their relative and absolute configurations were established by ROESY correlations, coupling constants, LC-MS analysis of thiocarbamoyl-thiazolidine carboxylate derivatives, and ECD calculation. Compounds 1–3 showed indoleamine 2,3-dioxygenase 1 (IDO1) inhibitory activity with IC50 values of 18.38, 9.17, and 8.81 μM. The molecular docking analysis suggested that all compounds act as heme-displacing inhibitors against IDO1 enzyme.
Introduction
Natural products from actinomycetes have been an excellent source for drug discovery due to their diversity of carbon skeletons and biological activities.1–4 Natural products can not only be used as drugs but also provide drug leads for the development of new drugs.3–6 A substantial number of natural product derived drugs have been released on the market.4,5 Hence, the discovery of novel natural products is important for drug development.Carbazole, dibenzopyrrole, consist of the 6/5/6 fused heterocyclic ring system, which was first extracted/derived from the anthracene fraction of coal tar in 1872.7,8 Natural carbazole alkaloid is categorized into two main groups according to their natural origin and carbon substituent at C-3. All known tricyclic 3,4-dioxygenated carbazole alkaloids, significantly, have originated from microorganisms.7 Tricyclic carbazole derivatives possess various biological activities7,9 such as antimicrobial,10 antimalarial,11 anti-inflammatory,12 neuroprotective,13 and free radical scavenging activities.14 Therefore, the structure diversity and a wide range of biological activities of carbazole have attracted much attention from chemists and biologists.
Indoleamine 2,3-dioxygenase 1 (IDO1) is an intracellular heme-containing enzyme that oxidizes L-tryptophan (L-Trp) to N-formyl-L-kynurenine (NFK), which initiates the first and rate-limiting step of the kynurenine pathway.15–18 Expression of IDO1 mediates the degradation of tryptophan and the accumulation of kynurenine that lead to local immunosuppression.16,19 Upregulation of IDO1 was observed in several cancer types and could promote tumor cell growth.18,20–22 IDO1 inhibition not only can terminate the immunosuppression but also increase the effectiveness of other treatments such as therapeutic vaccination, chemotherapy, or radiationtherapy.15,16,18,23,24 Thus, inhibition of the IDO1 enzyme is an attractive pharmaceutical target.
In our efforts to discover novel IDO1 inhibitor, we screened the culture extracts of microorganisms isolated from soil and marine samples from Jeju Island. The result of the in vitro enzymatic screening assay showed that Streptomyces sp. KCB15JA151 exhibited an inhibitory activity against the IDO1 enzyme. The bioassay-guided isolation from the culture extract of this strain provided three new active tricyclic carbazole glycosides. Herein, we describe the isolation and structure elucidation of jejucarbazoles A–C (1–3) (Fig. 1) and their inhibitory effect and molecular docking analysis against the IDO1 enzyme.
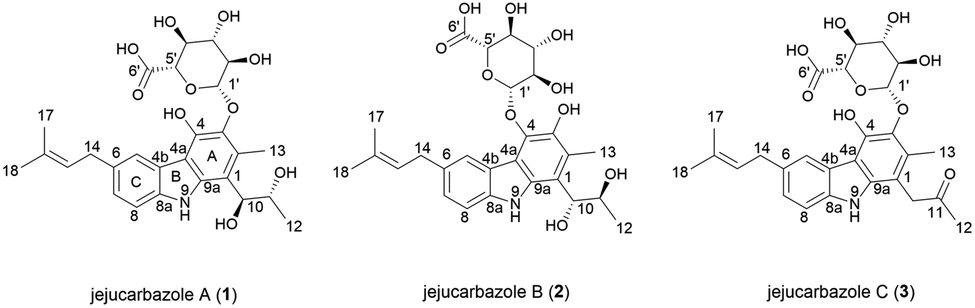 | ||
| Fig. 1 The chemical structures of 1–3. | ||
Results and discussion
Jejucarbazole A (1) was isolated as a brown amorphous powder, and its molecular formula was determined as C27H33NO10 by the HRESIMS analysis. The UV maxima absorption was at 226, 246, 268, 300, 339, and 354 nm, which is similar to those of neocarazostatins (Fig. S1†).14 The 1H NMR (800 MHz, acetone-d6) spectrum showed one exchangeable proton (δH 9.81), three aromatic protons (δH 8.05, 7.40, and 7.12), one olefinic proton (δH 5.45), seven methines (δH 4.95, 4.74, 4.17, 3.92, 3.71, 3.70, and 3.63), one methylene proton (δH 3.48), and four methyl protons (δH 2.51, 1.78, 1.74, and 1.04). The 13C NMR (200 MHz, acetone-d6) and HSQC-DEPT spectra showed 25 carbons including nine non-protonated sp2 carbons (δC 138.1, 137.5, 136.6, 131.6, 130.6, 127.7, 122.8, 113.4, and 110.7), one olefinic carbon (δC 125.0), three aromatic carbons (δC 124.9, 121.6, and 110.1), one anomeric carbon (δC 107.2), six oxygen-bearing methine carbons (δC 76.4, 75.3, 75.1, 74.2, 71.8, and 69.9), one methylene carbon (δC 34.3), and four methyl carbons (δC 24.9, 18.7, 17.0, and 13.2). Additionally, a carbonyl carbon (δC 169.9) was observed in the HMBC. NMR data were compared to chemical formula from the HRESIMS analysis. One carbon signal was not detected in the NMR data because the unstable nature of the compound in the NMR solvents led to the disappearance of several peaks in the NMR spectrum (Table 1).| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δCa | δHb, mult (J in Hz) | δCc | δHd, mult (J in Hz) | δCe | δHf, mult (J in Hz) | |
| a Recorded at 200 MHz in acetone-d6.b Recorded at 800 MHz in acetone-d6.c Recorded at 200 MHz in CD3OD.d Recorded at 800 MHz in CD3OD.e Recorded at 175 MHz in CD3OD.f Recorded at 700 MHz in CD3OD.g Not detected.h Overlapped signal.i Observed in HMBC.j Observed in HSQC. | ||||||
| 1 | 127.7 | 122.3 | 128.8 | |||
| 2 | 113.4 | 119.7 | 106.2 | |||
| 3 | 136.6 | 140.5 | 136.6 | |||
| 4 | N.Dg | 138.0 | 144.3 | |||
| 4a | 110.7 | 114.9 | 110.1 | |||
| 4b | 122.8 | 121.0 | 123.3 | |||
| 5 | 121.6 | 8.05, s | 122.8 | 8.61, s | 121.5 | 7.99, s |
| 6 | 131.6 | 131.4 | 131.8 | |||
| 7 | 124.9 | 7.12, d (8.16) | 125.1 | 7.11, d (8.31) | 124.7 | 7.12, d (7.96) |
| 8 | 110.1 | 7.40, d (8.17) | 109.4 | 7.30, d (8.22) | 109.5 | 7.29, d (8.19) |
| 8a | 138.1 | 138.3 | 138.3 | |||
| 9 | 9.81, s | |||||
| 9a | 137.5 | 133.8 | 138.5 | |||
| 10 | 75.3 | 4.95, d (7.70) | 75.1 | 5.02, d (7.65) | 42.8 | 3.99, s |
| 11 | 69.9 | 4.17, quintet (6.86) | 69.9 | 4.23, quintet (6.56) | 208.3 | |
| 12 | 18.7 | 1.04, d (6.30) | 18.1 | 1.07, d (6.36) | 27.7 | 2.18, s |
| 13 | 13.2 | 2.51, s | 12.0 | 2.40, s | 12.3 | 2.39, s |
| 14 | 34.3 | 3.48, d (6.06) | 34.0 | 3.49, m | 34.1 | 3.47, d (6.43) |
| 15 | 125.0 | 5.45, t (7.16) | 125.1 | 5.45, t (7.22) | 124.6 | 5.42, t (6.72) |
| 16 | 130.6 | 130.2 | 130.7 | |||
| 17 | 17.0 | 1.78, s | 16.6 | 1.80, s | 16.5 | 1.80, s |
| 18 | 24.9 | 1.74, s | 24.6 | 1.77, s | 24.6 | 1.77, s |
| 1′ | 107.2 | 4.74, d (7.88) | 105.5 | 4.99, d (7.61) | 107.0 | 4.65, br d (6.42) |
| 2′ | 74.2 | 3.71, ovlh | 74.1 | 3.79, t (8.45) | 74.0 | 3.66, m |
| 3′ | 76.4 | 3.63, ovlh | 76.3 | 3.56, m | 76.5 | 3.53, br s |
| 4′ | 71.8 | 3.70, ovlh | 72.0 | 3.70, br s | 72.2j | 3.65j, m |
| 5′ | 75.1 | 3.92, d (9.78) | N.Dg | N.Dg | N.Dg | N.Dg |
| 6′ | 169.9i | N.Dg | N.Dg | |||
The planar structure of 1 was elucidated by analyzing the NMR data and a literature study. The COSY correlation from H-7 to H-8 with HMBC correlations from H-5 to C-7/C-8a and from H-8 to C-4b/C-6 established ring C. The prenyl group was assigned based on the COSY correlation from H-14 to H-15 and the HMBC correlations from H-17 to C-15 and C-16 and from H-18 to C-15 and C-16. The HMBC correlation from H-14 to C-5, C-6, and C-7 implied that the prenyl group is located on C-6. The long range proton–proton coupling indicated the alkyl chain of a H-12/H-11/H-10 coupling system. The HMBC correlations of H-10 to C-1, C-2, and C-9a and of H-13 to C-1, C-2, and C-3 indicated that the alkyl side chain and the methyl group were attached to ring A, which partially established ring A. Ring B was determined by the cross peaks of the HMBC from NH-9 to C-4a/C-8a/C-9a. The H-5 to C-4a correlation of the HMBC showed a hint of the tricyclic backbone. Comparing the chemical shift with the data reported in the literature data suggests that the carbon signal, not detected in NMR data, is part of ring A (Fig. 2).14
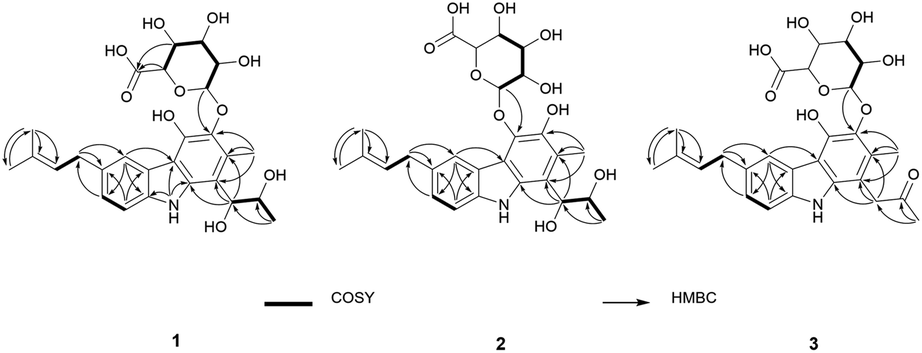 | ||
| Fig. 2 Key 2D NMR correlations of 1–3. | ||
The anomeric signals (δC 107.2/δH 4.74) suggested the presence of a sugar unit. The sugar unit was determined by the COSY correlation of H-1′/H-2′/H-3′/H-4′/H-5′. The HMBC correlation from H-4′ to C-6′ and from H-5′ to C-6′ established the carboxyl group of the sugar unit. The relative configuration of the sugar unit was elucidated by ROESY and the coupling constant. The large coupling constant (7.88 Hz) of H-1′ indicates a β-glycosidic bond and H-1′ and H-2′ in the axial position. The ROESY correlations of H-1′ to H-3′, H-1′ to H-5′, and H-3′ to H-5′ showed the same face between H-1′, H-3′, and H-5′. The coupling constant of H-5′ (9.78 Hz) implied an axial to axial coupling. Therefore, all the protons of the sugar unit occupied the axial positions, which was assigned to be β-glucuronic acid (Fig. S13†). The absolute configuration of the sugar unit was elucidated by HPLC.21,25 The acid hydrolysate of 1 was derivatized with D- and L-cysteine methyl ester followed by reaction with a o-tolyl isothiocyanate to prepare a thiocarbamoyl-thiazolidine carboxylate derivative. D- and L-derivative of 1 had the same retention time of the derivatives of an authentic sample (D-glucuronic acid) in LC-MS analysis. Therefore, the sugar unit of 1 was assigned as β-D-glucuronic acid. The same elution order of the derivatives as those of the reference (L-cysteine derivatives → D-cysteine derivatives) also supported the D-form of the sugar unit (Fig. S14†).25
Jejucarbazole B (2) was obtained as a brown amorphous powder. The UV maximum absorption, molecular formula, 1D NMR spectra, and 2D correlation patterns were very similar to those of 1, indicating that compound 2 possessed a similar backbone (Fig. S15†, Table 1, and Fig. 2). Unlike the anomeric proton (δH 4.74) and methyl proton (δH 2.51) showing cross peaks against the same carbon (δC 136.6) in the HMBC spectrum of 1, compound 2 had different correlations. The correlations of H-2 to C-3 and H-1′ to C-4 in the HMBC suggested that the sugar unit is bonded by a glycosidic bond at C-4. The ROESY cross peak signal between H-13 and H-1′ in 1 was absent in the NMR data of 2. Instead, The ROESY correlation from H-7 to H-1′ was present, which support the glycosylation position at C-4 (Fig. 3). A comparison of MS/MS fragmentation patterns also showed structural similarity between compounds 1 and 2 (Fig. S5 and S19†). Thus, the planar structure of compound 2 was determined to have the same skeleton as compound 1 except for the glycosylation position.
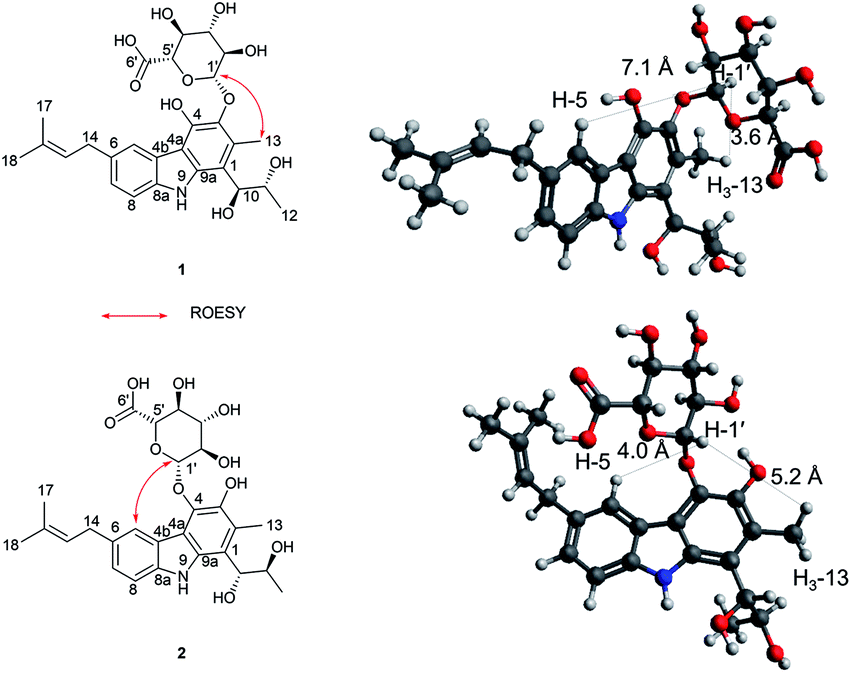 | ||
| Fig. 3 Key ROSEY correlation and interproton distances related to anomeric protons of 1 and 2. | ||
Jejucarbazole C (3) was isolated as a brown amorphous powder. Its molecular formula C27H31NO9 was assigned on the basis of the HRESIMS data. The UV maximum absorption and 1D NMR data suggested that the carbon skeleton of compound 3 is similar to those of compound 1 (Fig. S26†, Table 1, and Fig. 2). A ketone signal (δC 208.3) in the 13C NMR data and the HMBC correlations from H-12 to C-10/C-11 and H-10 to C-1/C-2/C-9a suggested that compound 3 possessed a butanone moiety instead of 2,3-butanediol group in compound 1. Moreover, the tandem mass fragmentation data supported the result of spectroscopic data (Fig. S6 and S30†).
Compounds 1 and 2 had the same aglycone. By contrast, the chiroptical properties (optical rotation and electronic circular dichroism) are dissimilar. Assigning the relative configuration of 1 and 2 was attempted using the ROESY data. However, it was difficult to determine the relative configuration unambiguously as a result of flexible side chains. Several chemical derivatization methods were attempted to establish the configuration, but it was also difficult due to low solubility and/or chemical instability of the compounds. The optical rotation of compound 1 compared with the data in the literature,14 which suggested that compound 1 and neocarazostatin A have the same configuration of the alkyl chain. Nevertheless, the absolute configuration of neocarazostatin A was not determined yet.26,27 Carquinostatin B consists of a carbazole-3,4-quinone core with a side chain, prenyl moiety, and an alkyl chain, which is attached to the same position of compound 1 and neocarazostatin A.13 The biosynthetic gene cluster of carquinostatin A showed a strong resemblance to those of neocarazostatin A.8 The putative biosynthetic gene cluster of compounds 1–3 is also similar to those of carquinostatin B and neocarazostatin A (Fig. S37, S38, and Table S1†). The 18O isotope labeling experiment of a neocarazostatin producing strain suggested the neocarazostatin biosynthetic gene cluster could synthesize the ortho-quinone type intermediate.27 Thus, an optical rotation value of 1 and literature studies hypothesized that the absolute configuration of the alkyl chain of 1 is 10S* and 11R*. To test this hypothesis about the absolute configuration of 1, the experimental electronic circular dichroism (ECD) data of 1 was compared with the calculated data. Compound 1 showed a positive cotton effect at 209, 221, and 337 nm with a negative effect at 255 nm, which were similar to those of the calculated data from the hypothesis (Fig. 4A). Thus, the absolute configuration of 1 was proposed as 10S and 11R.
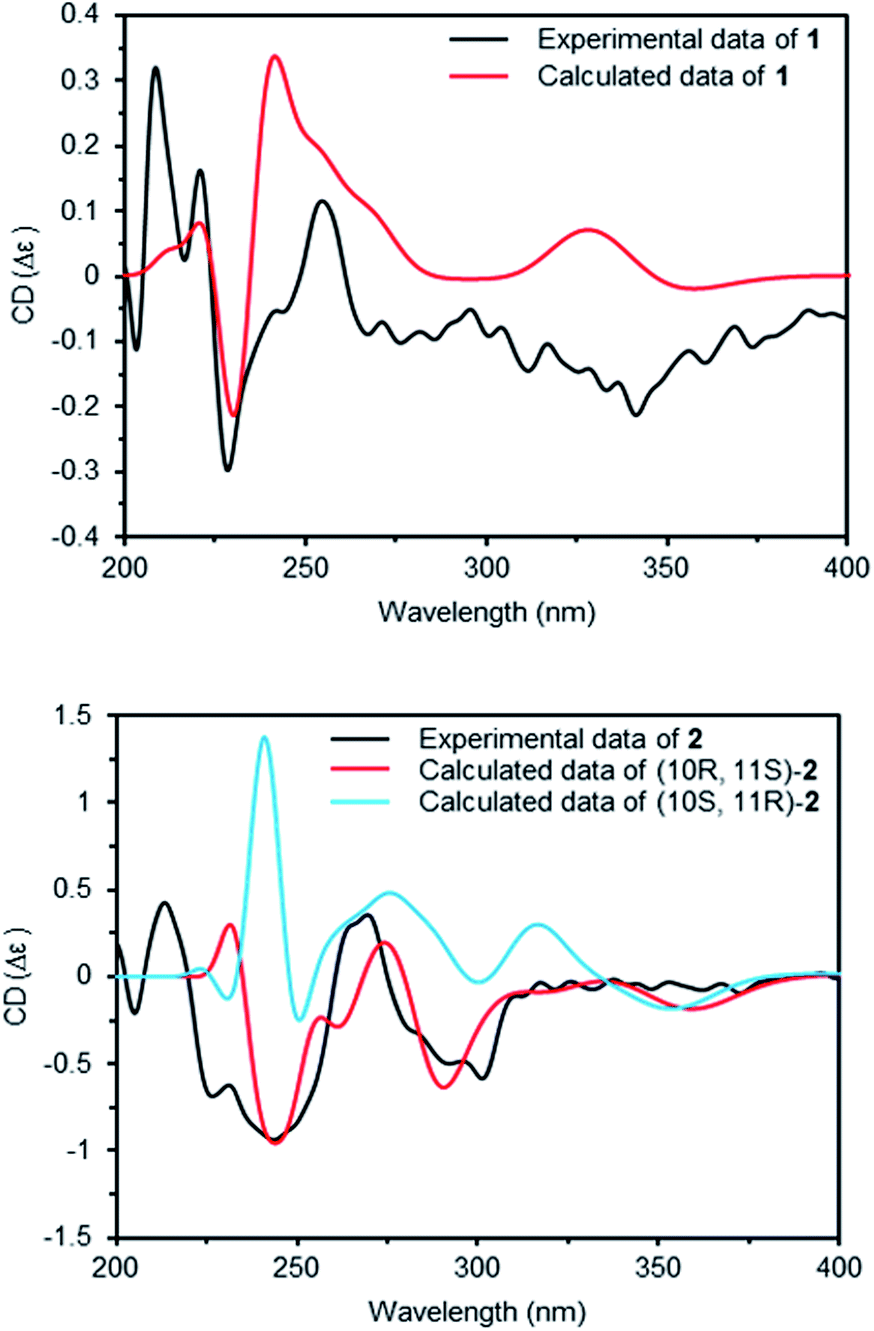 | ||
| Fig. 4 Experimental ECD and calculated ECD spectra of 1 and 2. | ||
The ECD curve and optical rotation of 2 were different to those of 1 despite of the fact that the ROESY signal patterns of 1 and 2 are similar; thus, we hypothesized that the alkyl chain of 2 has an opposite configuration compared to that of 1. To validate the enantiomeric relationships between the aglycone of 1 and 2, the experimental ECD spectrum of 2 was compared with the calculated spectrum of two candidates, (10R, 11S)-2 and (10S, 12R)-2. The result indicated that the calculated spectrum of (10R, 11S)-2 showed a similar curve to the experimental spectrum, which suggested that the absolute configuration of aglycone of 2 as 10R and 11S (Fig. 4B).
Compounds 1–3 were evaluated for inhibition of the indoleamine 2,3-dioxygenase 1 (IDO1) enzyme. Compounds 2 and 3 showed strong inhibition with IC50 values of 9.17 and 8.81 μM, while compound 1 exhibited a moderate activity of 18.38 μM (Fig. S39†). The positive control, menadione, showed IC50 values of 0.37 μM. To obtain the data about the putative binding site of the compounds, molecular docking analysis was conducted by Autodock vina28 using a crystal structure of holo-IDO1 and apo-IDO1 (PDB id: 6AZU and 6AZV).16 The lowest calculated binding affinity was −9.1 kcal mol−1, which was a result of compound 3 binding with apo-IDO1 enzyme (PDB id: 6AZV) (Table S2†). The docking analysis predicted that compound 3 binds to the A-pocket, tryptophan-binding site, and the abandoned heme pocket of apo-IDO1 enzyme.16,29–31 The sugar unit and ring A of compound 3 occupied A-pocket by hydrophobic interactions to the residues Phe-163, Phe-226, Arg-231, and Leu-234 with hydrogen bonds against Ser-235, Gly-236, Lys-238, Asn-240, and Gly-261. The prenyl group of compound 3 took up the abandoned heme pocket through hydrophobic interactions to the residues Ser-263, Phe-291, Leu-384, Phe-387, and Leu-388 (Fig. 5). Compounds 1 and 2 also had similar results in the docking analysis (Fig. S40, S41, and Table S2†).
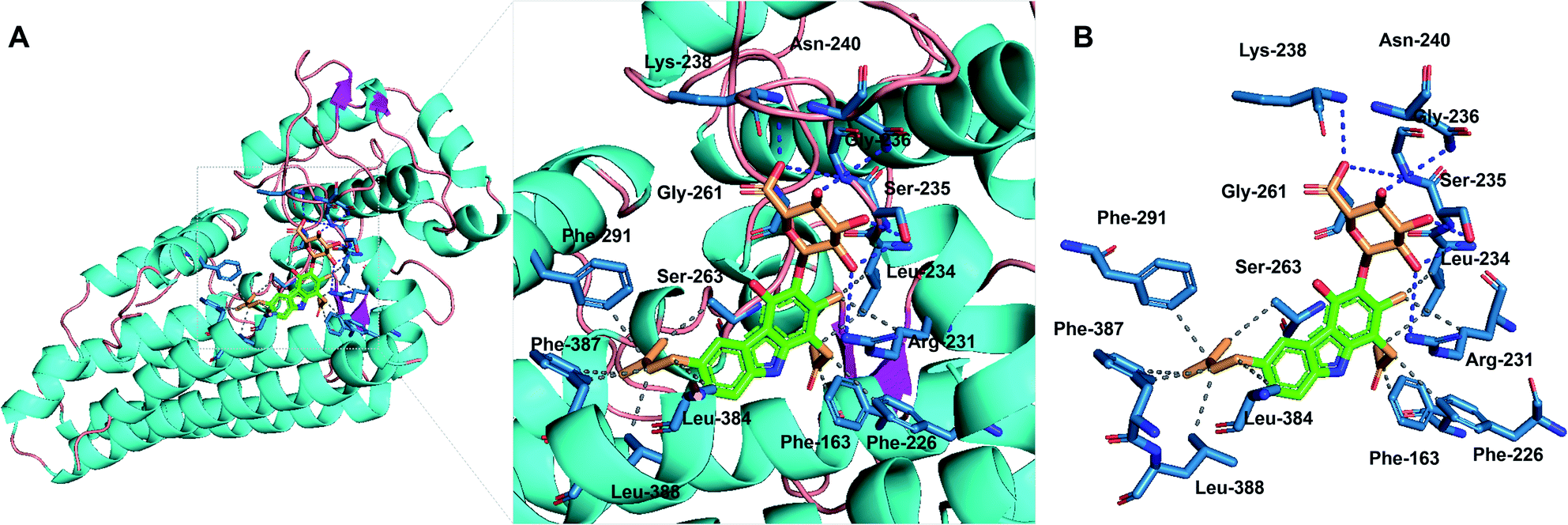 | ||
| Fig. 5 Molecular-docking analysis of compound 3 with the apo-IDO1 enzyme (PDB id: 6AZV). (A) Putative binding mode for compound 3 with apo-IDO1. (B) Protein–ligand interaction profile between compound 3 with IDO1. The gray dashed lines represent hydrophobic interactions; the blue dashed lines represent hydrogen bonds. | ||
Although bacterial tricyclic carbazole is classified into several subgroups according to oxidative status, glycosylated tricyclic carbazole has not been reported yet.7,27 The β-glucuronic acid moiety was reported in a few references, which is similar to the sugar unit of compounds 1–3.32–36 The β-glucuronic acid units, reported in the references, were mostly elucidated as the D-form by optical rotation analysis.33,34 The β-glucuronic acid is glycosylated at several positions on quinone analogue.36 In addition, regioisomers by glycosylation of another sugar unit was also reported.37 Therefore, to the best of our knowledge, compounds 1–3 are the first example of tricyclic carbazole glycoside comprised of dihydroxyl type carbazole and rare sugar unit, β-D-glucuronic acid.
A literature study revealed several modes of IDO1 inhibition18,31 through an allosteric regulator,38 heme-binder,23 and heme-displacer.16 Qinglin Pu et al. compared the activities of the IDO1 inhibitors in clinical trials, which showed that the inhibition by the heme-displacing mode is more potent due to its larger interaction interface with IDO1 enzyme by reason of the absence of the bulky heme moiety.18 Although various compounds of tryptophan or indole analogues were studied as IDO1 inhibitors, the carbazole class has not reported any inhibitory activity against IDO1 yet.23,39 The computational docking data between compounds 1–3 and apo-IDO1 enzyme (PDB id: 6AZV) were in good accordance with the results of IDO1 inhibition assay. Docking analysis proposed that compounds 1–3 interact with both the A-pocket and abandoned heme pocket of the IDO1 apo-enzyme by hydrogen bonds and hydrophobic interactions. Docking analysis using holo-IDO1 enzyme (PDB id: 6AZU) also predicted that compounds 1–3 show better affinity to modified enzyme structure (the heme moiety was discarded) than the original one (Table S2†). Thus, compounds 1–3 were considered to act to inhibit the IDO1 enzyme by displacing the heme.
Experimental
General experimental procedures
The specific rotations were measured on a JASCO P-1020 polarimeter that uses a 100 mm glass microcell. UV spectra were obtained on a NanoDrop 2000 spectrophotometer and UltiMate DAD-3000 connected to a Thermo Scientific Dionex Ultimate 3000 Rapid Separation LC system (UPLC-PDA) using a Waters HSS T3 column (Waters, 2.1 × 150 mm, 2.5 μm). IR spectra were recorded on a Bruker VERTEX 80v FT-IR spectrometer. The NMR spectra were recorded on Bruker AVANCE HD 700 and 800 NMR spectrometers at the Korea Basic Science Institute (KBSI) in Ochang, South Korea. Chemical shifts were referenced to a residual solvent signal (acetone-d6 δH 2.05, δC 206.68 and 29.92; CD3OD δH 3.31, δC 49.15). High-resolution electrospray ionization mass spectrometry (HRESIMS) data were acquired with a Q-TOF mass spectrometer on a SYNAPT G2 (Waters) at KBSI in Ochang, South Korea. The CD spectra were obtained on a JASCO J-1500 circular dichroism spectrophotometer at KBSI in Ochang, South Korea. Liquid chromatography-mass spectrometry (LC-MS) was performed with a Thermo LTQ XL linear ion trap attached to an ESI source that was connected to a Thermo Scientific Dionex Ultimate 3000 Rapid Separation LC system (ESI-LC-MS) using a Waters HSS T3 column (Waters, 2.1 × 150 mm, 2.5 μm). Open column chromatography was performed with a silica gel (Merck, silica gel 60 (0.063–0.200 mm)). Vacuum liquid column chromatography was carried out with an ODS (Cosmosil, 75 μm). Semi preparative C18 (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) columns were used for HPLC on a YL9100 HPLC system equipped with a photodiode array detector (YL9160) that uses HPLC grade solvents (Burdick & Jackson).Microorganism collection and identification
The strain KCB15JA151 was isolated from a soil sample from an agricultural field of garlic on Jeju Island, Korea. A soil sample was spread onto selective medium (24.0 g of Potato Dextrose Broth and 17 g of agar per 1 L of distilled water, pH 5.6) supplemented with chloramphenicol (50 ppm) and kanamycin (100 ppm). Strain KCB15JA151 was isolated from the selective media and transferred onto potato dextrose agar (PDA). Strain KCB15JA151 (GenBank accession no. MW282889) was identified as Streptomyces sp. by 16S rRNA gene sequencing, which show 99.7% similarity to Streptomyces roietensis WES2.Fermentation, extraction, and bioassay-guided isolation
The Streptomyces sp. KCB15JA151 was grown in potato dextrose broth (PDB) for 7 days at 28 °C on a rotary shaker operation at 165 rpm with baffled Erlenmeyer flask. The cultured broth (45 L) was divided into the filtered broth and mycelia by Büchner funnel. The filtered broth was extracted with an equal volume of ethyl acetate three times. The mycelia was extracted with an equal volume of 80% acetone in water followed by evaporation under reduced pressure to remove the organic solvent. The aqueous solution of the acetone extract was extracted three times with an equal volume of ethyl acetate. The combined extracts (14.49 g) from the filtered broth and mycelia were fractionated by silica gel column chromatography (70 i.d. × 320 mm), eluted with a stepwise gradient of CHCl3–MeOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 30:1, 20:1, 10:1, 7:1, 5:1, 3:1, 2:1, 1:1, and 0:100 (v/v)). The bioactive fraction 9 (CHCl3–MeOH, 1:1, 1.74 g) was separated by reversed-phase C18 vacuum liquid column chromatography (50 i.d. × 250 mm) eluted with a stepwise gradient of MeOH in water (20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, and 100%). The active fractions 9–3 (40% MeOH, 104.2 mg) was purified by reversed phase HPLC (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) that used a linear gradient condition (25–50% MeCN – H2O containing 0.025% formic acid, flow rate 3 mL min−1) to yield compound 2 (3.7 mg). The active fraction 9–4 (50% MeOH, 89.0 mg) was subjected to HPLC using a linear gradient condition (35–50% MeCN – H2O containing 0.025% formic acid, flow rate 3 mL min−1) with a semi preparative C18 column (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) to yield compound 1 (3.5 mg). The active fraction 9–5 (60% MeOH, 128.2 mg) was separated by reversed phase HPLC (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) that used a linear gradient condition (40–60% MeCN – H2O containing 0.025% formic acid, flow rate 3 mL min−1) to obtain compound 3 (2.1 mg).
:0, 30:1, 20:1, 10:1, 7:1, 5:1, 3:1, 2:1, 1:1, and 0:100 (v/v)). The bioactive fraction 9 (CHCl3–MeOH, 1:1, 1.74 g) was separated by reversed-phase C18 vacuum liquid column chromatography (50 i.d. × 250 mm) eluted with a stepwise gradient of MeOH in water (20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, and 100%). The active fractions 9–3 (40% MeOH, 104.2 mg) was purified by reversed phase HPLC (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) that used a linear gradient condition (25–50% MeCN – H2O containing 0.025% formic acid, flow rate 3 mL min−1) to yield compound 2 (3.7 mg). The active fraction 9–4 (50% MeOH, 89.0 mg) was subjected to HPLC using a linear gradient condition (35–50% MeCN – H2O containing 0.025% formic acid, flow rate 3 mL min−1) with a semi preparative C18 column (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) to yield compound 1 (3.5 mg). The active fraction 9–5 (60% MeOH, 128.2 mg) was separated by reversed phase HPLC (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) that used a linear gradient condition (40–60% MeCN – H2O containing 0.025% formic acid, flow rate 3 mL min−1) to obtain compound 3 (2.1 mg).
Characterization of compounds 1–3
ε) 226 (4.26), 246 (4.20), 268 (4.11), 300 (3.91), 339 (3.48), 354 (3.44) nm; CD (MeOH) Δε 209 (0.32), 221 (0.16), 228 (−0.30), 255 (0.12); IR (ATR) νmax 3360, 2923, 2854, 1611, 1411, 1062, 610 cm−1; 1H and 13C NMR spectroscopic data, see Table 1; HRESIMS m/z 530.2027 [M − H]− (calcd for C27H32NO10, 530.2026).ε) 227 (4.23), 249 (4.21), 270 (sh, 4.02), 290 (3.90), 326 (3.58), 343 (3.53) nm; CD (MeOH) Δε 205 (−0.21), 227 (−0.69), 243 (−0.94), 269 (0.36), 301 (−0.58); IR (ATR) νmax 3316, 2923, 1611, 1375, 1056, 607 cm−1; 1H and 13C NMR spectroscopic data, see Table 1; HRESIMS m/z 530.2028 [M − H]− (calcd for C27H32NO10, 530.2026).ε) 229 (4.08), 249 (4.15), 269 (sh, 3.88), 290 (3.80), 326 (3.35), 343 (3.36) nm; IR (ATR) νmax 3282, 2924, 1610, 1423, 1283, 1061, 805, 611 cm−1; 1H and 13C NMR spectroscopic data, see Table 1; HRESIMS m/z 536.1898 [M + Na]+ (calcd for C27H31NO9Na, 536.1897).Determination of the absolute configuration of the sugar unit of 1
Derivatization of sugar and LC-MS analysis was done as described previously with minor modifications.21,25 Compound 1 (0.7 mg) was hydrolyzed in 0.1 N H2SO4 (0.8 mL) at 100 °C for 4 h. The hydrolysate was neutralized by addition of 0.1 N KOH (0.8 mL). The neutralized mixture was partitioned by water (3.4 mL) with ethyl acetate (3 × 5 mL). The water layer was divided into two vials and dried under vacuum. D- or L-cysteine methyl ester hydrochloride in pyridine (0.2 mL, 2 mg mL−1) was added to each vials, which was heated at 60 °C for 1 h. O-Tolyl isothiocyanate in pyridine (0.2 mL, 2 mg mL−1) was added to reaction mixture and re-heated at 60 °C for 1 h. The reaction products were analyzed by LC-MS using a linear gradient condition (5–100% MeCN – H2O containing 0.05% formic acid over 25 min, flow rate 0.3 mL min−1) with a Waters HSS T3 column (Waters, 2.1 × 150 mm, 2.5 μm). D- and L-cysteine derivatives of 1 were detected at 9.58 and 9.63 min, respectively. The authentic D-glucuronic acid also prepared by the same method to yield the D- and L-cysteine derivatives, which were detected at 9.58 and 9.63 min.ECD calculation
The conformational search was conducted using avogadro 1.2.0 with the MMFF94 force field.40 The geometry optimization was performed for all conformers using the NWchem 7.0.0 with the B3LYP/6-31g(d) basis set.41 TD-DFT with the B3LYP/6-31g(d) basis set was used to calculate the ECD spectrum of each optimized conformers using Gaussian 09.42 The Boltzmann-weighted spectrum was obtained from each calculated spectrum using SpecDis 1.71.43 For a better comparison between calculated and experimental spectra, the calculated spectrum of (10S, 11R)-1 was adjusted (σ = 0.2 eV; scaled by 1/50 (y-axis); UV-shift = +15 nm), the calculated data of (10R, 11S)-2 and (10S, 11R)-2 were adjusted (σ = 0.2 eV; scaled by 1/35 (y-axis); UV-shift = +30 nm).Indoleamine 2,3-dioxygenase 1 inhibition assay
The IDO1 inhibition assay was done as described previously.38 The human IDO1 clone was provided from Korea Human Gene Bank, Medical Genomics Research Center, KRIBB, Korea. The 200 μL reaction mixture contained 50 mM potassium phosphate buffer (pH 6.5), 20 mM ascorbic acid, 10 μM methylene blue, 200 μM L-Trp, 20 μg mL−1 catalase, 5 μg mL−1 purified recombinant human IDO1 enzyme and 2 μL of the sample dissolved in dimethylsulfoxide (DMSO). The reaction was commenced by the addition of purified recombinant human IDO1 enzyme.38 The reaction mixture was incubated at 37 °C for 60 min. The reaction was stopped by the addition of 40 μL of 30% (w/v) trichloroacetic acid, which was heated at 65 °C for 15 min. An aliquot of supernatant (125 μL), obtained by centrifugation of the mixture, was mixed with the same volume of p-dimethylaminobenzaldehyde (2%, w/v) in acetic acid to produce the colored reaction product. The absorbance of the colored product was measured at 480 nm with a spectrophotometer (SpectraMax 190, Molecular Devices). Menadione (Sigma-Aldrich) was used as a positive control.Molecular docking analysis
Molecular docking analysis was performed as described previously.28 The energy-minimized structure of the compounds was generated using Avogadro 1.2.0.40 The protein structure of the IDO1 enzyme was obtained from the Protein Data Bank (https://www.rcsb.org/, PDB id: 6AZU and 6AZV). Protein Data Bank (PDB) file was modified using mgltools 1.5.6 (https://ccsb.scripps.edu/mgltools/). The docking simulations were carried out using Autodock vina.44 PLIP (https://projects.biotec.tu-dresden.de/plip-web/plip) was used to analyze the protein–ligand interaction patterns of the protein–ligand complex obtained from docking analysis between compounds and the IDO1 enzyme.45 The protein-ligand interactions were visualized using PyMOL.46Whole-genome sequencing analysis
Whole genome sequence of Streptomyces sp. KCB15JA151 was obtained from commercial service of Macrogen, Inc. (Korea). Whole-genome de novo sequencing was performed using PAcBio RSII and Illumina platform. Biosynthetic gene cluster of jejucarbazoles was assigned by antiSMASH 5.0 and reference study.8,27,47Conclusions
In this study, jejucarbazoles A–C (1–3) were isolated from Streptomyces sp. KCB15JA151 by the bioactivity-guided isolation. Their chemical structures were composed of the carbazole backbone and β-D-glucuronic acid, which is the first example of glycosylated tricyclic carbazole. Compounds 1–3 exhibited significant inhibitory against IDO1 enzyme with IC50 values ranging from 8.81 to 18.38 μM. In silico molecular docking analysis proposed that compounds 1–3 bound to the IDO1 enzyme by heme-displacing binding mode. Therefore, it is possible that tricyclic carbazole glycoside might be a promising novel scaffold for inhibitor development against IDO1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (No. NRF-2021M3H9A1030164) and the KRIBB Research Initiative Program funded by the Ministry of Science ICT (MSIT) of the Republic of Korea. We thank the Korea Basic Science Institute, Ochang, Korea, for providing the NMR (700 and 800 Hz), HRESIMS, and CD data.References
- O. Genilloud, Nat. Prod. Rep., 2017, 34, 1203–1232 RSC.
- Q. Zhao, L. Wang and Y. Luo, Eng. Life Sci., 2019, 19, 452–462 CrossRef CAS PubMed.
- A. Bauer and M. Bronstrup, Nat. Prod. Rep., 2014, 31, 35–60 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- M. S. Butler, A. A. Robertson and M. A. Cooper, Nat. Prod. Rep., 2014, 31, 1612–1661 RSC.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- A. W. Schmidt, K. R. Reddy and H. J. Knolker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- M. Kobayashi, T. Tomita, K. Shin-Ya, M. Nishiyama and T. Kuzuyama, Angew. Chem., Int. Ed., 2019, 58, 13349–13353 CrossRef CAS PubMed.
- A. Gluszynska, Eur. J. Med. Chem., 2015, 94, 405–426 CrossRef CAS PubMed.
- K. Sakano, K. Ishimaru and S. Nakamura, J. Antibiot., 1980, 33, 683–689 CrossRef CAS PubMed.
- J. Molette, J. Routier, N. Abla, D. Besson, A. Bombrun, R. Brun, H. Burt, K. Georgi, M. Kaiser, S. Nwaka, M. Muzerelle and A. Scheer, ACS Med. Chem. Lett., 2013, 4, 1037–1041 CrossRef CAS PubMed.
- B. P. Bandgar, L. K. Adsul, H. V. Chavan, S. S. Jalde, S. N. Shringare, R. Shaikh, R. J. Meshram, R. N. Gacche and V. Masand, Bioorg. Med. Chem. Lett., 2012, 22, 5839–5844 CrossRef CAS PubMed.
- K. Shin-ya, T. Kunigami, J. S. Kim, K. Furihata, Y. Hayakawa and H. Seto, Biosci. Biotechnol. Biochem., 1997, 61, 1768–1769 CrossRef CAS PubMed.
- S. Kato, K. Shindo, Y. Kataoka, Y. Yamagishi and J. Mochizuki, J. Antibiot., 1991, 44, 903–907 CrossRef CAS PubMed.
- S. Lob, A. Konigsrainer, H. G. Rammensee, G. Opelz and P. Terness, Nat. Rev. Cancer, 2009, 9, 445–452 CrossRef PubMed.
- M. T. Nelp, P. A. Kates, J. T. Hunt, J. A. Newitt, A. Balog, D. Maley, X. Zhu, L. Abell, A. Allentoff, R. Borzilleri, H. A. Lewis, Z. Lin, S. P. Seitz, C. Yan and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3249–3254 CrossRef CAS PubMed.
- A. Griglio, E. Torre, M. Serafini, A. Bianchi, R. Schmid, G. Coda Zabetta, A. Massarotti, G. Sorba, T. Pirali and S. Fallarini, Bioorg. Med. Chem. Lett., 2018, 28, 651–657 CrossRef CAS PubMed.
- Q. Pu, H. Zhang, L. Guo, M. Cheng, A. C. Doty, H. Ferguson, X. Fradera, C. A. Lesburg, M. A. McGowan, J. R. Miller, P. Geda, X. Song, K. Otte, N. Sciammetta, N. Solban, W. Yu, D. L. Sloman, H. Zhou, A. Lammens, L. Neumann, D. J. Bennett, A. Pasternak and Y. Han, ACS Med. Chem. Lett., 2020, 11, 1548–1554 CrossRef CAS PubMed.
- A. A. Badawy, Int. J. Tryptophan Res., 2017, 10, 1178646917691938 Search PubMed.
- J. Godin-Ethier, L. A. Hanafi, C. A. Piccirillo and R. Lapointe, Clin. Cancer Res., 2011, 17, 6985–6991 CrossRef CAS PubMed.
- S. Son, S. K. Ko, M. Jang, J. K. Lee, M. C. Kwon, D. H. Kang, I. J. Ryoo, J. S. Lee, Y. S. Hong, B. Y. Kim, J. H. Jang and J. S. Ahn, J. Nat. Prod., 2017, 80, 1378–1386 CrossRef CAS PubMed.
- M. Platten, W. Wick and B. J. Van den Eynde, Cancer Res., 2012, 72, 5435–5440 CrossRef CAS PubMed.
- U. F. Rohrig, S. R. Majjigapu, P. Vogel, V. Zoete and O. Michielin, J. Med. Chem., 2015, 58, 9421–9437 CrossRef CAS PubMed.
- C. Uyttenhove, L. Pilotte, I. Theate, V. Stroobant, D. Colau, N. Parmentier, T. Boon and B. J. Van den Eynde, Nat. Med., 2003, 9, 1269–1274 CrossRef CAS PubMed.
- T. Tanaka, T. Nakashima, T. Ueda, K. Tomii and I. Kouno, Chem. Pharm. Bull., 2007, 55, 899–901 CrossRef CAS PubMed.
- R. Czerwonka, K. R. Reddy, E. Baum and H. J. Knolker, Chem. Commun., 2006, 711–713, 10.1039/b515674b.
- Y. Liu, L. Su, Q. Fang, J. Tabudravu, X. Yang, K. Rickaby, L. Trembleau, K. Kyeremeh, Z. Deng, H. Deng and Y. Yu, J. Org. Chem., 2019, 84, 16323–16328 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- C. White, M. A. McGowan, H. Zhou, N. Sciammetta, X. Fradera, J. Lim, E. M. Joshi, C. Andrews, E. B. Nickbarg, P. Cowley, S. Trewick, M. Augustin, K. von Koenig, C. A. Lesburg, K. Otte, I. Knemeyer, H. Woo, W. Yu, M. Cheng, P. Spacciapoli, P. Geda, X. Song, N. Smotrov, P. Curran, M. R. Heo, P. Abeywickrema, J. R. Miller, D. J. Bennett and Y. Han, ACS Med. Chem. Lett., 2020, 11, 550–557 CrossRef CAS PubMed.
- U. F. Rohrig, L. Awad, A. Grosdidier, P. Larrieu, V. Stroobant, D. Colau, V. Cerundolo, A. J. Simpson, P. Vogel, B. J. Van den Eynde, V. Zoete and O. Michielin, J. Med. Chem., 2010, 53, 1172–1189 CrossRef CAS PubMed.
- A. Lewis-Ballester, K. N. Pham, D. Batabyal, S. Karkashon, J. B. Bonanno, T. L. Poulos and S. R. Yeh, Nat. Commun., 2017, 8, 1693 CrossRef PubMed.
- S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591–7697 RSC.
- T. Hida, S. Tsubotani, N. Katayama, H. Okazaki and S. Harada, J. Antibiot., 1985, 38, 1128–1140 CrossRef CAS PubMed.
- S. Takahashi, M. Takeuchi, M. Arai, H. Seto and N. Otake, J. Antibiot., 1983, 36, 226–228 CrossRef CAS PubMed.
- K. A. Shaaban, M. Shaaban, I. Grun-Wollny, A. Maier, H. H. Fiebig and H. Laatsch, J. Nat. Prod., 2007, 70, 1545–1550 CrossRef CAS PubMed.
- G. Carr, E. R. Derbyshire, E. Caldera, C. R. Currie and J. Clardy, J. Nat. Prod., 2012, 75, 1806–1809 CrossRef CAS PubMed.
- X. Wang, A. R. Reynolds, S. I. Elshahawi, K. A. Shaaban, L. V. Ponomareva, M. A. Saunders, I. S. Elgumati, Y. Zhang, G. C. Copley, J. C. Hower, M. Sunkara, A. J. Morris, M. K. Kharel, S. G. Van Lanen, M. A. Prendergast and J. S. Thorson, Org. Lett., 2015, 17, 2796–2799 CrossRef CAS PubMed.
- M. Kwon, S. K. Ko, M. Jang, G. H. Kim, I. J. Ryoo, S. Son, H. W. Ryu, S. R. Oh, W. K. Lee, B. Y. Kim, J. H. Jang and J. S. Ahn, J. Enzyme Inhib. Med. Chem., 2019, 34, 1481–1488 CrossRef CAS PubMed.
- X. X. Wang, S. Y. Sun, Q. Q. Dong, X. X. Wu, W. Tang and Y. Q. Xing, Med. Chem. Comm., 2019, 10, 1740–1754 RSC.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminform., 2012, 4, 17 CrossRef CAS PubMed.
- E. Apra, E. J. Bylaska, W. A. de Jong, N. Govind, K. Kowalski, T. P. Straatsma, M. Valiev, H. J. J. van Dam, Y. Alexeev, J. Anchell, V. Anisimov, F. W. Aquino, R. Atta-Fynn, J. Autschbach, N. P. Bauman, J. C. Becca, D. E. Bernholdt, K. Bhaskaran-Nair, S. Bogatko, P. Borowski, J. Boschen, J. Brabec, A. Bruner, E. Cauet, Y. Chen, G. N. Chuev, C. J. Cramer, J. Daily, M. J. O. Deegan, T. H. Dunning Jr, M. Dupuis, K. G. Dyall, G. I. Fann, S. A. Fischer, A. Fonari, H. Fruchtl, L. Gagliardi, J. Garza, N. Gawande, S. Ghosh, K. Glaesemann, A. W. Gotz, J. Hammond, V. Helms, E. D. Hermes, K. Hirao, S. Hirata, M. Jacquelin, L. Jensen, B. G. Johnson, H. Jonsson, R. A. Kendall, M. Klemm, R. Kobayashi, V. Konkov, S. Krishnamoorthy, M. Krishnan, Z. Lin, R. D. Lins, R. J. Littlefield, A. J. Logsdail, K. Lopata, W. Ma, A. V. Marenich, J. Martin Del Campo, D. Mejia-Rodriguez, J. E. Moore, J. M. Mullin, T. Nakajima, D. R. Nascimento, J. A. Nichols, P. J. Nichols, J. Nieplocha, A. Otero-de-la-Roza, B. Palmer, A. Panyala, T. Pirojsirikul, B. Peng, R. Peverati, J. Pittner, L. Pollack, R. M. Richard, P. Sadayappan, G. C. Schatz, W. A. Shelton, D. W. Silverstein, D. M. A. Smith, T. A. Soares, D. Song, M. Swart, H. L. Taylor, G. S. Thomas, V. Tipparaju, D. G. Truhlar, K. Tsemekhman, T. Van Voorhis, A. Vazquez-Mayagoitia, P. Verma, O. Villa, A. Vishnu, K. D. Vogiatzis, D. Wang, J. H. Weare, M. J. Williamson, T. L. Windus, K. Wolinski, A. T. Wong, Q. Wu, C. Yang, Q. Yu, M. Zacharias, Z. Zhang, Y. Zhao and R. J. Harrison, J. Chem. Phys., 2020, 152, 184102 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Rev. E.01, Gaussian Inc, 2013 Search PubMed.
- T. Bruhn, A. Schaumloffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, W443–W447 CrossRef CAS PubMed.
- W. L. DeLano, J. Vertrees, B. Bell, T. Holder, P. Rotkiewicz, T. Colvin, G. Marques, J. Sampson, M. G. Lerner, and J. Johnson, The PyMOL Molecular Graphics System, Version 2.3.0, Schrödinger, LLC, 2019 Search PubMed.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic data of compounds 1–3; putative biosynthetic gene cluster; docking analysis result of compounds 1 and 2. See DOI: 10.1039/d1ra02895b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |