
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, oligomerization and catalytic studies of a redox-active Ni4-cubane: a detailed mechanistic investigation†
Saroj Kumar Kushvaha‡
a,
Maria Francis‡ b,
Jayasree Kumara,
Ekta Nagb,
Prathap Ravichandrana,
Sudipta Roy*b and
Kartik Chandra Mondal*a
b,
Jayasree Kumara,
Ekta Nagb,
Prathap Ravichandrana,
Sudipta Roy*b and
Kartik Chandra Mondal*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: csdkartik@iitm.ac.in
bDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Tirupati 517507, India. E-mail: roy.sudipta@iisertirupati.ac.in
First published on 30th June 2021
Abstract
A robust tetrameric nickel complex [Ni4((Oal−)2L-Me)4(s)4] (3) (s = solvent) with cubane-like Ni4O4 core topology was isolated as a light greenish-orange crystalline solid in excellent yield. The mechanism of formation of 3 involving the two chloride-containing precursors [Ni4((Oal−)2L-Me)4(s)4]·2MeOH (1) and [Ni4((O−)2L-Me)3((Oal−)(OH)L-Me)Cl] (2) was studied by ESI mass spectrometry and confirmed by the solid state isolation and single-crystal X-ray diffraction. The challenging ligand fields containing mono/di-anionic O2N donating atoms and/or chloride ions stabilized the pentacoordinate Ni(II) ions in 1–2 upon controlling the experimental conditions. Complexes 1–3 have been characterized by NMR, UV-Vis and mass spectrometric analysis. Complex 3 was found to be redox active by cyclic voltammetry (CV) studies. Theoretical calculations were carried out to shed light on the effects of ligand fields on the stability of complexes 1–3. Complex 3 was found to be a potential catalyst for the diastereoselective cyclopropanation of heteroarenes with good to excellent yields. The ESI mass spectrometric analysis revealed the existence of solution dynamics and oligomerization of 3 in solution. Mechanistic investigation of the catalytic cycle revealed that complex 3 and its various oligomers bind to the diazoester employed, followed by dissociative insertion of the respective carbene moieties to the C2–C3 double bond of the involved aromatic heterocycle, leading to the diastereoselective cyclopropanation.
1 Introduction
Nickel-based coordination complexes have been known to be utilized as catalysts in several organic transformations.1 The oxidation state and the coordination environment around the metal center play an essential role in the respective complex's catalytic efficiency. The penta-coordinate Ni(II) complexes2–17 have been reported in the literature as excellent catalysts for alkene oligomerization due to the available vacant binding site for the approaching substrates, followed by catalytic polymerization of the olefin.4–12 In this regard, several neutral organic ligands containing N3 donor sets have been employed to synthesize the respective complexes with penta-coordinate Ni(II)X2 (X = Cl, Br) units.2–4,11–17 There are also a few reports where N3 donor ligands have been further functionalized with S-donor atoms.11 Functionalization of ligands with bulky organic groups around the N-donor favoring the Ni(II) ion's five-coordination is also reported.9 Sterically crowded, neutral N2-donor stabilized dinuclear Ni(II) complexes have been shown to efficiently catalyze ethylene polymerization.5 Neutral N2P-donor bis-(oxazolinyl)phenylphosphonite ligand stabilized Ni-complexes with penta-coordinate Ni(II) ions also have been shown to be an active catalyst for oligomerization of ethylene.6 Neutral ON2 and O2N donor ligands have been shown to stabilize Ni(II) with a five coordination geometry.7 Interestingly, the neutral N3-donor stabilized penta-coordinate Ni(II) fluoroalkyl complex has been shown to produce F3C˙ radicals upon oxidation with ferrocenium cations.14 The neutral bis(benzimidazolin-2-ylidene)pyridine pincer ligands have been shown to stabilize Ni(II) ions with both tetra- and penta-coordinate geometries.15 However, the choice of the ligand is so far limited to the aforementioned classes of neutral ligands. On the other hand, the two chelating pockets (P-I, P-II)-containing di-anionic ligand, [(O−)2L2−] [(OH)2L-Me = (E)-2-((2-hydroxy-3-methoxybenzylidene)amino)-4-methylphenol] with O2N donor set (Scheme 1) was known in the literature to stabilize Ni(II)-ions with only octahedral coordination geometry to date.18–20 This di-anionic ligand was also shown to stabilize Ni(II)-ions with square planar geometry when PPh3 was employed as a co-ligand and reported as an excellent pre-catalyst for the chemodivergent C–H functionalization and cyclopropanation of aromatic heterocycles.21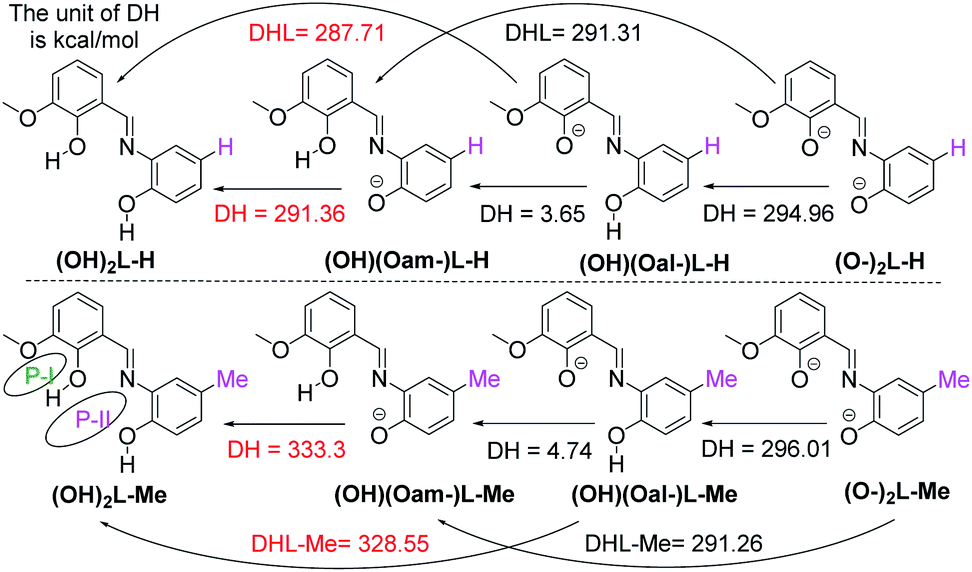 | ||
| Scheme 1 Proton affinity values of (OH)2L-H and (OH)2L-Me ligands. O2 (P-I) and O2N (P-II) pockets. | ||
Herein, we report the syntheses and isolation of three tetranuclear Ni(II)-complexes [Ni4((O−)2L-Me)2((Oal−)(OH)L-Me)2Cl2(MeOH)(MeCN)]·2MeOH (1), [Ni4((O−)2L-Me)3((Oal−)(OH)L-Me)Cl] (2) and [Ni4((Oal−)2L-Me)4(s)4] (3) containing penta-coordinate Ni(II) ions stabilized by partially and fully deprotonated ligands [(Oal−)(OH)L-Me, (O−)2L-Me]. The formation of the final complex 3 is rationalized by the initial formation of the chloride-containing H-bonded tetrameric complex 1 followed by the elimination of HCl to form the intermediate 2 and its successive deprotonation. The ferromagnetically coupled complex 3 was isolated in excellent yield and shown to catalyze the cyclopropanation of aromatic N/O-containing heterocycles in good to excellent yield and diastereoselectivity.
2 Experimental section
2.1. Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of NiCl2·6H2O (10 mmol, 2.38 g) and (H2L-Me) (10 mmol, 2.57 g) was dissolved in 150 mL of MeOH and stirred for 15 min to obtain a clear solution. Et3N (20 mmol, 2.0 g) was added dropwise over 5 min to obtain a clear orange-brown solution. Dark orange-brown precipitate was isolated in 94% yield (2.94 g). Block shaped greenish-orange single crystals were obtained when the reaction was performed in double volume of MeOH (300 mL). However, several attempts failed to produce the single crystals of 3 with good diffraction quality. Elemental analysis (Calcd) of [Ni4((O−)2L-Me)4(s)4] (3): C 54.82 (54.74), H 5.31 (5.29), N 3.73 (3.87), Cl < 0.02 (0.0), which closely matches with [Ni4((O−)2L-Me)4(MeOH)4]·2MeOH. IR (KBr, cm−1): 2965, 2917, 2851, 1601, 1535, 1486, 1460, 1433, 1381, 1323, 1297, 1258, 1231, 1202, 1170, 1143, 1121, 1100, 1020, 971, 858, 822, 729, 668, 594. ESI-MS (m/z): 1253 as [M + H]+, 1275 as [M + Na]+, 627 as [M + 2H]2+; M = [Ni4((O−)2L-Me)4], (O−)2L-Me = C15H13NO3.:1 by volume of MeOH and CH3CN) for 10 min to obtain a golden-yellow color solution. Upon addition of 0.15 mmol (15 mg) of triethylamine as a base into this solution changed the color to brownish. The reaction mixture was further stirred for 10 min and was left for slow evaporation. After six days of slow evaporation yellowish-orange color crystals were formed. A suitable yellowish-orange block-shape crystal was mounted for single crystal X-ray diffraction. Elemental analysis (Cald) of [Ni4((O−)2L-Me)3((Oal−)(OH)L)Cl] (2): C 55.45 (55.76), H 4.25 (4.13), N 4.48 (4.34), Cl 2.41 (2.74). Selected IR bands (KBr; cm−1): 2919(s), 2338(s), 1606(m), 1502(s), 1299(s), 726(m) (see ESI† for detailed characterization of 1–3).
:1 molar mixture of NiCl2·6H2O (10 mmol, 2.38 g) and (H2L-Me) (10 mmol, 2.57 g) was dissolved in 150 mL of MeOH and stirred for 15 min to obtain a clear solution. Et3N (20 mmol, 2.0 g) was added dropwise over 5 min to obtain a clear orange-brown solution. Dark orange-brown precipitate was isolated in 94% yield (2.94 g). Block shaped greenish-orange single crystals were obtained when the reaction was performed in double volume of MeOH (300 mL). However, several attempts failed to produce the single crystals of 3 with good diffraction quality. Elemental analysis (Calcd) of [Ni4((O−)2L-Me)4(s)4] (3): C 54.82 (54.74), H 5.31 (5.29), N 3.73 (3.87), Cl < 0.02 (0.0), which closely matches with [Ni4((O−)2L-Me)4(MeOH)4]·2MeOH. IR (KBr, cm−1): 2965, 2917, 2851, 1601, 1535, 1486, 1460, 1433, 1381, 1323, 1297, 1258, 1231, 1202, 1170, 1143, 1121, 1100, 1020, 971, 858, 822, 729, 668, 594. ESI-MS (m/z): 1253 as [M + H]+, 1275 as [M + Na]+, 627 as [M + 2H]2+; M = [Ni4((O−)2L-Me)4], (O−)2L-Me = C15H13NO3.:1 by volume of MeOH and CH3CN) for 10 min to obtain a golden-yellow color solution. Upon addition of 0.15 mmol (15 mg) of triethylamine as a base into this solution changed the color to brownish. The reaction mixture was further stirred for 10 min and was left for slow evaporation. After six days of slow evaporation yellowish-orange color crystals were formed. A suitable yellowish-orange block-shape crystal was mounted for single crystal X-ray diffraction. Elemental analysis (Cald) of [Ni4((O−)2L-Me)3((Oal−)(OH)L)Cl] (2): C 55.45 (55.76), H 4.25 (4.13), N 4.48 (4.34), Cl 2.41 (2.74). Selected IR bands (KBr; cm−1): 2919(s), 2338(s), 1606(m), 1502(s), 1299(s), 726(m) (see ESI† for detailed characterization of 1–3).2.1.3.1. Synthesis of 2-(tert-butyl)-6-ethyl-2-azabicyclo[3.1.0]hex-3-ene-2,6-dicarboxylate (8a). Following general procedure (GP-4), starting from N-Boc pyrrole, 6a (0.334 g, 2 mmol 1.0 equiv.), ethyl 2-diazoacetate, 7a (0.342 g, 3 mmol, 1.5 equiv.) and complex 3 (L-Me)4Ni4(MeOH) (0.032 g, 1.25 mol%), 8a was isolated as colorless gummy solid (0.197 g, 39%). Rf = 0.55 (Hex/EA = 10
:1). 1H NMR (400 MHz, CDCl3, ppm) δ: 6.53–6.37 (m, 1H, Boc-NCsp2H), 5.33–5.28 (m, 1H), 4.37–4.21 (m, 1H), 4.07–4.04 (m, 2H, –OCH2CH3), 2.73 (s, 1H), 1.44 (s, 9H, –C(CH3)3), 1.19–1.18 (m, 3H, –CH2CH3), 0.87 (s, 1H) (signal broadening due to rotamers);25 13C NMR (101 MHz, CDCl3, ppm) δ: 173.1, 172.8, 151.2, 150.9, 129.7, 129.5, 109.9, 81.6, 60.6, 44.2, 44.0, 32.1, 30.9, 28.2, 28.1, 23.0, 22.8, 14.2 (signal doubling due to rotamers);25 FT-IR νmax (neat)/cm−1: 2980, 2936, 1707, 1629, 1585, 1459, 1397, 1368, 1336, 1287, 1249, 1158, 1065, 1013, 935, 833, 762, 724; HRMS (ESI): m/z calculated for C13H20NO4 [M + H]+: 254.1387; found: 254.1390.
2.1.3.2. Synthesis of di-tert-butyl-2-azabicyclo[3.1.0]hex-3-ene-2,6-dicarboxylate (8b). Following GP-4, starting from N-Boc pyrrole, 6a (836 mg, 5 mmol, 1.0 equiv.), tert-butyl 2-diazoacetate, 7b (0.1066 g, 7.5 mmol, 1.5 equiv.) and complex 3 (L-Me)4Ni4(MeOH) (0.081 g, 3.12 mol%), 8b was isolated as a colorless gummy solid (0.548 g, 39%). Rf = 0.45 (Hex/EA = 10
:1); mp: 70–72 °C; 1H NMR (400 MHz, CDCl3, ppm) δ: 6.50–6.35 (m, 1H, ArH), 5.31–5.27 (m, 1H), 4.32–4.14 (m, 1H), 2.66 (s, 1H), 1.44 (s, 9H, –C(CH3)3), 1.37 (s, 9H, –C(CH3)3), 0.90 (s, 1H) (signal broadening due to rotamers);25 13C NMR (101 MHz, CDCl3, ppm) δ: 172.4, 172.0, 151.1, 150.9, 130.1, 129.4, 110.0, 81.5, 80.7, 40.0, 43.7, 32.1, 30.6, 28.2, 28.1 (signal doubling due to rotamers);25 FT-IR νmax (neat)/cm−1: 2977, 2933, 1704, 1585, 1477, 1457, 1392, 1366, 1341, 1291, 1254, 1133, 1020, 939, 896, 851, 829, 761, 720; HRMS (ESI): m/z calculated for C15H24NO4 [M + H]+: 282.1700; found: 282.1704.
2.1.4.1. 2-(tert-Butyl)1-methyl-6b-methyl-1-phenyl-1a,6b-dihydrocyclopropa[b]indole-1,2(1H)-dicarboxylate (8i, major product). Following the alternative procedure (GP-5), starting from N-Boc indole, 6c (0.434 g, 2 mmol, 1.0 equiv.), methyl-2-diazo-2-phenylacetate, 7c (0.529 g, 3 mmol, 1.5 equiv.) and complex 3 (L-Me)4Ni4(MeOH) (0.032 g, 1.25 mol%), 8i was isolated as a colorless gummy solid (0.394 g, 54%). Rf = 0.25 (Hex/EA = 9
:1); 1H NMR (400 MHz, CDCl3, ppm) δ: 7.43–7.38 (m, 2H), 7.05–6.90 (m, 7H), 4.91 (dd, J = 43.0, 5.3 Hz, 1H), 3.73 (s, 1H), 3.66 (d, J = 10.3 Hz, 3H), 1.65–1.53 (m, 9H); 13C NMR (101 MHz, CDCl3, ppm) δ: 173.65, 151.64, 142.42, 141.34, 132.34, 132.01, 30.30 128.39, 127.87, 127.59, 125.57, 125.11, 122.37, 114.62, 81.86, 52.79, 50.57, 35.50, 28.45; HRMS (ESI): m/z calculated for C22H23NNaO4 [M + Na]+: 388.1519; found: 388.1520; FT-IR νmax (neat)/cm−1: 2981, 1712, 1478, 1392, 1354, 1309, 1242, 1195, 1153, 1084, 1049, 729 (see ESI† for details).
2.2. Computational details
The wavefunction for performing Atoms in Molecules (AIM)26 analysis was generated at M06-2X/def2-SVPP to study the topology and structural features of the ligands and the complexes. AIMALL software package27 (Version 10.05.04, Professional) was used to generate the electron density and Laplacian of electron density from molecular wavefunction data (see ESI† for details).
2.3. HRESI-MS measurement
HRESI-MS measurements were conducted at a capillary temperature of 225 °C. Aliquots of the solution were injected into the device at 2.00 μL min−1. The mass spectrometer used for the measurements was an Agilent Technologies 6545 Q-TOF LC/MS and the data were collected in positive ion modes. The spectrometer was previously calibrated with the standard tune mix to give a precision of ca. 2.0 ppm within the region of 100–3000 m/z. The capillary voltage was 2.5 kV, the tube lens voltage was 1.0 kV, and the skimmer voltage was 65 V. The GC vials were evacuated using a needle which was connected to the vacuum line. The vials were purged with argon. This process was repeated three times to make it air free. MeOH (1.4 mL) was injected inside the GC vial and reaction solution (0.2 mL) was also injected after wards. Separately the needle/syringe was made oxygen free (before use) by purging argon into it. The methanol was boiled under argon atmosphere for 2 h in Schlenk flask to make it O2 free. Freshly distilled toluene (boiled with Na/K alloy in the presence of benzophenone under argon flow) and DCM (boiled with CaCl2 under argon) were utilized for reaction with studying the binding of diazoester (7b) in DCM and toluene. Catalyst 3 (0.1 mmol) which was isolated as orange precipitate from MeOH was reacted with 7b (0.2 mmol) in toluene and/or DCM (10 mL) at room temperature. The orange brown solution was sampled over different time intervals to study binding with the nickel centers of the catalyst.3 Results and discussion
The ligand, (OH)2L-Me [(E)-2-((2-hydroxy-3-methoxybenzylidene)amino)-4-methylphenol] was previously utilized as a deprotonated di-anionic ligand for the syntheses of different coordination metal clusters.19,20 To understand the electronic effect of the para-substitution at the aminophenol part of the ligand on the successive deprotonation and thereby on the overall complexation method, we have calculated (at M06-2X/def2-SVPP level of theory) the first and second proton affinity values of the di-anionic [(O−)2L-Me] and mono-anionic [(O−)(OH)L-Me] ligands which are in the range of 290–334 kcal mol−1. Significantly, the phenolic Oaminophenol moiety's proton affinity value was found to be nearly 5 kcal mol−1 higher than that of the Ovanillin moiety of the ligand (Scheme 1). The electron density distributions and QTAIM analysis of the neutral, mono- and di-anionic (OH)2L-Me ligands are given in the ESI.† Theoretical calculations suggest that the presence of a methyl group (instead of an H-atom in (OH)2L-H) at the para-position of the aromatic ring (w.r.t phenolic O-atom) led to the significant increase of the second proton affinity value by 38–43 kcal mol−1 (Scheme 1).Utilizing this crucial difference in proton affinity values,19 three novel tetranuclear Ni(II) complexes (1–3) with five/six-coordinate Ni(II) centers were synthesized, isolated and characterized.
3.1. Synthetic procedures
A 1:1:2 molar mixture of NiCl2·6H2O, (OH)2L-Me and Et3N reacted in methanol at room temperature to obtain a dark orange-brown solution. The ESI mass spectrometric analysis of the reaction solution after 10 min showed the formation of [Ni4((O−)2L-Me)3((Oal−)(OH)L-Me)Cl] (2) and [Ni4((Oal−)2L-Me)4(s)4] (3) [s = solvent = H2O/MeOH] (Scheme 2). The later became the major product with the progress of time. Finally, this reaction afforded the crystalline greenish-orange crystalline solid of complex 3 in 94% yield, which was recrystallized from DMF as 3·2DMF (Fig. 4, bottom; the structure of 3 is shown only for atom connectivity). Several attempts to grow good quality single crystals from various solvents and a mixture of solvents suitable for X-ray diffraction were failed. The single crystals of 3 were found to be very weakly diffracting, limiting the high-resolution structure determination (Fig. 4, bottom; see ESI†). It is important to note that all efforts to produce 1 and 2 exclusively remained unsuccessful even though several reactions were performed by varying stoichiometry of the base used, cooling the reaction solution, and changing the base from Et3N to NaOMe. In all the cases, 3 was found to be the final product indicating its high thermodynamic stability. Complex 3 was characterized by ESI mass spectrometry as [3 + Na]+ and [3 + H]+ (Table 1). The bulk purity of 3 was also confirmed by elemental (C, H, N, Cl) analysis. Complex [Ni4((O−)2L-Me)3((Oal−)(OH)L-Me)Cl] (2) could be isolated en route to the structural characterization of 3 only when the similar reaction was performed in a much lower concentration of the metal ions and the ligands in a mixture of solvents (MeOH:MeCN = 3:1) at room temperature.
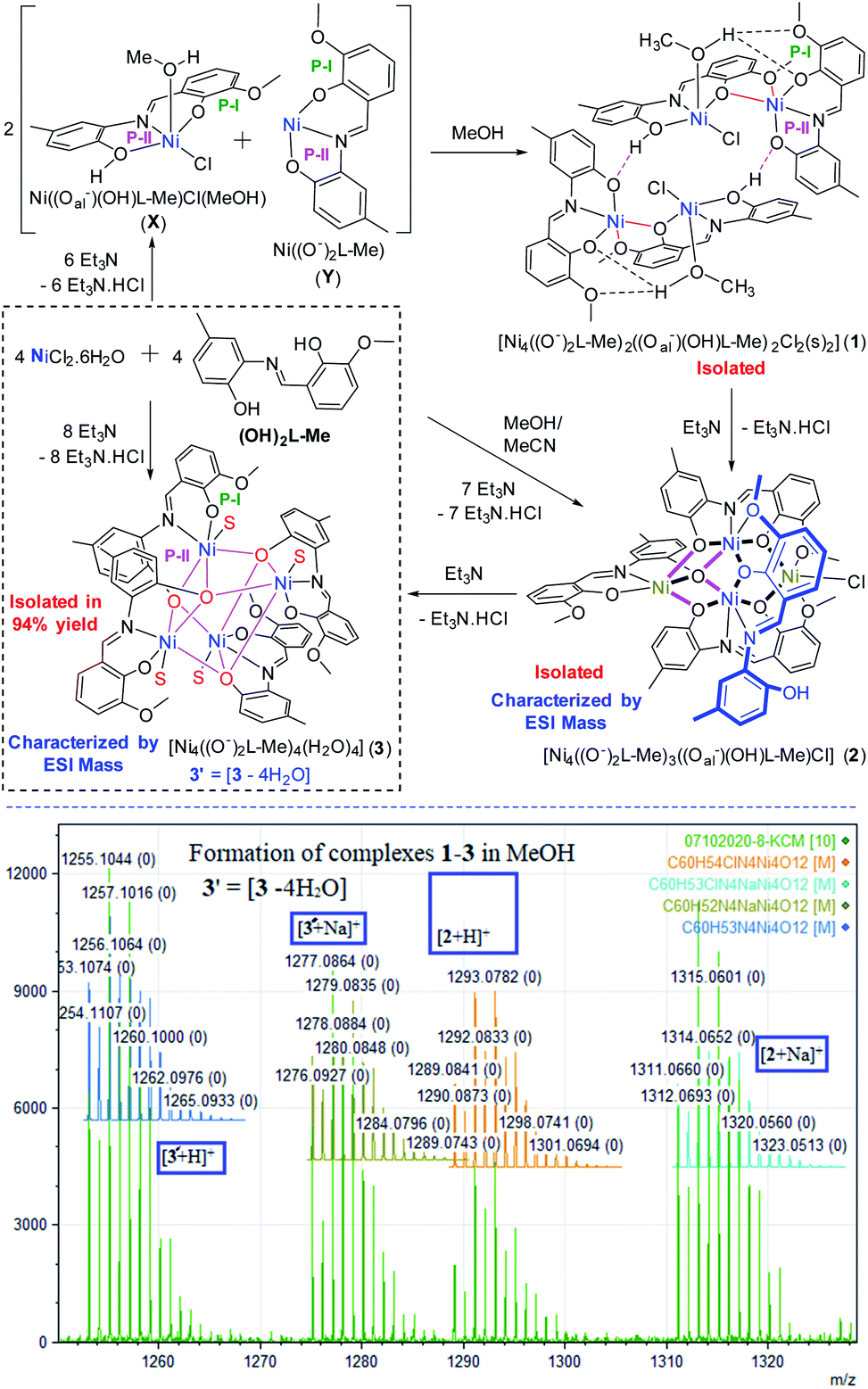 | ||
| Scheme 2 Syntheses of complexes 1–3 in MeOH rationalizing the mechanism of their formation (top). ESI mass spectrometric analysis of reaction solution (bottom). S = solvent (MeOH/H2O). | ||
| Complexes | Compositions of cation | m/z | z |
|---|---|---|---|
| a For general conditions of mass spectrometric analysis see Experimental section and ESI. | |||
| [2 + H]+ | [Ni4(L-Me)3(HL-Me)Cl + H]+ | 1289.08 | 1 |
| [2 + Na]+ = [3′(HCl)+Na]+ | [Ni4(L-Me)3(HL-Me)Cl + Na]+ | 1311.07 | 1 |
| [9 + Na]+ | [Ni3(L-Me)3(KCl) + H]+ | 1014.02 | 1 |
| [5′′+2H]+ | [Ni3(L-Me)3(CHCO2tBu)(N2CHCO2tBu) + 2H]+ | 1197.23 | 1 |
| [3′ + H]+ | [Ni4(L-Me)4 + H]+ | 1253.11 | 1 |
| [3′ + Na]+ | [Ni4(L-Me)4 + Na]+ | 1275.09 | 1 |
| [3′ +NaCl + Na]+ | [Ni4(L-Me)4(NaCl)+ Na]+ | 1333.05 | 1 |
| [3′ + 2H]2+ | [Ni4(L-Me)4 + 2H]2+ | 627.05 | 2 |
| [4]+ | [Ni4(L-Me)4(CHCO2tBu)(N2CHCO2tBu)+ 2H]+ | 1510.26 | 1 |
| [5 + H]+ | [Ni5(L-Me)5 + H]+ | 1566.13 | 1 |
| [5 + N2]+ | [Ni5(L-Me)5 + N2]+ | 1593.13 | 1 |
| [5′ + 2H]+ | [Ni5(L-Me)5(CHCO2tBu)(N2CHCO2tBu)+ 2H]+ | 1823.28 | 1 |
| [5 + 2H]2+ | [Ni5(L-Me)5 + 2H]+ | 783.57 | 2 |
| [5′′′ + H]+ | [Ni6(L-Me)6 + H]+ | 1879.15 | 1 |
| [5′′′ + 2H]+ | [Ni6(L-Me)6 + 2H]+ | 940.08 | 2 |
| [5′′′ + H]+ | [Ni6(L-Me)6NaCl(CH3CN)Na–H]+ | 1879.15 | 1 |
| [10 + Na]+ | [Ni8(L-Me)8 + Na]+ | 2527.19 | 1 |
The bulk purity of 2 was studied by elemental (C, H, N) analysis including halogen (Cl: 2.50%) analysis. The formation of complex 2 was confirmed by single-crystal X-ray diffraction and ESI mass spectrometric analysis of the reaction solution as the mono-cations [2 − Cl]+ and [2 + Na]+ (Fig. 1). X-ray single-crystal analysis revealed that 2 possesses a near defect-dicubane Ni–O core topology (Fig. 2, top).
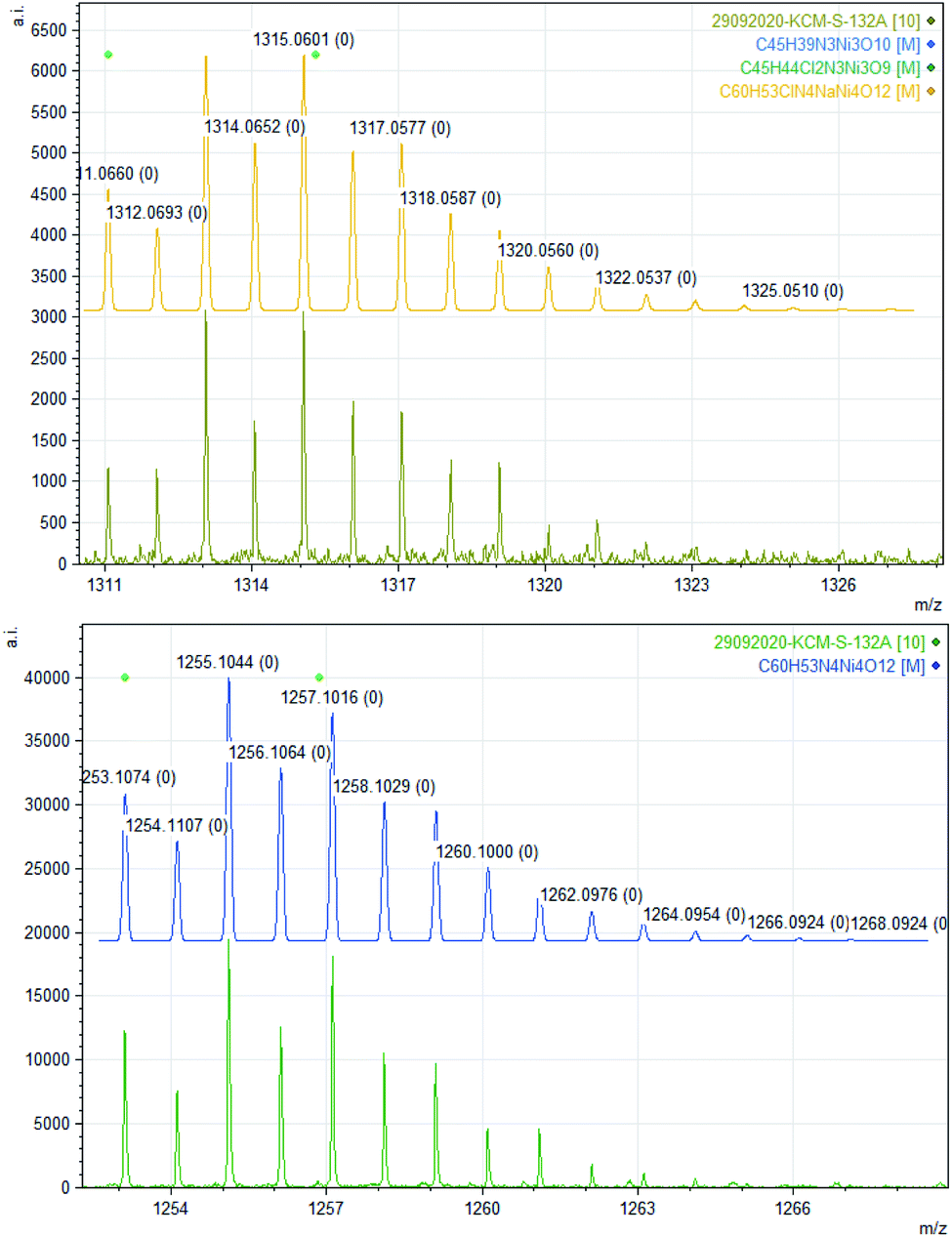 | ||
| Fig. 1 Experimental and calculated ESI mass spectra of de-halogenated complex 2 [Ni4((O−)2L-Me)3((Oal−)(OH)L-Me)Cl] as [(2 + Na)+] (top) and 3′ [Ni4((O−)2L-Me)4] as [3′ + Na]+ (bottom). | ||
Another controlled experimental condition with 1:1:1 molar ratio of NiCl2·6H2O, (OH)2L-Me and Et3N in methanol afforded the complex [Ni4((O−)2L-Me)2((Oal−)(OH)L-Me)2Cl2(MeOH) (MeCN)]·2MeOH (1·2MeOH). The absolute structure of 1 was determined by single-crystal X-ray diffraction as an H-bonded dimer of the di-nuclear units [Ni2((O−)2L-Me)((Oal−)(OH)L-Me)Cl(s)] (Scheme 2; s = MeOH/MeCN). To have an insight into the reaction progress, we have performed the time-dependent ESI mass spectrometric analysis of the two different reaction solutions containing 1:1:1 and 1:1:2 molar ratios of NiCl2·6H2O, (OH)2L-Me, Et3N in methanol which revealed the formation of 1–3 after 10 min of initiation of reaction in both the cases. However, irrespective of the various molar ratios taken, complex 3 was always isolated as the final product in excellent yield and therefore concluded 3 is the thermodynamically most stable product among 1–3. The highest yield of the complex 3 was obtained when the reaction was performed in 1:1:2 molar ratio. The complex 3 was isolated as crystalline yellow-orange solid, washed with cold methanol and dried in open air. This dry crystalline solid of 3 was utilized for further catalytic studies.
The formation of hydrogen chloride (Cl−) free complex, 3′ could be rationalized by eliminating one equiv. of HCl from the unsymmetrical complex 2 in the presence of Et3N. The formation of 2 was further rationalized by the isolation of 1 under controlled experimental conditions upon elimination of one equiv. of HCl. The tetrameric complex 1 possesses two acidic H-atoms in between the phenolic O-atoms and the Cl-atoms bonded to Ni(II) centers, which undergoes the loss of one HCl molecule in the presence of Et3N in solution leading to the formation of 2. Presumably, the formation of 2 also involved a rearrangement with the migration of one of the Ni((O−)2L-Me) sub-unit of 1 (see ESI† for a plausible mechanism).
3.2. Crystallography
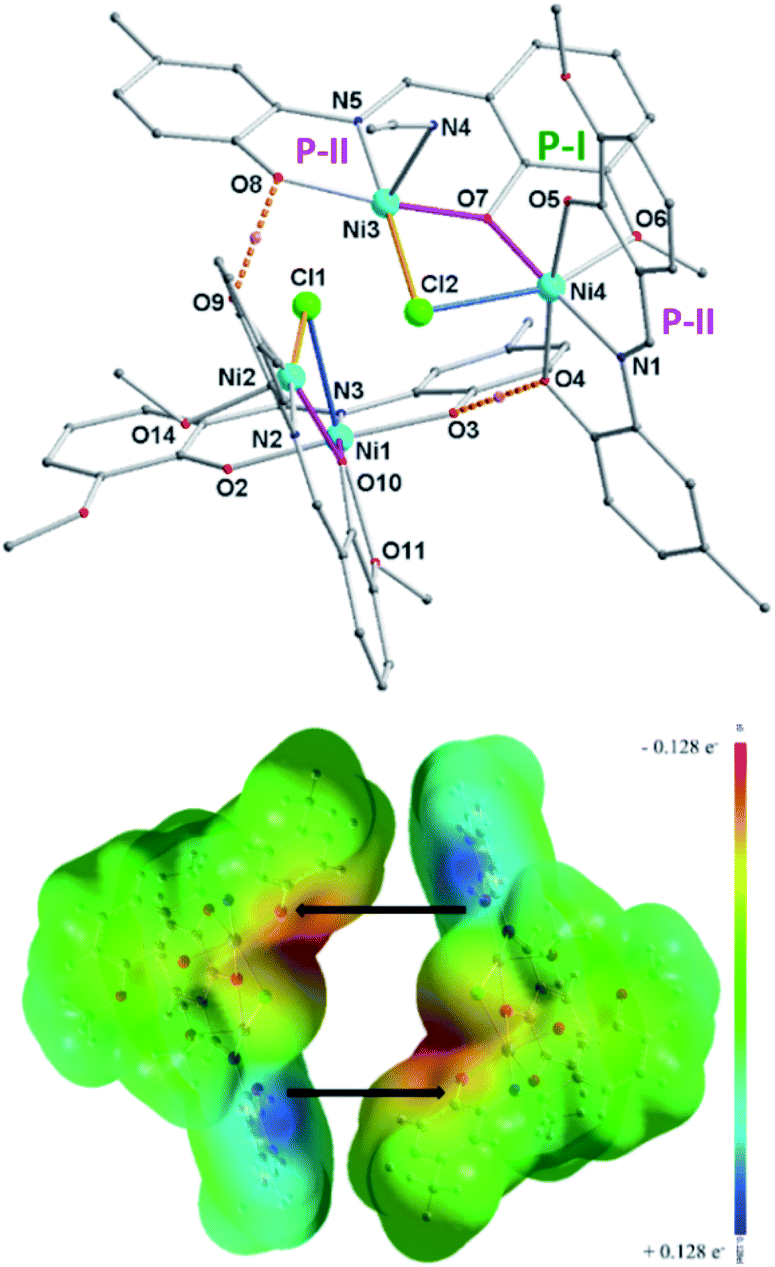 | ||
| Fig. 2 (Top) Molecular structure of H-bonded tetranuclear complex (1·2MeOH) [Ni4((O−)2L-Me)2((Oal−)(OH)L-Me)2Cl2]·2MeOH. H-atoms and MeOH were omitted for clarity except the two between O8 and O9, O3 and O4. H-bonding was shown with dotted lines. Blue colored Ni–Cl bond lengths are greater than 2.50 Å. Selected bond lengths (Å) and angles (°): Ni1–Cl1 2.5267(16), Ni2–Cl1 2.4088(16), Ni3–Cl2 2.4035(18), Ni4–Cl2 2.5416 (18); Ni2–Cl1–Ni1 82.05(6), Ni3–Cl2–Ni4 81.50(5), Ni3–O7–Ni4 108.66(15), Ni2–O10–Ni1 109.33(15). (Bottom) MEP plots (computed at iso-density surface value of 0.0004 a.u) of two dimeric units [Ni2((O−)2L-Me)((Oal−)(OH)L-Me)Cl(MeOH/MeCN)] showing the H-bond positions. | ||
The ligand containing N5-atom is bonded to two Ni(II)-ions (Ni3, Ni4) in two pockets (P-II and P-I) connected via O7-phenolate and Cl2-chloride bridges. The Ni3-center is further coordinated by a solvent molecule (MeCN/MeOH) to adopt a five-coordination number, while, the octahedral Ni4-center is chelated by another ligand via P-II, leaving P-I free. Similarly, another dimeric unit of 1 contains two Ni(II)-ions (Ni1, Ni2). Ni3–Cl2/Ni2–Cl1 and Ni4–Cl2/Ni1–Cl1 bond distances are 2.4035(18)/2.4088(16) and 2.5416(18)/2.5267(16) Å, respectively. Two sub-dimer units of 1 are firmly held together by hydrogen bonding (O8⋯H⋯O9/O4⋯H⋯O3 2.41 Å) and weak Ni3⋯Cl1/Ni2⋯Cl2 interaction (2.7037(18)/2.609(2) Å) (Fig. 2, bond with blue color). Additionally, one lattice methanol molecule is H-bonded to one P-I. The self-assembly process leading to the formation of 1 is favoured by H-bonding and Ni⋯Cl weak interactions. The driving force for the formation of 1 could be explained by the favourable Ni–O bonds (−61 kcal mol; a combination of X and Y subunits of 1; Fig. 3) with additional two H-bonds (Fig. 2, bottom). The base (Et3N) mediated elimination of HCl might have dramatically changed the structure of 1 into 2 in solution.
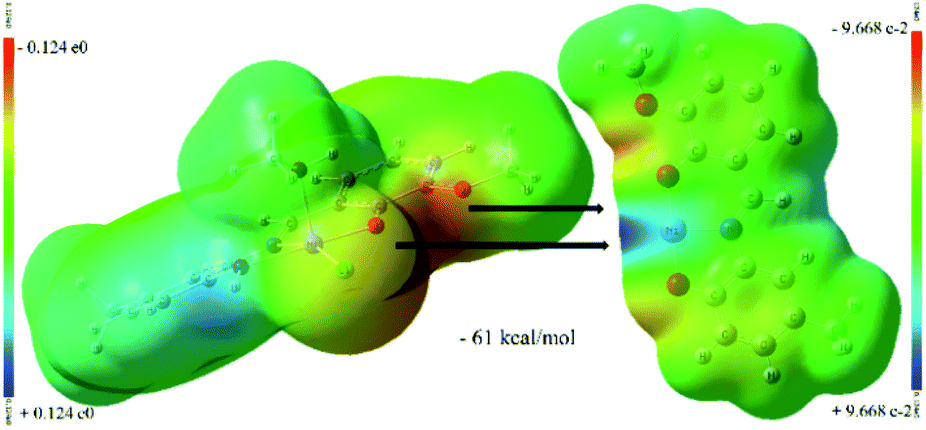 | ||
| Fig. 3 Hypothetical MEP plots (computed at iso-density surface value of 0.0004 a.u) of X and Y fragments (Scheme 1) of [Ni4((O−)2L-Me)2((Oal−)(OH)L-Me)2Cl2]·MeOH (1). | ||
Complex 2 crystallizes in the monoclinic P21/c space group. The unsymmetrical complex 2 contains four Ni(II) centres with defect dicubane type Ni–O core topology (Fig. 4, top). Two octahedral Ni(II) ions occupy the body positions and the other two penta-coordinate Ni(II) ions are at the wing positions. The formation of the monomeric three coordinate intermediate species Y [((O−)2L-Me)Ni] was evident from the ESI mass spectrometric analysis.21b The assembly of two ((O−)2L-Me)Ni units occurs in cis-fashion (ligand containing O6, O7, Ni3 and another ligand with O4, O8, Ni2; Fig. 4, top). The third ((O−)2L-Me)Ni unit (with O12, O5, Ni1) resides in between those two units. The mono-anionic ligand (Oal−)(OH)L-Me bridges between Ni2 and Ni3 centers utilizing both P-I and P-II. The OH-group (O3) is H-bonded (O3⋯O8 2.99 Å) with another bridging phenolate anion (O8). The Ni–Cl unit (Ni4) is chelated in between two P-I pockets at the right-wing position (Fig. 4).
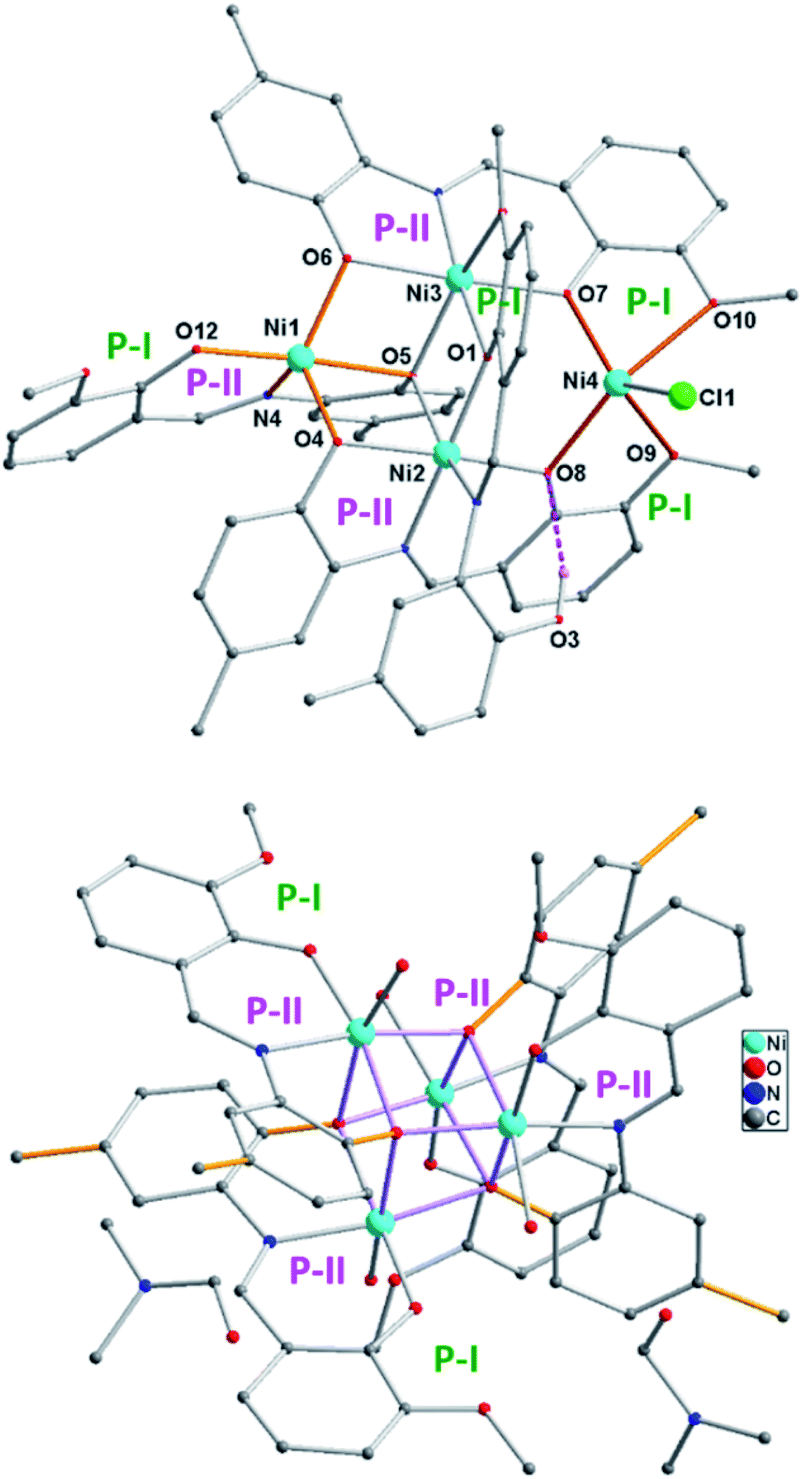 | ||
| Fig. 4 Molecular structures of 2 [Ni4((O−)2L-Me)3((Oal−)(OH)L-Me)Cl] (top) and the de-halogenated complex 3·2DMF [Ni4((Oal−)2L-Me)4(H2O)4]·2DMF (bottom) (explicitly shown for the atom connectivity) [2–HCl = 3′]. Penta-coordinate Ni(II) centers (Ni1 and Ni4) are at wing positions. H-atoms were omitted for clarity except between O3 and O8. H-bonding was shown with dotted lines. Selected bond lengths (Å) and angles (°): Ni1–O12 1.955(5), Ni1–O6 1.987(4), Ni1–O4 1.989(5), Ni1–N4 2.035(6), Ni1–O5 2.252(5), Ni4–O7 1.982(5), Ni4–O8 2.025(4), Ni4–Cl1 2.188(2), Ni4–O9 2.220(5), Ni4–O10 2.388(5). | ||
The de-hydrohalogenation of 2 can easily lead to the formation of 3. We assume that the Et3N mediated elimination of HCl from 2 must have led to the formation of 3 (Fig. 4, bottom) with major rearrangement of four Ni((O−)2L-Me) units (3′). Coordination of solvents at Ni(II)-centers might have led to the formation of 3. The Ni4O4 of 3 possesses a distorted cubane core topology (Fig. 4, bottom). Four μ3-Oaminophenol atoms of four doubly deprotonated (O−)2L-Me ligands bridge (with η0:η1:η1:η3:μ3 bridging mode) among four Ni(II) ions. Each Ni(II) ion is coordinated by a water ligand to adopt octahedral coordination geometry. In contrast, the (O−)2L dianion of previously reported [Ni4((O−)2L)4(MeOH)2] cubane19 displayed two different bridging modes η0:η1:η1:η3:μ3 and η1:η3:η1:η1:μ3. Theoretical calculations showed that the negative NPA charge (−0.888/−0.881) on Oaminophenol atom of (O−)2L-Me/(O−)2L-H is slightly higher than that of Oaminophenol (−0.855/−0.855) which rationalizes the preference for only one bridging mode of (O−)2L-Me in 3 (see ESI†).
The magnetic susceptibility measurements of 3 show that the Ni(II) ions are ferromagnetically coupled below 100 K to give rise to the ground state spin, S = 4 (see ESI†).18,19 Thermogravimetric analysis (TGA) of 3 suggests that loss of methanol ligands of 3 to produce 3′ which is stable till 350 °C.
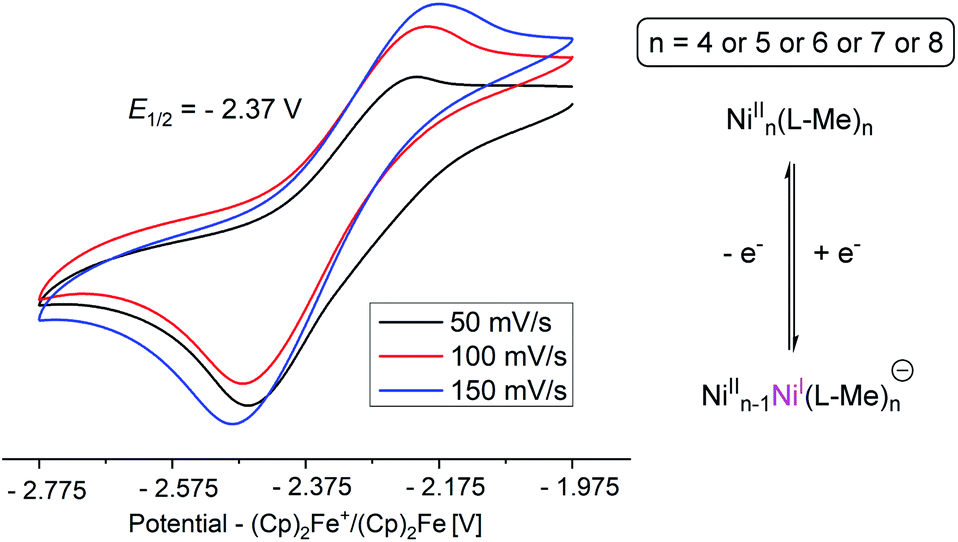 | ||
| Fig. 5 Cyclic voltammograms of complex 3 in THF containing 0.1 M [n-Bu4N]ClO4 as the electrolyte (CE: Pt, WE: GC, RE: Ag). | ||
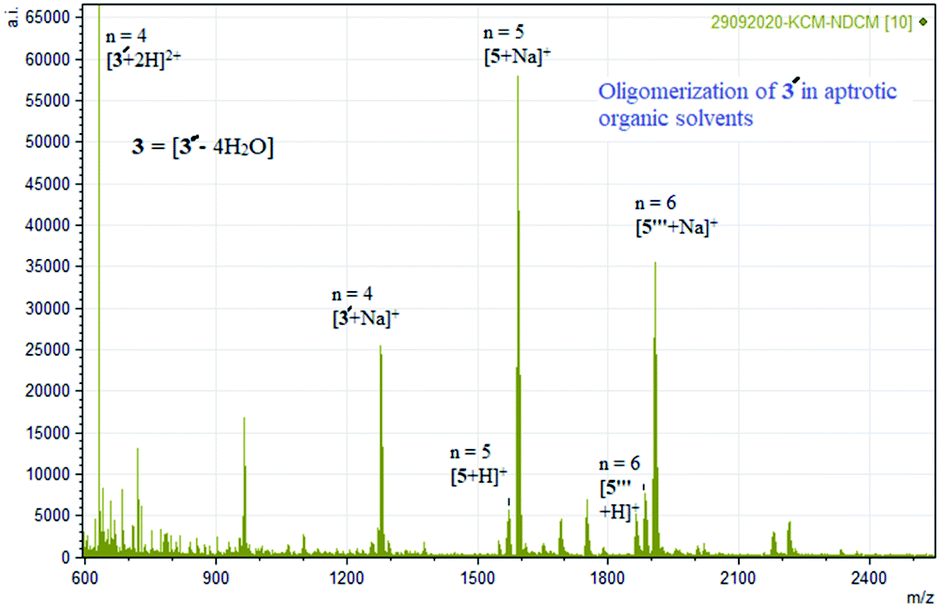 | ||
| Fig. 6 Experimental mass spectrum of 3 representing oligomerization in aprotic solvent [Ni4((O−)2L-Me)4] = 3′ = [3 – 4H2O] (Table 1). | ||
The formation of higher oligomers is entropically unfavourable. The polarity of the solvents used was found to play an important role in displaying the oligomerization phenomenon. The temperature-dependent 1H NMR of 3 studies show resonances from +40 to −40 ppm. The 1H NMR resonances of 3 are broad and weak in intensities which increase on warming up the CDCl3 solution (non-coordinating polar solvent) and cooling down the DMSO-D6 solution (coordinating polar solvent), respectively (see ESI†).
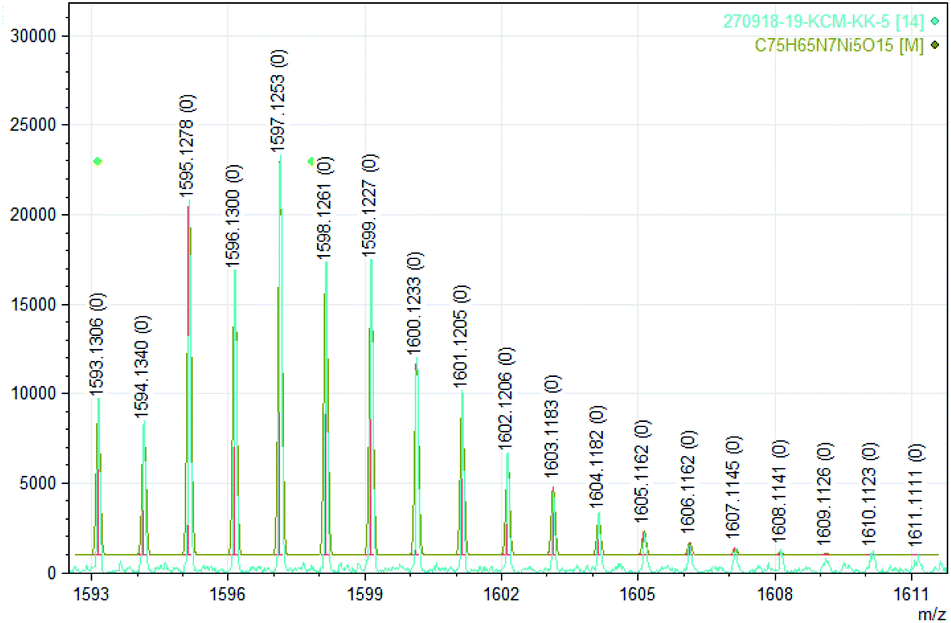 | ||
| Fig. 7 Experimental (bottom) and calculated (top) ESI mass of dinitrogen bonded tetranuclear nickel complex [Ni5((Oal−)2L-Me)5 + N2]+ (5 + N2)+ (see Table 1). | ||
We initiated the catalytic studies of 3 (see Table S29† for complete optimization table) by choosing N-Boc pyrrole 6a as the substrate and ethyl diazoacetate 7a as the carbene source in DCM at room temperature in presence of 0.25 mol% of the complex 3 (1.25 mol% per nickel). To our delight, after 24 h, we could isolate the expected cyclopropane carboxylate derivative 8a in 28% yield as the single diastereomer (Table S29,† entry 1). Increasing the catalyst loading to 1.25 mol% under similar reaction condition increased the product yield to 39% (with 76% conversion) without any loss of diastereoselectivity (Table S29,† entry 2).
However, further increase in the catalyst loading up to 2.5 mol% significantly reduced the product yield to 25%. Increasing the reaction temperature to 100 °C did not improve the reaction yield. In this context, it is noteworthy to mention that the previously reported Ni-catalyst21b from our group could not yield any desired products under similar reaction conditions showing a relatively better catalytic activity of the present system. When we employed other multi-centered hexa-coordinate nickel complexes developed in our group, e.g., Ni4(L-H)4(MeOH)2 and Ni2Dy2(L-Me)4(NO3)2(DMF)2 (Fig. S31†) as catalysts,19,20 in the former case, the corresponding cyclopropanated product was obtained in low yield (16%); whereas in the latter case no product formation was observed and the starting materials was recovered in more than 90% yields. When a similar reaction was carried out in DMF, no product formation was seen (Table S29,† entry 6). When a similar reaction was performed in the absence of complex 3, no conversion of the substrate (6a) to product (8a) could be observed, confirming the catalytic activity of complex 3 (Table S29,† entry 8).
After optimizing the reaction conditions, we moved towards studying the substrate scope of the present catalytic system by using various substituted aromatic heterocycles (6) and diazoesters (7) in the presence of 1.25 mol% of complex 3 as catalyst (Scheme 3).
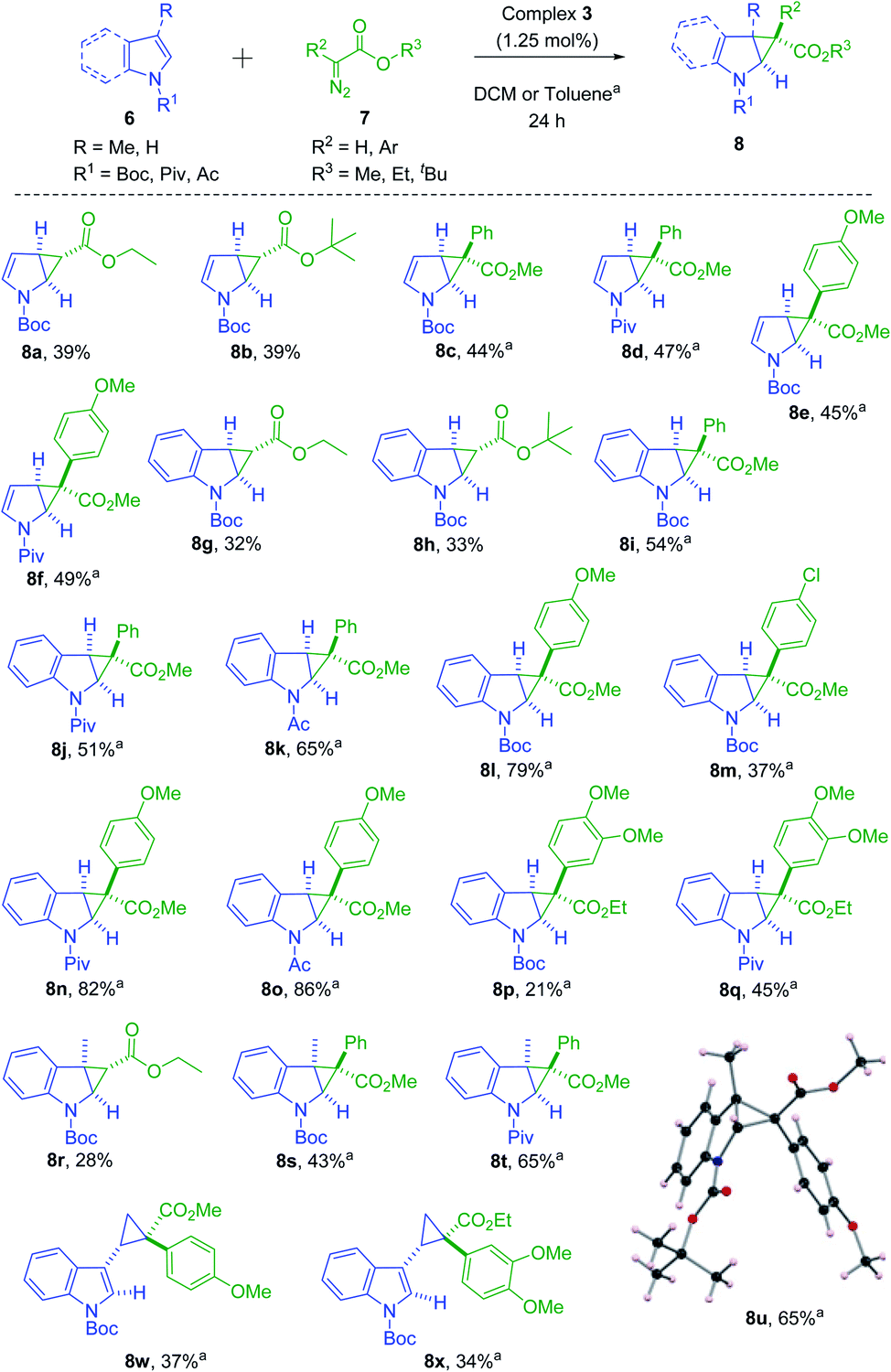 | ||
| Scheme 3 Substrate scope for cyclopropanation of aromatic heterocycles catalyzed by complex 3. aThe reported yields are for reactions carried out at 100 °C using an oil bath. | ||
When N-Boc indole (6c) was employed as the aromatic heterocycle in presence of ethyl 2-diazoacetate (7a) under similar reaction conditions, the corresponding exo-cyclopropanated product 8g was obtained in 32% yield. In presence of tert-butyl diazoacetate (7b) both N-Boc pyrrole (6a) and N-Boc indole (6c) afforded the corresponding cyclopropanated products 8b and 8h in 39% and 33% isolated yields, respectively. When a more sterically hindered substrate, 3-methyl N-Boc indole (6f) was employed, under similar reaction conditions the diazoesters 7a and 7b yielded the corresponding cyclopropanated products in lower yields (28% (8r) and 23% (8r′), see ESI†). However, employing the substituted pyrrole and/or indole derivatives and donor–acceptor type of diazoesters, e.g., methyl-2-diazo-2-phenylacetate (7c) and 4-methoxypheneylmethyl diazoester (7d) we obtained the corresponding cyclopropanated products (8c–8f, 8i–8l, 8m–8q, 8s–8u, 8w, 8x) in increased the yields (34% to 86%) at a higher temperature of 100 °C with excellent diastereoselctivity. It is important to mention here that at this temperature, no cyclopropane ring-opened products were obtained. However, the solvent molecule (toluene) got activated and the corresponding 3-methyl cyclohexene–cyclopropane–carboxylates were isolated as minor products when substituted indoles were employed as the heteroarene partner (see ESI†). Performing the similar reactions with 7c–7d at room temperature yielded the expected cyclopropanated products in lower yields. The relative stereochemistry of the products, 8 were unambiguously confirmed by the single crystal X-ray diffraction of 8u. Notable to mention that in all the above cases single diastereomers were obtained although the product yields were found to be relatively lower due to the overall sterically crowded nickel centres.
To have an insight into the reaction mechanism, we performed the ESI mass spectrometric analysis of the reaction solution containing 1:2 molar ratios of the pre-catalyst 3 and N2CHCO2tBu (7b) in toluene under an argon atmosphere at rt after 1 h, which revealed the formation of the diazoester bound various oligomeric complexes 4 [Ni4((O−)2L-Me)4(:CHCO2tBu)(N2CHCO2tBu)]+, 5′ and 5′′ (for 4, n = 4, for 5′ and 5′′, n = 5) (Scheme 4).
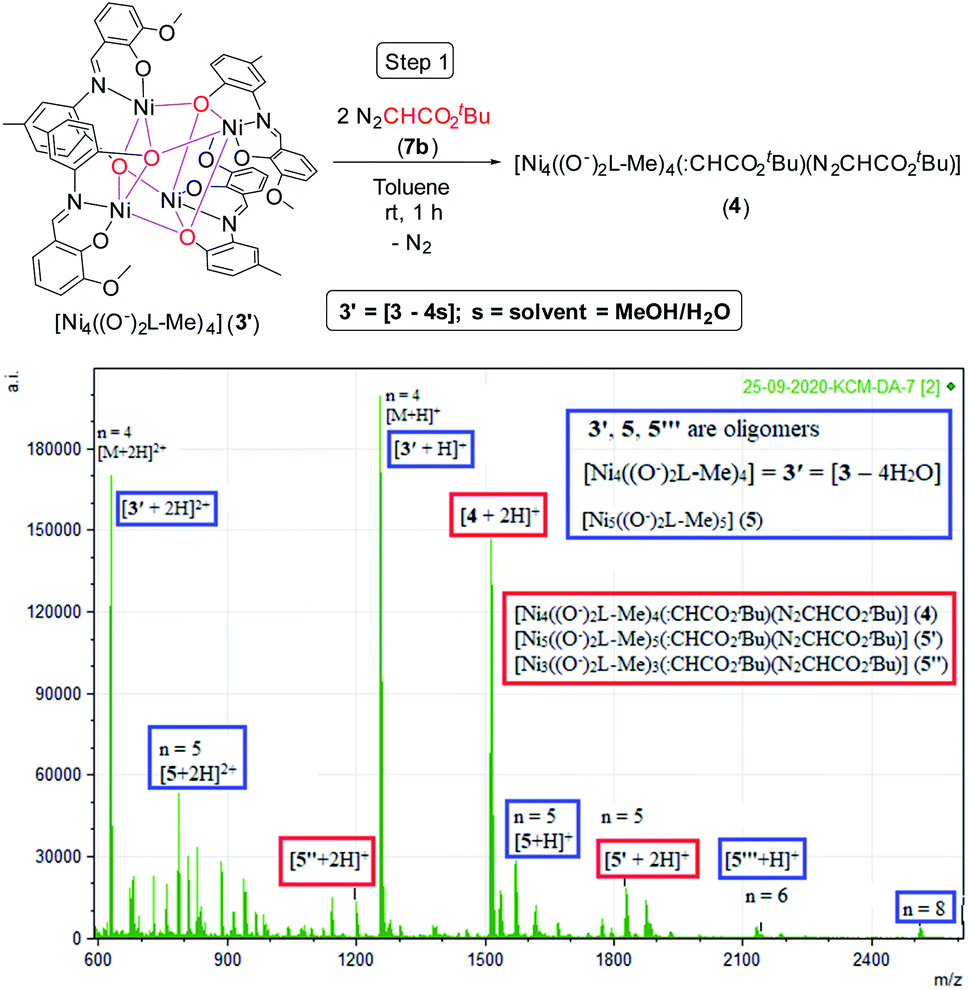 | ||
| Scheme 4 Formation of the tetrameric species 4 (top). ESI-MS spectrum of the reaction solution containing 3 and 7b after 1 h (n = degree of oligomerization) (bottom) (see Table 1 for compositions of different species). | ||
The Ni(II) centers of 3′ are expected to have five-coordinate geometries as in 2 after losing weakly coordinated solvent molecules of 3. A UV-vis absorption band was observed at 370 nm due to the binding of 7b with the catalyst 3/3′ in DCM (see ESI†). 3′ having four Ni(II) centers with five-coordinate geometry can bind with two diazoesters (7b) to form 4 (tetrameric), 5′ (pentameric) and 5′′ (trimeric) after 1 h with the formation of bubbles of N2 gas (see ESI,† Scheme 4, Table 1).
Based on these experimental results, we proposed a plausible mechanism for cyclopropanation catalyzed by in situ generated oligomeric {Ni(L-Me)}n species (n = 5 to 8), which initially bind to the diazoesters to produce the bis-nickel-carbenoids followed by the carbene transfer to the C2–C3 double bond of the aromatic heterocycle leading to the formation of the corresponding cyclopropanated product 8 (Scheme 5, Table 1).
 | ||
| Scheme 5 Proton affinity values of (OH)2L-H and (OH)2L-Me ligands. O2 (P-I) and O2N (P-II) pockets. | ||
4 Conclusions
In conclusion, we have synthesized a ferromagnetically coupled, redox active tetranuclear Ni(II)-complex (3) with a Ni4O4 core topology and relatively challenging O2N-donating mono-/di-anionic Schiff base ligand in excellent yield. The mechanism of formation of 3 has been proposed based on the structurally characterized two chloride-containing intermediate complexes (1, 2) with unique penta-coordinate Ni(II) ions and further supported by theoretical calculations. The complex 3-catalyzed cyclopropanation of various aromatic heterocycles has been achieved at room temperature and/or at an elevated temperature of 100 °C depending upon the diazoester involved utilizing the weakly coordinated solvent binding sites of Ni(II) ions of 3. Complex 3 displayed oligomerization in aprotic organic solvents to produce the species {Nin(L-Me)}n (n = 4, 5, 6, 8), which were characterized by mass spectrometric analysis. We have shown that the oligomeric metamer can bind with dinitrogen (5 + N2), and the species has been characterized by ESI mass spectrometry. Three of such diazoester-bound oligomers (n = 3, 4, 5) have been identified by ESI-MS (see Table 1).Conflicts of interest
There are no conflicts to declare.Acknowledgements
KCM and SR gratefully acknowledge SERB, New Delhi for their respective ECR grants (ECR/2016/000890 for KCM; ECR/2016/000733 for SR). SK, JK and PR thank IIT Madras for their respective SRFs and JRF. MF thanks CSIR for JRF. EN thanks IISERT for SRF.References
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Recent Advances in Nickel Catalysis, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- (a) E. C. Alyea and D. W. Meek, Four- and Five-Coordinate Nickel (II) Complexes with Monodentate Phosphines, J. Am. Chem. Soc., 1969, 91, 5761–5768 CrossRef CAS; (b) J. C. Cloyd Jr and D. W. Meek, Four- and Five-Coordinate Nickel(II) Complexes of the Flexible Triphosphine Bis(2-diphenylphosphinoethyl)phenylphosphine, Inorg. Chim. Acta, 1972, 6, 607–612 CrossRef.
- A. Escuer, R. Vicente, J. Ribas, R. Costa and X. Solans, New Nickel(II)-Copper(II) Heterodinuclear Complexes with Hexa- and Pentacoordinated Nickel(II) Ions. Magneto-structural Correlations, Inorg. Chem., 1992, 31, 2627–2633 CrossRef CAS.
- M. Zhang, S. Zhang, P. Hao, S. Jie, W.-H. Sun, P. Li and X. Lu, Nickel Complexes Bearing 2-(Benzimidazol-2-yl)-1,10-phenanthrolines: Synthesis, Characterization and Their Cata-lytic Behavior Toward Ethylene Oligomerization, Eur. J. Inorg. Chem., 2007, 3816–3826 CrossRef CAS.
- L. Zhang, X. Hao, W.-H. Sun and C. Redshaw, Synthesis, Characterization, and Ethylene Polymerization Behavior of 8-(Nitroarylamino)-5,6,7-trihydroquinolylnickel Dichlorides: Influence of the Nitro Group and Impurities on Catalytic Ac-tivity, ACS Catal., 2011, 1, 1213–1220 CrossRef CAS.
- F. Speiser, P. Braunstein and L. Saussine, Nickel and Iron Com-plexes with Oxazoline- or Pyridine-Phosphonite Ligands; Syn-thesis, Structure and Application for the Catalytic Oligomeri-zation of Ethylene, Dalton Trans., 2004, 1539–1543 RSC.
- (a) A. Boudier, P.-A. R. Breuil, L. Magna, H. Olivier-Bourbigou and P. Braunstein, J. Organomet. Chem., 2012, 718, 31–37 CrossRef CAS; (b) N. Ajellal, M. C. A. Kuhn, A. D. G. Boff, M. Hörner, C. M. Thomas, J.-F. Carpentier and O. L. Casagrande Jr, Nickel Complexes Based on Tridentate Pyrazolyl Ligands for Highly Efficient Dimerization of Ethylene to 1-Butene, Organometallics, 2006, 25, 1213–1216 CrossRef CAS; (c) A. H. P. S. Ulbrich, A. L. Bergamo Jr and O. L. Casagrande, Oligomerization of Ethylene using Tridentate Nickel Catalysts bearing Ether-Pyrazol Ligands with Pendant O- and S-Donor Groups, Catal. Commun., 2011, 16, 245–249 CrossRef CAS and references therein..
- R. Gao, M. Zhang, T. Liang, F. Wang and W.-H. Sun, Nickel(II) Complexes Chelated by 2-Arylimino-6-benzoxazolylpyridine: Syntheses, Characterization, and Ethylene Oligomerization, Organometallics, 2008, 27, 5641–5648 CrossRef CAS.
- S. O. Ojwach, I. A. Guzei, L. L. Benade, S. F. Mapolie and J. Darkwa, (Pyrazol-1-ylmethyl)pyridine Nickel Complexes: Ethylene Oligomerization and Unusual Friedel-Crafts Alkylation Catalysts, Organometallics, 2009, 28, 2127–2133 CrossRef CAS.
- M. D. Santana, G. García, M. Julve, F. Lloret, J. Pérez, M. Liu, F. Sanz, J. Cano and G. López, Oxamidate-Bridged Dinuclear Five-Coordinate Nickel(II) Complexes: A Magneto-Structural Study, Inorg. Chem., 2004, 43, 2132–2140 CrossRef CAS PubMed.
- A. Boudier, P.-A. R. Breuil, L. Magna, H. Olivier-Bourbigou and P. Braunstein, Nickel(II) Complexes with Imino-Imidazole Chelating Ligands bearing Pendant Donor Groups (SR, OR, NR2, PR2) as Pre-catalysts in Ethylene Oligomerization, J. Or-ganomet. Chem., 2012, 718, 31–37 CrossRef CAS.
- A. C. Pinheiro, A. H. Virgili, T. Roisnel, E. Kirillov, J.-F. Carpentier and O. L. Casagrande Jr, Ni(ii) Complexes bearing Pyrrolide-Imine Ligands with Pendant N-, O- and S-Donor Groups: Synthesis, Structural Characterization and use in Ethylene Oligomerization, RSC Adv., 2015, 5, 91524–91531 RSC.
- A. A. Lozano, M. Sáez, J. Pérez, L. García, L. Lezama, T. Rojo, G. López, G. García and M. D. Santana, Structure and Magnetic Properties of Carbonate-Bridged Five-Coordinate Nickel(II) Complexes Controlled by Solvent Effect, Dalton Trans., 2006, 3906–3911 RSC.
- C.-P. Zhang, H. Wang, A. Klein, C. Biewer, K. Stirnat, Y. Yamaguchi, L. Xu, V. Gomez-Benitez and D. A. Vicic, A Five-Coordinate Nickel(II) Fluoroalkyl Complex as a Precursor to a Spectroscopically Detectable Ni(III) Species, J. Am. Chem. Soc., 2013, 135, 8141–8144 CrossRef CAS PubMed.
- D. H. Brown and B. W. Skelton, Nickel Complexes of a Bis(benzimidazolin-2-ylidene)pyridine Pincer Ligand with Four- and Five-Coordinate Geometries, Dalton Trans., 2011, 40, 8849–8858 RSC.
- M. D. Santana, R. García-Bueno, G. García, J. Pérez, L. García, M. Mongec and A. Lagunad, Luminescence of Five-Coordinated Nickel(II) Complexes with Substituted-8-Hydroxyquinolines and Macrocyclic Ligands, Dalton Trans., 2010, 39, 1797–1806 RSC.
- F. Gutzeit, M. Dommaschk, N. Levin, A. Buchholz, E. Schaub, W. Plass, C. Näther and R. Herges, Structure and Properties of a Five-Coordinate Nickel(II) Porphyrin, Inorg. Chem., 2019, 58, 12542–12546 CrossRef CAS PubMed.
- R. Vinayak, A. Harinath, C. J. Gómez-García, T. K. Panda, S. Benmansour and H. P. Nayek, Solvent Modulated Assembly of Two Ni(II) Complexes: Syntheses, Structures and Magnetic Properties, ChemistrySelect, 2016, 1, 6532–6539 CrossRef.
- S. K. Kushvaha, S. Arumugam, B. Shankar, R. S. Sarkar, V. Ramkumar and K. C. Mondal, Isolation and Characterization of Different Homometallic and Heterobimetallic Complexes of Nickel and Zinc Ions by Controlling Molar Ratios and Solvents, Eur. J. Inorg. Chem., 2019, 2871–2882 CrossRef CAS.
- S. M. N. V. T. Gorantla, P. G. Reddy, S. M. A. Shakoor, R. Mandal, S. Roy and K. C. Mondal, Tetranuclear 3d/4f Coor-dination Complexes as Homogeneous Catalysts for Bis(indolyl)methane Syntheses, ChemistrySelect, 2019, 4, 7722–7727 CrossRef.
- (a) S. Saha, S. Jana, S. Gupta, A. Ghosh and H. P. Nayek, Syntheses, Structures and Biological Activities of Square Planar Ni(II), Cu(II) Complexes, Polyhedron, 2016, 107, 183–189 CrossRef CAS; (b) E. Nag, S. M. N. V. T. Gorantla, S. Arumugam, A. Kulkarni, K. C. Mondal and S. Roy, Tridentate Nickel(II)-Catalyzed Chemodivergent C-H Functionalization and Cyclopropana-tion: Regioselective and Diastereoselective Access to Substituted Aromatic Heterocycles, Org. Lett., 2020, 22, 6313–6318 CrossRef CAS PubMed.
- (a) F. Weinhold and C. Landis, Valency and Bonding, A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed; (b) C. R. Landis and F. Weinhold, The NBO View of Chemical Bonding, in The Chemical Bond: Fundamental Aspects of Chemical Bonding, ed. G. Frenking and S. Shaik. Wiley, 2014, pp. 91–120 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: Natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- K. B. Wiberg, Application of the Pople-santry-segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- G. A. Zhurko and D. A. Zhurko, Chemcraft, 2005, available from: http://www.chemcraftprog.com Search PubMed.
- R. F. W. Bader, A Quantum Theory of Molecular Structure and its Applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. A. Keith, AIMAll (Version 10.05.04, Professional), Copyright © 1997-2013, http://aim.tkgristmill.com Search PubMed.
- M. P. Doyle, Catalytic Methods for Metal Carbene Trans-formations, Chem. Rev., 1986, 86, 919–939 CrossRef CAS.
- (a) F. Gnad, M. Poleschak and O. Reiser, Stereoselective Synthesis of Novel Conformationally Restricted β- and γ-Amino Acids, Tetrahedron Lett., 2004, 45, 4277–4280 CrossRef CAS; (b) G. Özüduru, T. Schubach and M. M. K. Boysen, Enantioselective Cyclopropanation of Indoles: Construction of All-Carbon Qua-ternary Stereocenters, Org. Lett., 2012, 14, 4990–4993 CrossRef PubMed; (c) L. K. Pilsl, T. Ertl and O. Reiser, Enantioselective Three-Step Synthesis of Homo-β-proline: A Donor-Acceptor Cyclopropane as Key Intermediate, Org. Lett., 2017, 19, 2754–2757 CrossRef CAS PubMed.
- (a) H.-U. Reissig and R. Zimmer, Donor-Acceptor-Substituted Cyclopropane Derivatives and their Application in Organic Synthesis, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed; (b) H. M. L. Davies and J. R. Denton, Application of Donor/Acceptor-Carbenoids to the Synthesis of Natural Products, Chem. Soc. Rev., 2009, 38, 3061–3071 RSC; (c) T. F. Schneider, J. Kaschel and D. B. Werz, A New Golden Age for Donor-Acceptor Cyclopropanes, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed.
- (a) J. Xu, N. B. Samsuri and H. A. Duong, Nickel-Catalyzed Cyclopropanation of Electron-Deficient Alkenes with Diiodo-methane and Diethylzinc, Chem. Commun., 2016, 52, 3372–3375 RSC; (b) A. Nakamura, T. Yoshida, M. Cowie, S. Otsuka and J. A. Ibers, Cyclopropanation Reactions of Diazoalkanes with Substituted Olefins in the Presence and Absence of Nickel(0) and Palladium(0) Catalysts. The Structure of (Diazofluo-rene)bis(tert-butylisocyanide)nickel(0); A Complex Containing a π-Bonded Diazofluorene Molecule, J. Am. Chem. Soc., 1977, 99, 2108–2117 CrossRef CAS.
- H. Liu, Y. Wei and C. Cai, Hypervalent-Iodine(III) Oxidation of Hydrazones to Diazo Compounds and One-pot Nickel(II)-Catalyzed Cyclopropanation, New J. Chem., 2016, 40, 674–678 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses of complexes 1–3, general catalytic method for cyclopropanation; NMR; UV-vis; mass spectrometric analysis; single crystal X-ray diffraction of complexes 1–2; TGA, magnetic properties of complex 3; computational details; spectral data for cyclopropanated products 8; copies of 1H, 13C NMR spectra (PDF). CCDC 2006271, 2006270, 2072516 and 2092320. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03071j |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |