
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of PET tracers designed for imaging of calcium activated potassium channel 3.1 (KCa3.1) channels in vivo†
Kathrin Brömmel‡
a,
Christian Paul Konken‡ *b,
Frederik Börgela,
Henry Obeng-Darkoa,
Sonja Schelhaasc,
Etmar Bulkd,
Thomas Buddeef,
Albrecht Schwabdf,
Michael Schäfersbcf and
Bernhard Wünsch‡af
*b,
Frederik Börgela,
Henry Obeng-Darkoa,
Sonja Schelhaasc,
Etmar Bulkd,
Thomas Buddeef,
Albrecht Schwabdf,
Michael Schäfersbcf and
Bernhard Wünsch‡af
aInstitute for Pharmaceutical and Medicinal Chemistry, Westphalian Wilhelms-University Münster, Corrensstraße 48, D-48149 Münster, Germany
bDepartment of Nuclear Medicine, University Hospital Münster, Albert-Schweitzer-Campus 1, Building A1, D-48149 Münster, Germany. E-mail: Paul.Konken@uni-muenster.de; Fax: +49-8347363; Tel: +49-251-8344791
cEuropean Institute for Molecular Imaging (EIMI), Westphalian Wilhelms-University Münster, Waldeyerstraße 15, D-48149 Münster, Germany
dInstitute for Physiology II, University Hospital Münster, Robert-Koch-Straße 27b, D-48149 Münster, Germany
eInstitute for Physiology I, University Hospital Münster, Robert-Koch-Straße 27a, D-48149 Münster, Germany
fCells-in-Motion Interfaculty Center, Westphalian Wilhelms-University Münster, Waldeyerstraße 15, D-84149 Münster, Germany
First published on 10th September 2021
Abstract
Expression of the Ca2+ activated potassium channel 3.1 (KCa3.1) channel (also known as the Gàrdos channel) is dysregulated in many tumor entities and has predictive power with respect to patient survival. Therefore, a positron emission tomography (PET) tracer targeting this ion channel could serve as a potential diagnostic tool by imaging the KCa3.1 channel in vivo. It was envisaged to synthesize [18F]senicapoc ([18F]1) since senicapoc (1) shows high affinity and excellent selectivity towards the KCa3.1 channels. Because problems occurred during 18F-fluorination, the [18F]fluoroethoxy senicapoc derivative [18F]28 was synthesized to generate an alternative PET tracer targeting the KCa3.1 channel. Inhibition of the KCa3.1 channel by 28 was confirmed by patch clamp experiments. In vitro stability in mouse and human serum was shown for 28. Furthermore, biodistribution experiments in wild type mice were performed. Since [18F]fluoride was detected in vivo after application of [18F]28, an in vitro metabolism study was conducted. A potential degradation route of fluoroethoxy derivatives in vivo was found which in general should be taken into account when designing new PET tracers for different targets with a [18F]fluoroethoxy moiety as well as when using the popular prosthetic group [18F]fluoroethyl tosylate for the alkylation of phenols.
1. Introduction
Dysregulation of the KCa3.1 channel is directly associated with many different diseases such as asthma,1,2 sickle cell anemia,3–5 coronary restenosis after angioplasty,6 kidney fibrosis7,8 and atherosclerosis.9,10 It has a a place in corneal11 and conjunctival fibrosis12 and was shown to be vastly overexpressed in Alzheimers disease.13–15 Furthermore, the KCa3.1 channel is a modulator in hepatic injury.16 The KCa3.1 channel plays an important role in cancer diseases: Along with other ion channels it contributes to features of essential “cancer hallmarks”, such as proliferation, migration, invasion and extravasation.17–19 Consequently, the ion channel plays a critical role in the metastatic cascade. Inhibition of the KCa3.1 channel reduces proliferation, migration and metastasis in different tumor entities.20–22 The direct correlation between overexpression of the KCa3.1 channel and tumor grade as well as metastatic status highlights the importance of this ion channel in cancer diseases.23 Furthermore, overexpression of KCa3.1 channels is often related to poor prognosis of tumor patients and high lethality rates.24,25 Therefore, the KCa3.1 channel could serve as predictive marker with respect to patient survival.23–25 Additionally, pro-tumor functions of endogenously expressed KCa3.1 channels have been shown in a mouse model.18,26The impact of the potassium channel on various diseases is rather outstanding, please also refer to the available comprehensive reviews.27–29
Our aim is to enable in vivo visualization of the KCa3.1 channel via PET technology to offer the opportunity of a potential diagnostic tool in different diseases. We decided to focus on KCa3.1 channels in cancer diseases since essentially all cells of the tumor stroma express KCa3.1 channels.30 A signal from a potential PET tracer binding to this ion channel should therefore be very robust. Thus, KCa3.1 channels in tumor cells are particularly suitable as a first model for testing PET tracers targeting this ion channel.
Herein, we present the design and radiosynthesis of potential PET tracers for imaging of the KCa3.1 channel in vivo.
The KCa3.1 channel inhibitor senicapoc (1) served as starting point for the development of PET tracers for in vivo imaging of the ion channel. Senicapoc (1) shows a high affinity towards the KCa3.1 channel with an IC50 value of 11 ± 2 nM (measured on human erythrocytes) and excellent selectivity over related ion channels.31 These properties are promising for selective imaging of the KCa3.1 channel in vivo. Senicapoc (1) contains two fluorine atoms. Replacement of one fluorine atom by fluorine-18 results in [18F]senicapoc ([18F]1) bearing an authentic label (Fig. 1). Moreover, senicapoc (1) was already evaluated in phase III clinical trials for potential therapy of sickle cell anemia, so data regarding toxicity and tolerability are available reducing the effort for first in vivo studies using the new tracer in humans. Herein, different strategies for the radiosynthesis of [18F]senicapoc, the synthesis of suitable precursors as well as advantages and disadvantages of the chosen routes are presented.
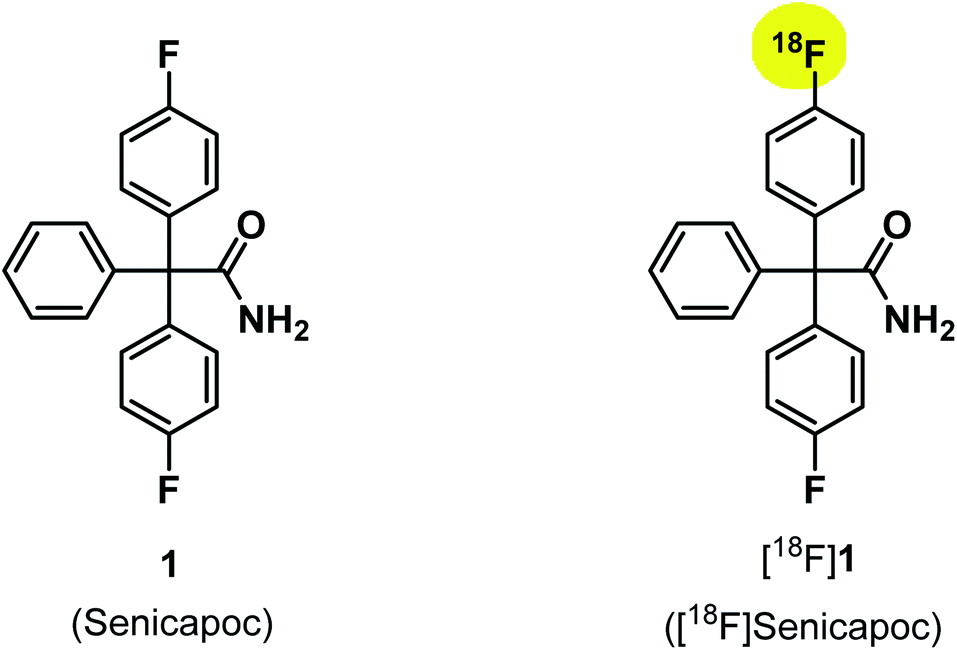 | ||
| Fig. 1 Structures of senicapoc (1) and [18F]senicapoc ([18F]1). | ||
2. Synthesis
To identify the [18F]labeled compounds by co-injection on the radio-HPLC system after radiosynthesis, non-radioactive [19F]1 reference compound had to be synthesized first. Therefore, senicapoc (1) was prepared on the basis of previously described procedures.32 (see ESI, Scheme S1†). The literature-known synthesis of senicapoc (1) is not suitable for radiosynthesis as a multi-step protocol is following the introduction of the fluorine atoms. In PET tracer synthesis the fluorine has to be introduced in the last steps due to the short half-life of [18F]fluorine.In 2016, Neumann et al. published the deoxyfluorination of phenols via concerted nucleophilic aromatic substitution (CSNAr), which was also performed with [18F]fluoride.33 Uronium salts were used as precursors for the radiosynthesis. We started our efforts to prepare [18F]senicapoc ([18F]1) using this method, which required the uronium salt 10 via the phenolic derivative 9. (Scheme 1): Synthesis of the uronium salt precursor 10 from phenol 9: reagents and reaction conditions: (a) 2-chloro-1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-3-ium chloride (8), Ag2CO3, CH2Cl2, 60 °C, 4 h, 68%. The phenol 9 was synthesized in five steps as recently published in connection with the synthesis of fluorescently labeled probes for in vitro imaging of KCa3.1 channels.34–37
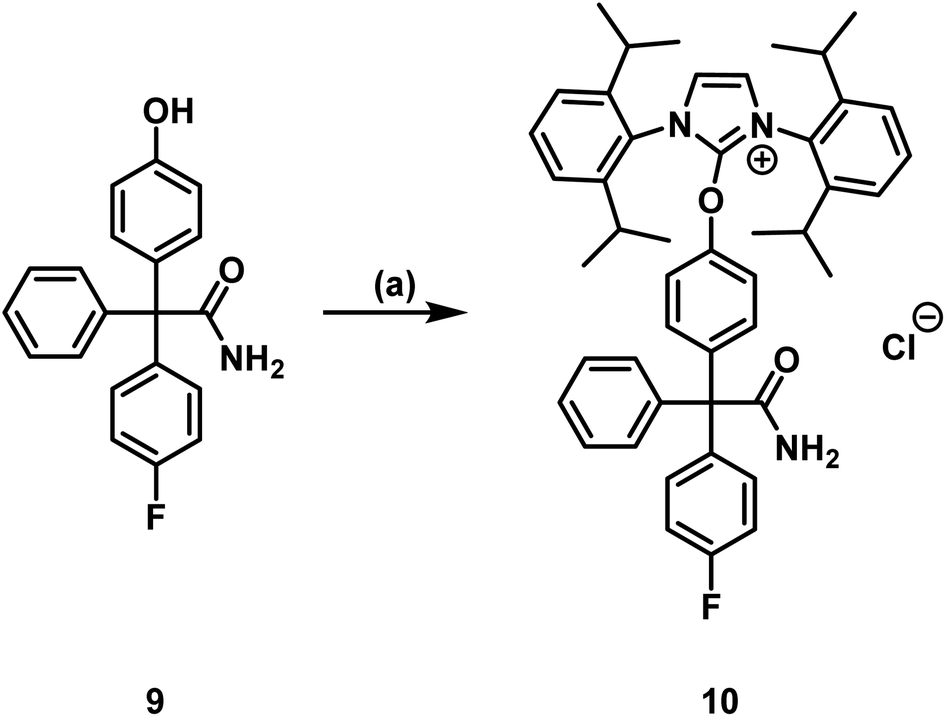 | ||
| Scheme 1 Synthesis of the uronium salt precursor 10 from phenol 9: reagents and reaction conditions: (a) 2-chloro-1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-3-ium chloride (8), Ag2CO3, CH2Cl2, 60 °C, 4 h, 68%. | ||
Neumann et al. used the uronium salts prepared by this method directly for 18F-deoxyfluorination without further purification.33 Since the purity of the prepared uronium salt 10 was only 78% (HPLC), we decided to purify 10 by column chromatography. Thus, uronium salt 10 was isolated with 99.2% purity (HPLC) and was fully characterized. The purified as well as the unpurified uronium salts 10 were used in radiosynthesis test reactions in order to obtain [18F]senicapoc ([18F]1) (Scheme 2).
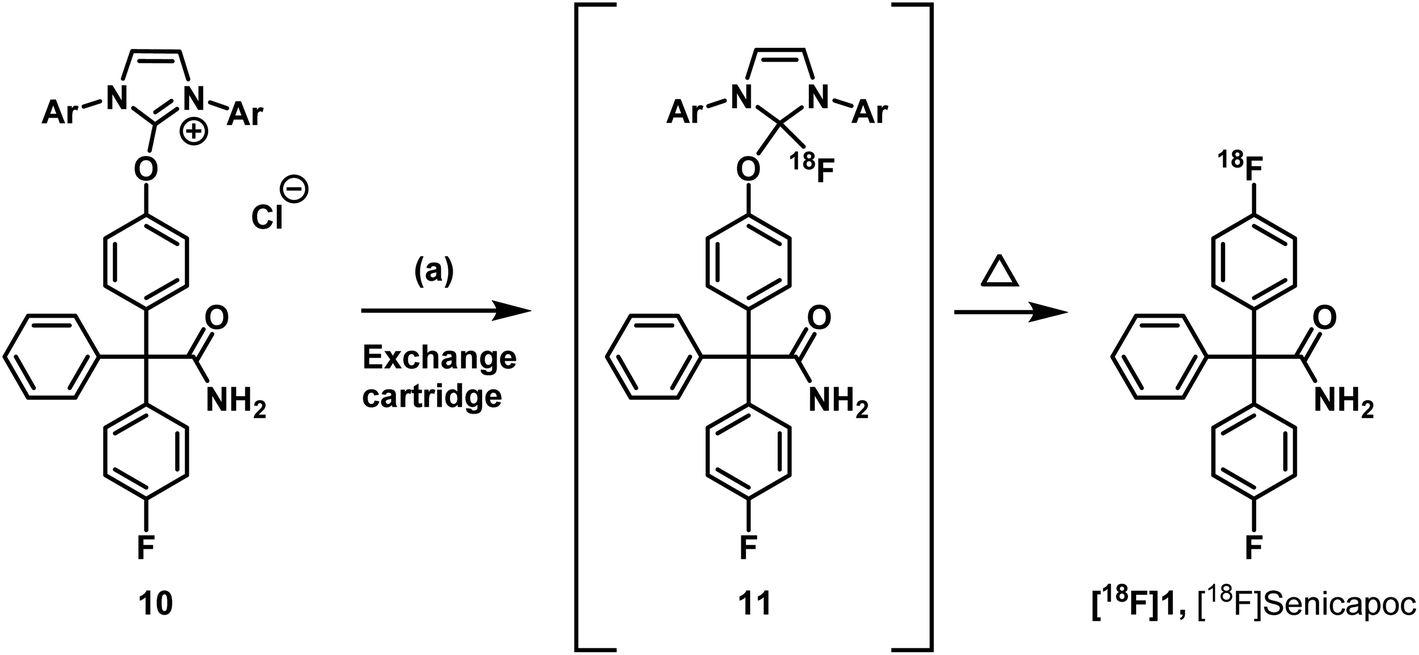 | ||
Scheme 2 Radiosythesis of [18F]senicapoc ([18F]1). Reagents and reaction conditions: (a) [18F]F−, butan-2-one![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH:Bu3N (100:10:1), 130 °C, 20 min. RCY (d.c.) = 0.4% (n = 1). :EtOH:Bu3N (100:10:1), 130 °C, 20 min. RCY (d.c.) = 0.4% (n = 1). | ||
The radiosynthesis was performed semiautomatically according to Neumann et al.33 An aqueous solution of [18F]fluoride was flushed through an anionic exchange cartridge. Different cartridges were used during evaluation, starting with Chromabond® PS-HCO3 cartridges containing 45 mg or 10 mg sorbent identical with the sorbent of the Chromafix PS-HCO3 cartridges used in the original publication.33 After trapping of [18F]fluoride, the cartridge (45 mg) was flushed with butan-2-one:ethanol (1:1, 1 mL). Thereafter, the cartridge was reversed manually using a luerlock® system as suggested in the original procedure. Since this step is highly critical in terms of radiation protection, we only used maximum starting activities of <1 GBq. After reversing the cartridge it was eluted with 5 ± 1 mg of the uronium salt 10 in butan-2-one:ethanol:tributylamine (100:10:1, 1 mL). In an average synthesis 40 ± 10% of the starting activity remained on the cartridge. After a reaction time of 20 min at 130 °C, only a small fraction of the theoretical activity was found in the reactor solution. This is a second critical step in the protocol, as the only explanation for this reproducible result is the formation of volatile 18F-labeled compounds. The protocol led to [18F]senicapoc ([18F]1). Unfortunately, the yield was much lower than in comparable published labelling approaches (0.4% RCY d.c.). Moreover, the radiochemical purity was insufficient (87%) (for chromatogram see ESI: Fig. S1†).
The very low radiochemical yield in combination with the very low starting activity (due to radiation protection regulations) and the technical problems mentioned above rendered in vivo experiments impossible. The use of cartridges with lower sorbent amounts (10 mg) resulted in even lower elution efficacy combined with considerably lower radiochemical yields (<0.1% RCY d.c.). The use of our standard cartridges Sep-Pak® Light QMA Fa. Waters® produced even worse results. However, in these experiments the cartridges were not reversed in the elution process to avoid the risk of contamination. 67% of the starting activity remained on the cartridge and it was not possible to isolate [18F]senicapoc ([18F]1). Further experiments combining the elution with potassium hydrogencarbonate (KHCO3) (5 mg) as well as different elution solutions did not improve the initial result. For these reasons, the synthesis of [18F]senicapoc ([18F]1) using the uronium salt precursor 10 was no longer pursued.
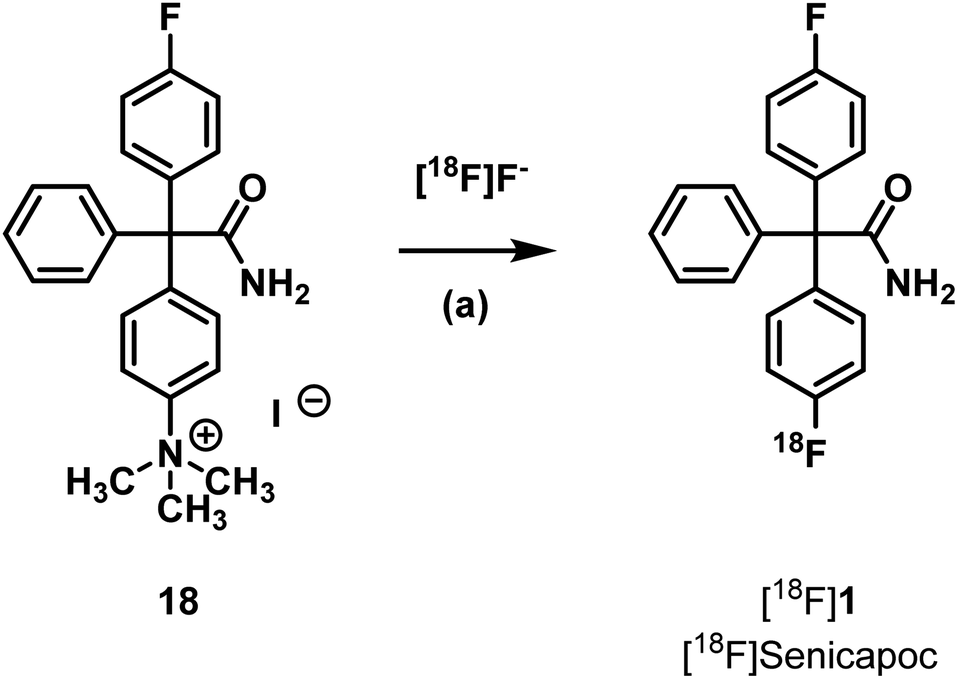
Another strategy is the direct nucleophilic substitution of a suitable precursor with [18F]fluoride to produce [18F]senicapoc ([18F]1). A well-established leaving group to achieve aromatic nucleophilic substitution is the trimethylammonium group.38,39 Although the aromatic system is not specially activated by electron withdrawing functional groups, the 18F-labelling of the trimethylammonium precursor 18 was envisaged. The synthesis of such a precursor is similar to the synthesis of senicapoc (1) itself (see ESI, Scheme S3†).32
Radiosynthesis by nucleophilic aromatic substitution with [18F]fluoride was performed using various conditions summarized in Table 1 (Scheme 3 and Table 1).
| Eluent solution (in CH3CN/H2O) | Solvent | Temperature [°C] | Time [min] | Product |
|---|---|---|---|---|
| K2CO3 (aq), K222 | Acetonitrile (CH3CN) | 120 | 15 | — |
| K2CO3 (aq), K222 | Dimethylformamide (DMF) | 150 | 15 | — |
| K2CO3 (aq), K222 | DMF | 155 | 15 | — |
| K2CO3 (aq), K222 | Dimethylsulfoxide (DMSO) | 165 | 30 | — |
| K2C2O4 (aq), K222 | DMF | 155 | 30 | — |
| Bu4NHCO3 (aq) | DMF | 150 | 15 | ? |
| Bu4NHCO3 (aq) | DMSO | 150 | 15 | ? |
| Bu4NHCO3 (aq) | DMSO | 140 | 15 | ? |
 | ||
| Scheme 3 Radiosynthesis of [18F]senicapoc ([18F]1). Reagents and reaction conditions: (a) cartridge: Sep-Pak® Light QMA Fa. Waters®, conditions see Table 1. | ||
The investigated reaction conditions using carbonate or oxalate as base did not lead to [18F]senicapoc ([18F]1). The use of tetrabutylammonium hydrogen carbonate as base led to the formation of tetrabutylammonium-[18F]-fluoride ([18F]TBAF) as fluorination agent. All reactions with [18F]TBAF provided a product with very similar polarity compared to [18F]senicapoc ([18F]1). The retention times on the semipreparative as well as on the analytical radio-HPLC were very similar to those determined for senicapoc (1) (Fig. 2). Due to the high similarity, we initially assumed a successful transformation.
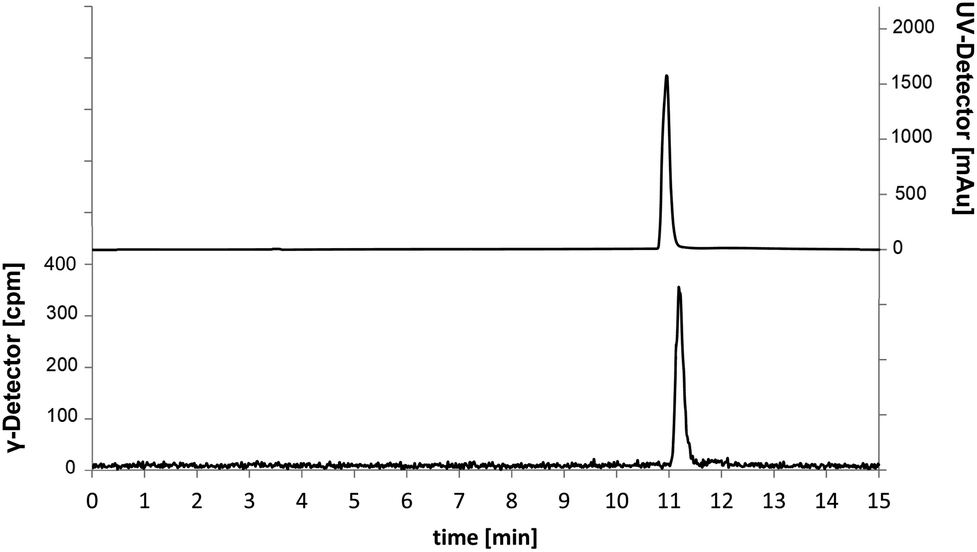 | ||
| Fig. 2 Chromatograms of the analytical radio-HPLC. Traces for 18F-labelled (γ-detector [cpm], bottom) compound and co-injection of [19F]senicapoc (1) as reference compound (UV-detector [mAu], top). | ||
In order to prepare a solution for injection after HPLC purification, the solvents were removed under reduced pressure. Afterwards, only a small fraction of the theoretical radioactivity was left. It was postulated that the reduced amount of radioactivity is due to removal of a volatile 18F-labelled compound. As [18F]senicapoc ([18F]1) should not be able to release a volatile 18F-species, the result of the synthesis was questioned. To get a better insight into the decomposition of the 18F-labelled compound obtained during radiosynthesis, samples were taken during the removal of the solvent and analyzed by analytical radio-HPLC and radio-TLC (Fig. 3; for further chromatograms see ESI: Fig. S2 and S3†).
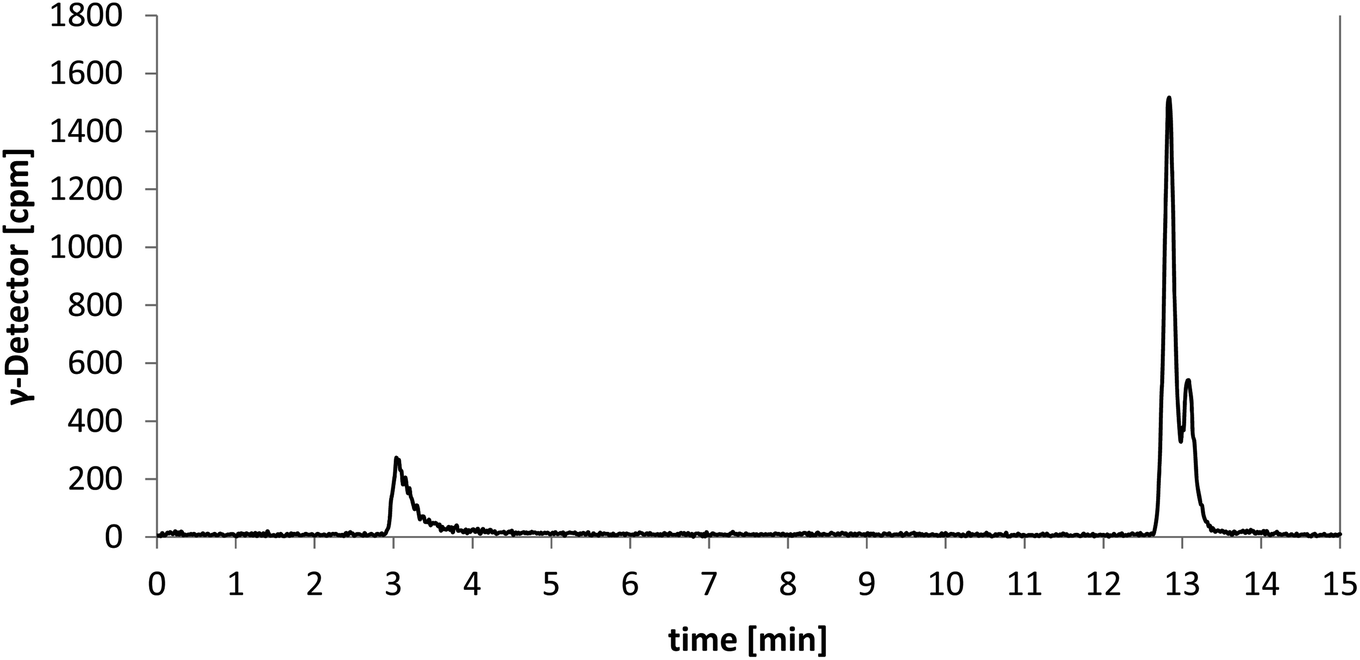 | ||
| Fig. 3 Chromatogram of the analytical radio-HPLC after 30 min of removal of the solvent. | ||
It was shown that decomposition occurred leading to a less polar compound. The peak with a retention time of 3 min (analytical radio-HPLC; Fig. 3), which increased over time, as well as the baseline peak in the radio-TLC (see ESI: Fig. S4†) are a strong hint towards the formation of [18F]fluoride. Since senicapoc (1) is stable even at high temperature and under reduced pressure, it was suggested that a different product had been formed. To get an idea of the formed product, the fluorination of the quaternary ammonium compound 18 was performed with [19F]fluoride under the conditions of the radiosynthesis. Indeed, the reaction of 18 with TBAF in DMF at 155 °C for 10 min resulted in the less polar compound 19, which was purified and fully characterized. Spectroscopic analysis showed the formation of triarylmethane 19, which was formed by decarboxylation and demethylation of the quaternary ammonium precursor 18 (Scheme 4).
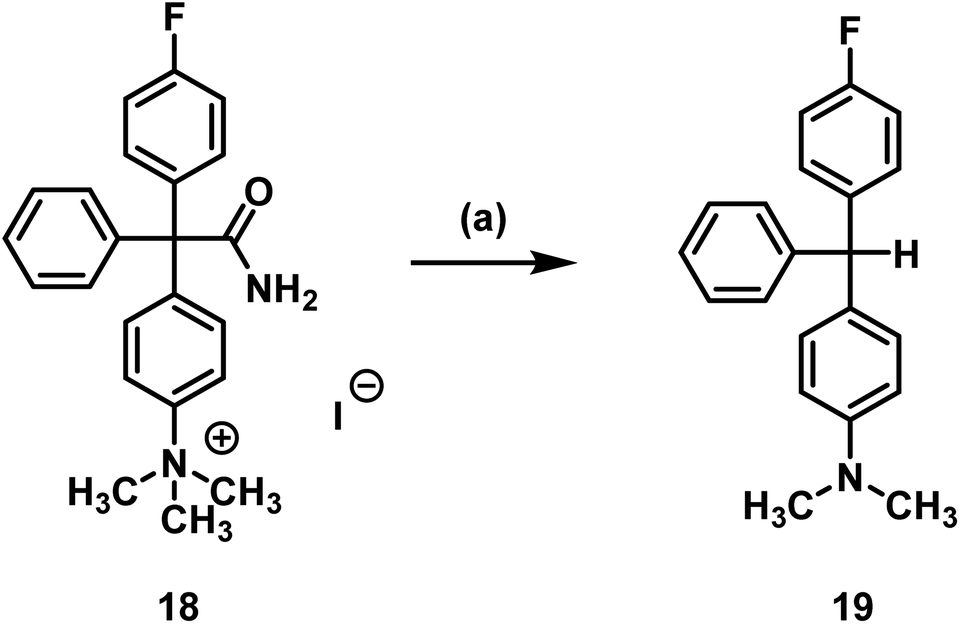 | ||
| Scheme 4 Decarboxylation and demethylation of trimethylarylammoninium precursor 18. Reagents and reaction conditions: (a) TBAF, DMF, 155 °C, 10 min, 78%. | ||
The same reaction occurred in dry DMF used in the radiosynthesis. The formation of [18F]fluoride during radiochemistry may be explained by radical formation of the 18F-substituted triarylmethane, which would lead to loss of fluoride upon dimerization forming a quinoidic system. The dimerization would form a structure that is a substituted Gomberg's dimer.
The titanium nanoparticle catalyzed nucleophilic substitution of aryl tosylates to obtain [18F]aryl compounds was successfully established by Sergeev et al.40 The required tosylate precursor 20 was obtained by reaction of phenol 9 with tosyl chloride (Scheme 5).
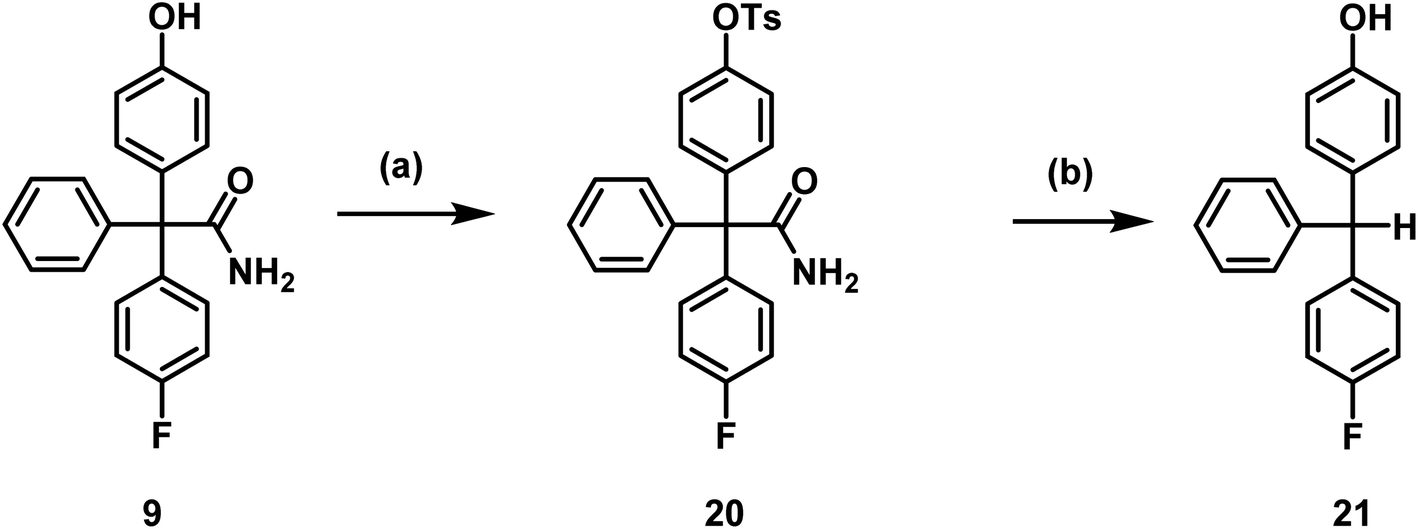 | ||
| Scheme 5 Toslyation of phenol 9 and decomposition of tosylate 20. Reagents and reaction conditions: (a) TsCl, CH2Cl2, RT, 2 h, 98%, (b) TBAF, DMF, 130 °C, 20 min, 80%. | ||
Since stability issues occurred using the trimethylarylammonium salt 18 as precursor in 18F-labelling reactions, the stability of the prepared tosylate precursor 20 was evaluated before starting radiosynthesis. Usually, the titanium nanoparticle catalyzed nucleophilic substitution takes place at temperatures between 110 °C and 140 °C with reaction times between 7 and 12 min.40 The conditions required for the radiosynthesis were tested and are summarized in Table 2.
| Base (5 eq.) | Temperature [°C] | Stability (TLC) |
|---|---|---|
| — | 130 | Stable |
| TBAF | 130 | Full conversion |
| TBAF | 110 | Full conversion |
| TBAF | 90 | Full conversion |
| K2CO3 | 130 | Full conversion |
| K2CO3 | 110 | Partial conversion |
| K2CO3 | 90 | Partial conversion |
| (NH4)2C2O4 | 130 | Stable |
| (NH4)2C2O4 | 110 | Stable |
| (NH4)2C2O4 | 90 | Stable |
Tosylate 20 was not stable in the presence of carbonate or tetrabutylammoniumfluoride (TBAF), as a less polar compound was formed. To characterize the decomposition product, the tosylate 20 was heated up to 130°C for 20 min with TBAF in DMF and the product 21 was isolated by flash chromatography. The obtained decomposition product 21 indicated that hydrolysis of the tosylate and loss of the carbamoyl moiety at the quaternary C-atom had taken place. Hydrolysis and decarboxylation of the amide 20 could lead to the triarylmethane derivative 21 (Scheme 5).
Since the titanium nanoparticle catalyzed nucleophilic substitution of aryl tosylates requires activation of [18F]fluoride as [18F]TBAF, it was concluded that the tosylate 20 is not a useful precursor for 18F-labelling. Furthermore, it is impossible to use the heterogeneous system in the automatic synthesis modules, since the solids would clog the thin tubing resulting in limitations in the further transfer of the radiosynthesis into clinical environment.
Literature protocols for 18F-labelling of aryl pinacol boronic esters use [18F]TBAF or tetraethylammonium [18F]fluoride ([18F]TEAF) and temperatures above 100 °C.41 It is very unlikely that these conditions are tolerated by the triaryl acetamide structure of the required precursor, since similar conditions led to decomposition of both precursors 18 and 20. Because the solvent system differs and the reaction time is relatively short, the pinacol boronic ester 24 should nevertheless be tested as precursor. Therefore, aryl bromide 22 was prepared in four steps analogous to the synthesis of the trimethylammonium precursor 18 using 1,4-dibromobenzene instead of 4-bromo-N,N-dimethylaniline (12) as starting material (see ESI, Scheme S4†). Finally, the boronic ester 24 was prepared by reaction of aryl bromide 22 with diboronate 23 and ferrocene-1,1′-(diyl)bis(diphenylphosphane)palladium(II)chlorid (Pd(dppf)Cl2) (Scheme 6).
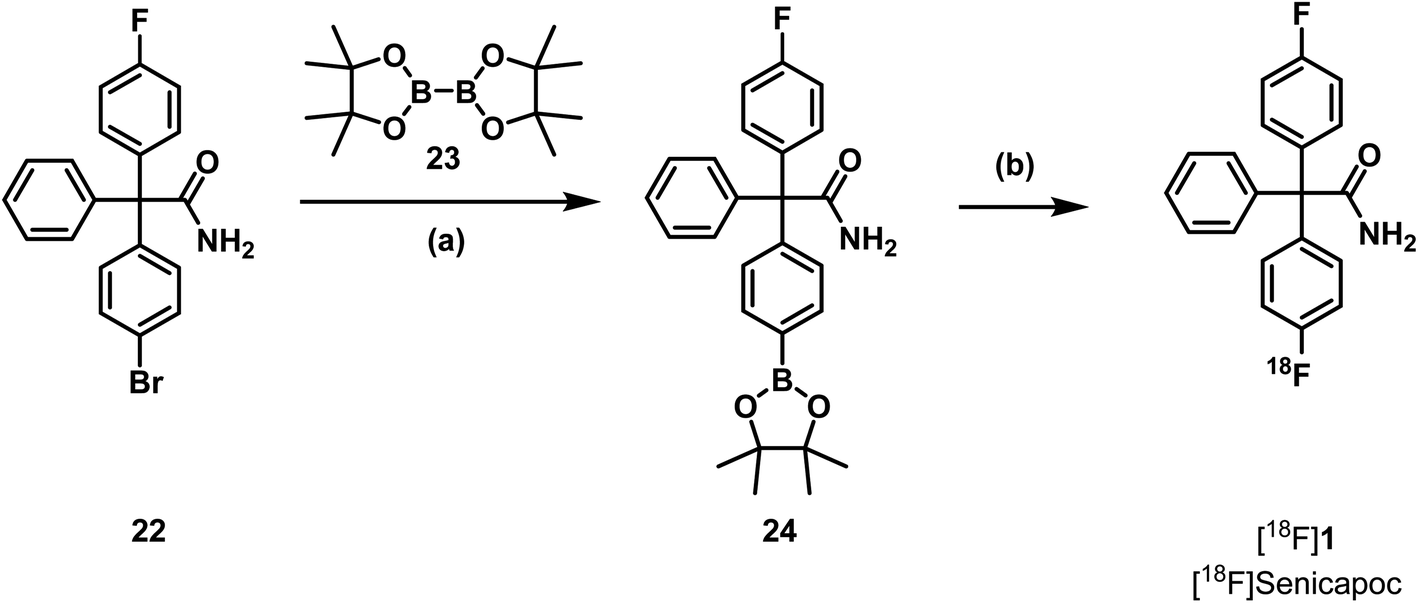 | ||
| Scheme 6 Last step of the synthesis of the pinacol boronic ester 24 and radiosynthesis of [18F]senicapoc ([18F]1). Reagents and reaction conditions: (a) Pd(dppf)Cl2, KOAc, dioxane, RT- 50 °C, 5 h, 15%. (b) cartridge: Sep-Pak® Light QMA Fa. Waters®, [18F]TEAF, butan-1-ol, Cu(OTf)2py4, DMF, 110 °C, 10 min, 0%. | ||
For Cu(II)-catalyzed transformations of areneboronates into aryl fluorides, azeotropic drying was not reported.41 Thus, a solution of precursor 24 and tetrakis(pyridine)copper(II)triflate (Cu(OTf)2py4) in DMF was added to a solution of [18F]TEAF in butan-1-ol and the mixture was heated to 110 °C for 10 min. The copper catalyst was removed by a C18+ cartridge and the reaction mixture was analyzed by semipreparative and analytical radio-HPLC. However, a peak showing the formation of [18F]senicapoc ([18F]1) could not be detected. It was concluded that this strategy is not useful for the synthesis of [18F]senicapoc ([18F]1).
Senicapoc (1) contains the same triarylacetamide structure as the unstable precursors 18, 20 and 24. Since it is essential that senicapoc (1) is stable under the conditions used to prepare [18F]senicapoc ([18F]1), different reaction conditions reasonable for 18F-labelling were evaluated. The results are summarized in Table 3.
| Base (5 eq.) | Temperature [°C] | Stability (TLC) |
|---|---|---|
| — | 130 | Stable |
| TBAF | 130 | Complete decomposition |
| TBAF | 110 | Complete decomposition |
| TBAF | 90 | Complete decomposition |
| K2CO3 | 130 | Complete decomposition |
| K2CO3 | 110 | Partial decomposition |
| K2CO3 | 90 | Partial decomposition |
| (NH4)2C2O4 | 130 | Stable |
| (NH4)2C2O4 | 110 | Stable |
| (NH4)2C2O4 | 90 | Stable |
Senicapoc (1) showed very similar reactivity under the conditions tested as the precursors 18 and 20. As reported for 18 and 20, triarylmethane 25 was formed as decomposition product. Removal of the carbamoyl moiety, presumably by hydrolysis and decarboxylation, is an obvious inherent instability of this type of triarylacetamide derivatives under basic conditions (K2CO3, TBAF) and elevated temperature (Scheme 7).
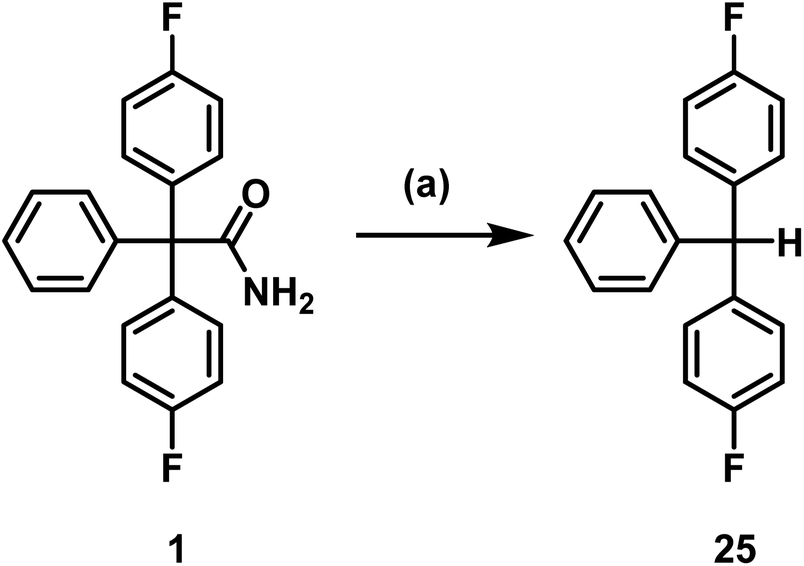 | ||
| Scheme 7 Decomposition of senicapoc (1). Reagents and reaction conditions: (a) TBAF, DMF, 130 °C, 10 min, 75%. | ||
Analysis of the data summarized in Table 3 showed that oxalate is the preferred base to perform a direct nucleophilic fluorination, as decomposition of senicapoc to the methane derivative 25 could not be observed. The base carbonate can be used if the reaction temperature does not exceed 90 °C.
Since aliphatic nucleophilic substitution with [18F]fluoride is possible using rather mild conditions, the [18F]fluoroethoxy derivative [18F]28 of senicapoc (1) was envisaged for imaging of KCa3.1 channels in vivo. At first, the non-radioactive 19F-containing reference compound 28 was synthesized to perform in vitro experiments addressing the desired interaction of 28 with the KCa3.1 channel and to identify unequivocally the labelled compound by co-injection on the radio-HPLC system. 28 was synthesized by alkylation of phenol 9 with (2-fluoroethyl) tosylate (27),42 which was prepared from 2-fluoroethanol (26) and tosyl chloride (Scheme 8).
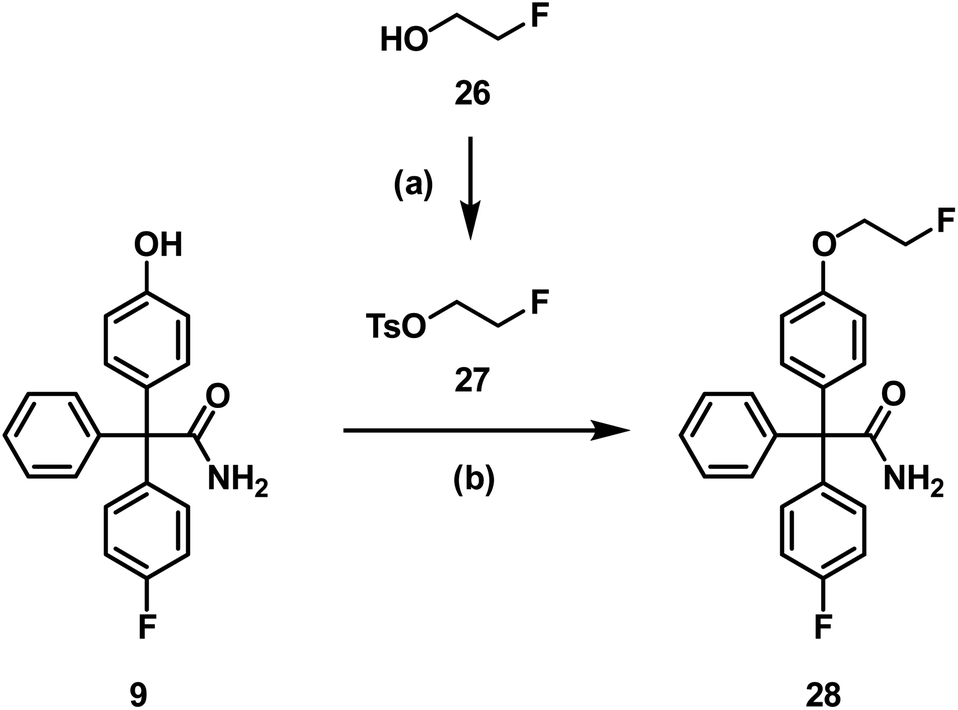 | ||
| Scheme 8 Synthesis of the fluoroethoxy derivative 28. Reagents and reaction conditions: (a) TsCl, Et3N, CH2Cl2, RT, 24 h, 88%. (b) DMF, Cs2CO3, 100 °C, 10 °C/h, 84%. | ||
At first, the interaction of 28 with the KCa3.1 channel had to be analyzed. For this purpose, patch clamp experiments were performed using the non-small-cell lung cancer cell line A549-3R, which expresses a high number of the KCa3.1 channels (Fig. 4).
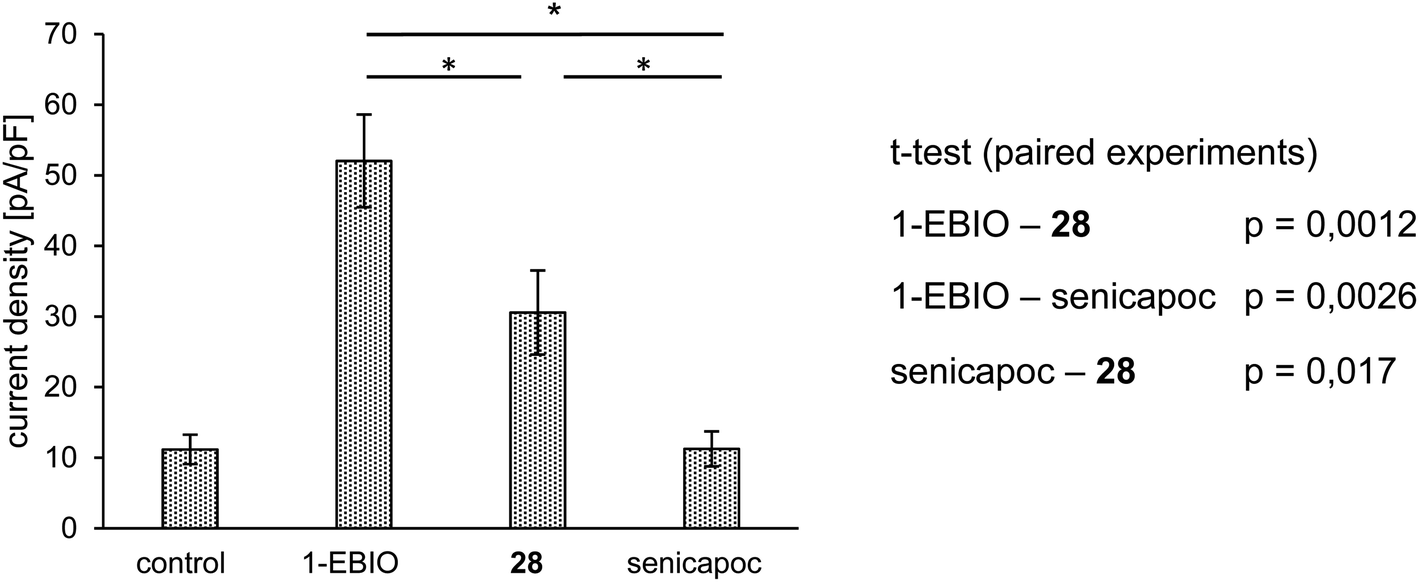 | ||
| Fig. 4 Summary of patch-clamp experiments. Average current density [pA/pF] ± SEM (n = 5). KCa3.1 mediated current is activated with 1-EBIO (50 μM). It is inhibited by the consecutive application of 28 (10 μM) and senicapoc (1 μM). All compounds were dissolved in DMSO and added to the standard extracellular solution and applied for a period of 2–5 min (1-EBIO = 1-ethyl-2-benzimidazolone). | ||
A549-3R cells were activated with 1-EBIO leading to an increased outward current in the patch clamp experiments. Then, the fluoroethoxy derivative 28 was added, which led to reduced ionic current. Finally, senicapoc was added resulting in a complete inhibition of the current increase induced by 1-EBIO. The experiment was performed in a paired fashion and showed that 28 was able to inhibit the KCa3.1 channel, although not as efficiently as senicapoc (1). The effect of 28 on the ionic current was regarded as interaction with the ion channel. Therefore, imaging of the KCa3.1 channel in vivo using [18F]38 was assumed to be possible.
Consequently, radiosynthesis of [18F]28 using precursor 31 bearing a tosyloxy leaving group for aliphatic nucleophilic substitution with [18F]fluoride under mild conditions was planned. Thus, phenol 9 was directly alkylated with ethylene ditosylate (30),43 which was obtained by ditosylation of ethylene glycol (29) with an excess of tosyl chloride (Scheme 9).
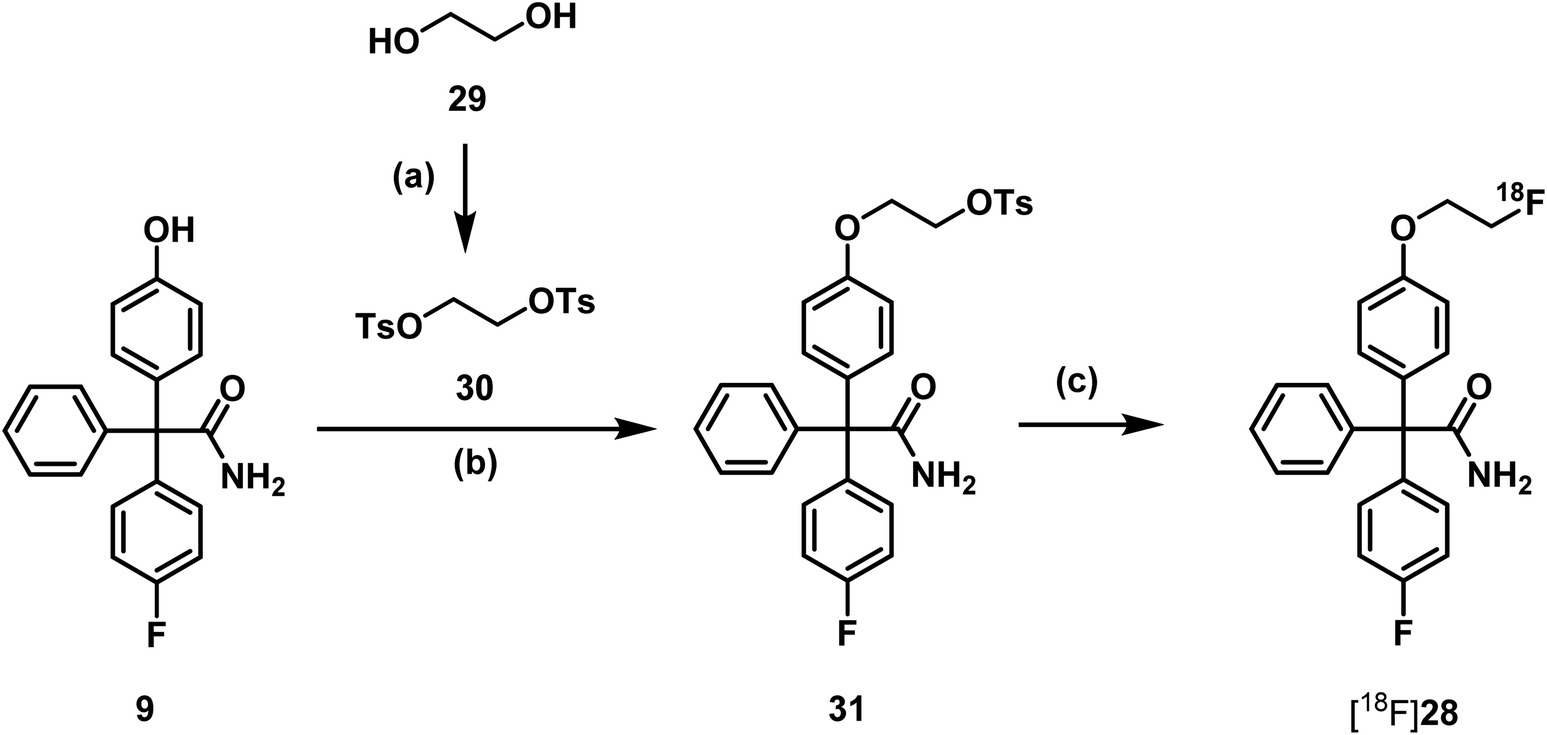 | ||
| Scheme 9 Synthesis of precursor 31 and radiosynthesis of [18F]fluoroethoxy senicapoc [18F]28. Reagents and reaction conditions: (a) TsCl, Et3N, CH2Cl2, RT, 12 h, 97%. (b) DMF, Cs2CO3, RT, 24 h, 49%. (c) [18F]KF.K222, CH3CN, 90 °C, 15 min, RCY (d.c.) = 4 ± 1.5%. | ||
Since it had been found that carbonate leads to degradation of triarylacetamides, oxalate was used as base in the first 18F-labelling experiments. The radiosynthesis was carried out in a semiautomated radiosynthesizer. The aqueous solution of [18F]fluoride ions was rinsed through a preconditioned QMA-light cartridge (Waters®). The trapped [18F]fluoride ions were eluted with a solution of K222 and K2CO3 in CH3CN and H2O. After azeotropic drying a solution of the precursor 31 in DMF was added to the radiosynthesizer. The mixture was heated to 90 °C for 15 min in the closed reactor (Scheme 9).
The desired product [18F]28 was isolated by semi-preparative radio-HPLC. Identity was confirmed by coinjection of the corresponding reference compound 28 using the analytical HPLC-system (for chromatogram see ESI: Fig. S5†).
After removal of the solvent under reduced pressure, the product [18F]28 was reconstituted with ethanol. PBS was added to achieve an ethanol concentration below 10%. This ready-to-inject solution was controlled again by radio-HPLC as well as radio-TLC. The decay corrected radiochemical yield of the formulated PET tracer was 4.0 ± 1.5%, radiochemical purity was found to be >96%.
In vitro serum stability was tested using murine and human blood [18F]28 showed high stability over the complete test period of 120 min. The corresponding chromatograms are shown in the ESI (Fig. S6 and S7†). The logD7,4-value was determined to be 1.84 ± 0.5 (±SEM) using the shake-flask method by Prante et al.44
Before performing biodistribution experiments in vivo, the plasma protein binding (PPB) of senicapoc (1) and its fluoroethoxy derivative 28 were determined in vitro using a LC-MS method with external calibration. The PPB has a considerable influence on both distribution and metabolic stability of a drug in vivo. A high PPB ensures good distribution and long circulation in the blood as well as low metabolic degradation in vivo. On the other side, only the unbound fraction of the drug can enter the tissue and cells and thus bind to the respective target. It was found that both senicapoc (1) and its fluoroethoxy derivative 28 have a PPB of >99%.
Due to the high PPB, a good biodistribution of [18F]28 in vivo was expected. Biodistribution studies with the PET tracer [18F]28 in mice will show undesired possible accumulation in different tissues, which e.g. also physiologically express the target of the PET tracer, or non-specific binding to other proteins. Moreover, biodistribution studies show the route of excretion of the tracer. Altogether, analysis of the in vivo biodistribution of a PET tracer provides important data, which are crucial for the further development of a PET tracer for imaging studies.
In this study, three 12 week-old C57BL/6 wild-type mice (n = 3) were used. The experiments were approved by the State Office for Nature, Environment and Consumer Protection North Rhine-Westphalia under license Az. 84–02.04.2015.A410. The mice were anesthetized with isoflurane and placed on a warmed PET scanner bed. Important parameters such as breathing were constantly monitored. The formulated PET tracer [18F]28 was administered via a tail vein catheter and with the aid of an injection pump. After 90 min acquisition time in the quadHIDAC PET scanner, a CT scan of the mouse was performed. The data and images were evaluated with the in-house developed software MEDgical (Fig. 5; please also refer to Fig. S8 in the ESI†).
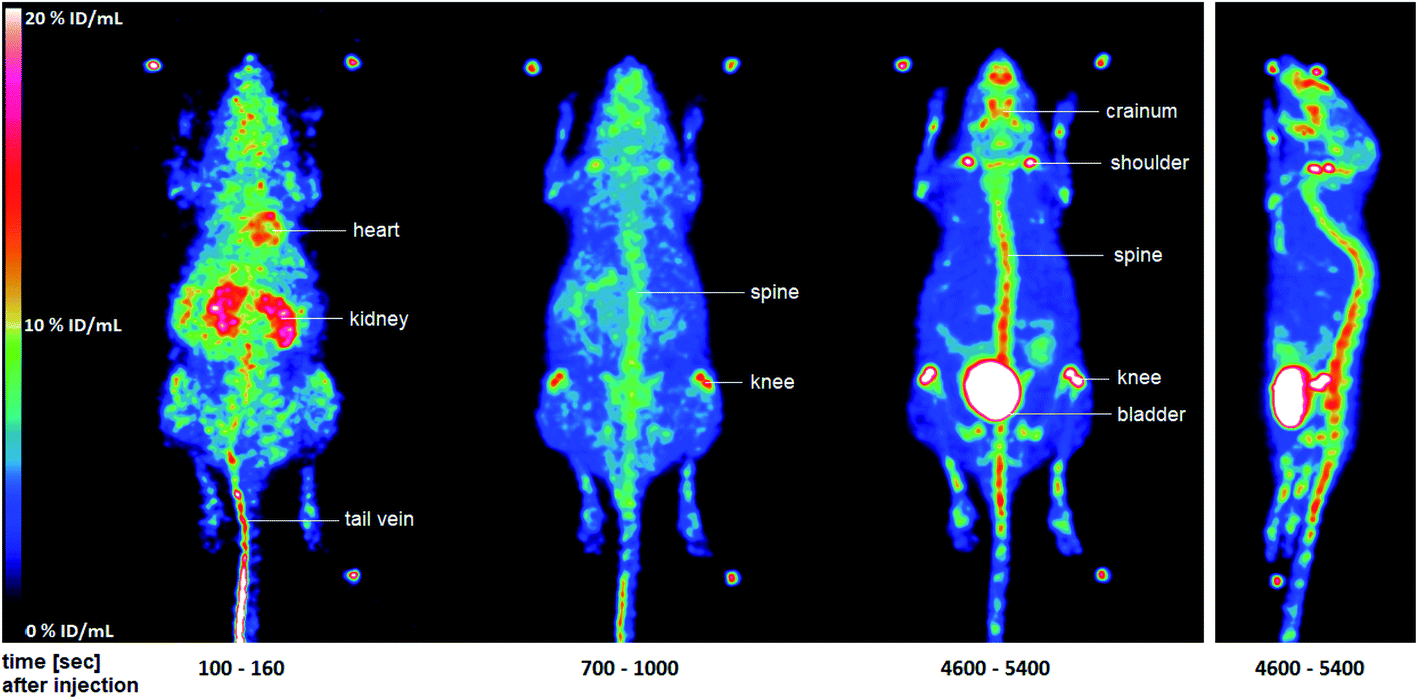 | ||
| Fig. 5 Maximum-intensity-projection of the activity distribution of selected time intervals in a C57BL/6 wild-type mouse after injection of [18F]28. The two additional images of mice measured may be found in the ESI (Fig. S8†). | ||
Fig. 5 shows the distribution of radioactivity expressed as percentage of the injected dose per mL (% ID/mL) in whole-body images after injection of [18F]28 in a representative mouse. After a few seconds, a large amount of the injected dose (ID) was still found in the tail vein [18F]28 also reached the heart via the blood. A significant amount of ID was already seen in the kidneys at this early stage. After 700–1000 s, accumulation in the bones, especially in the spine and knees, was detected. This accumulation increased substantially over the time. After 90 min, the highest amount of the ID was found in the bladder indicating renal elimination of the radioactive compound. However, large amounts of ID were also found in the spine, knees, shoulders, and cranium. The accumulation in the bones is probably due to the formation of [18F]fluoride. This finding indicates a metabolic instability of the tracer [18F]28 in vivo, which was not observed during the in vitro serum stability tests. Due to this metabolic instability, the PET tracer [18F]28 was not suitable for further tumor studies.
In order to analyze the metabolic instability of the tracer [18F]28 in more detail and provide information about possible degradation pathways, an in vitro metabolism study was conducted with the non-radioactive reference compound 28. Therefore, 28 was incubated with mouse liver microsomes and NADPH for 90 min at 37 °C. The transformation was stopped by the addition of CH3CN/CH3OH (1:1), the solution was cooled and then centrifuged to remove the protein precipitate. The supernatant was analyzed by LC-MS/MS (qToF) (Fig. 6).
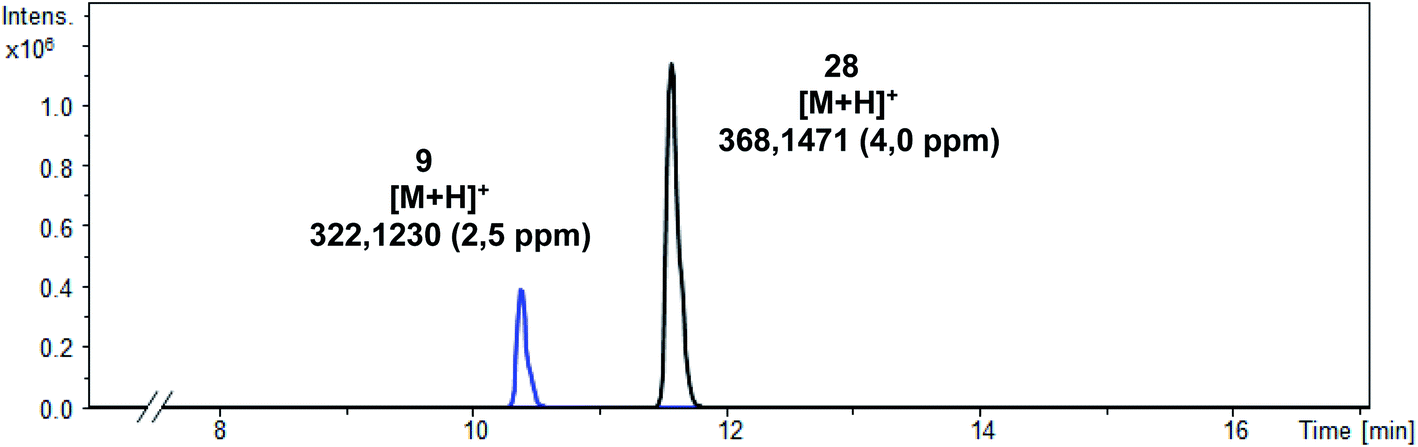 | ||
| Fig. 6 LC-MS/MS chromatogram of the supernatant. | ||
In addition to the intact parent compound 28, a more polar metabolite with m/z = 322.1230 was detected. Since ESI is a soft ionization method, only limited fragmentation reactions were expected. Therefore, the peak of the metabolite probably corresponds to [M + H] indicating the formation of phenol 9 (calculated exact mass of [M + H]+: 322.1238) as main metabolite (Scheme 10).
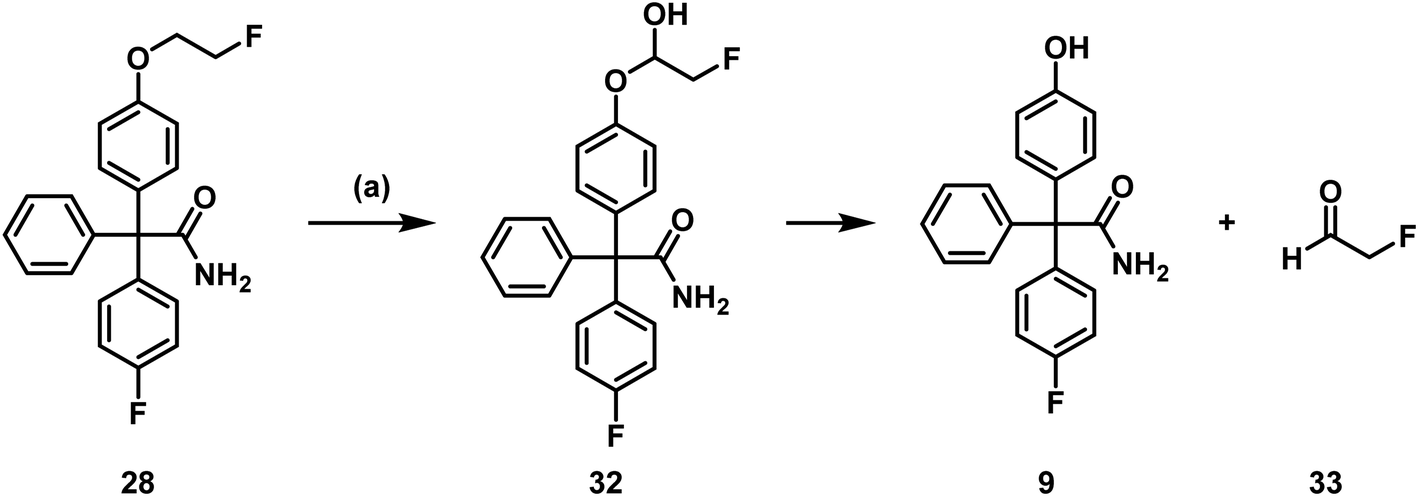 | ||
| Scheme 10 Postulated metabolism pathway of 28. (a) Incubation with NADPH and murine liver microsomes, 37 °C, 90 min. | ||
Phenol 9 can be formed by oxidation of the fluoroethoxy side chain of 28 by CYP450 enzymes and NADPH resulting in the labile hemi acetal 32, which releases the phenol 9 and 2-fluoroacetaldehyde (33) spontaneously. 2-Fluoroacetaldehyde or its oxidation product 2-fluoroacetic acid can release fluoride due to the high reactivity of the F-atom in α-position of the carbonyl moiety.
3. Conclusion
Introduction of [18F]fluoride in the aromatic ring resulting in [18F]senicapoc ([18F]1) required high temperature and strong bases. Unfortunately, the triarylacetamide moiety was not stable under these conditions leading to triarylmethane derivatives. Thus, only traces of [18F]senicapoc ([18F]1) were obtained by concerted nucleophilic substitution using the uronium salt precursor 10.As an alternative PET tracer targeting KCa3.1 channels the [18F]fluoroethoxy derivative [18F]28 was designed for imaging in vivo. Affinity towards the ion channel was shown in patch clamp experiments using the non-radioactive reference compound 28. Radiosynthesis of [18F]28 was performed successfully by aliphatic nucleophilic substitution of the tosylated precursor 31 with [18F]fluoride under mild conditions [18F]28 was obtained with a decay corrected radiochemical yield of 4 ± 1.5% (n = 4) within 106 ± 11.5 min (n = 4) and a radiochemical purity of >96% by analytical radio-HPLC and radio-TLC.
Over a period of 120 min, [18F]28 was stable in both human and mouse serum. The logD7.4 value of 1.84 lies in a promising range. More than 99% of the drug was bound to human serum albumin. During biodistribution studies with [18F]28 in vivo using C57/BL6 wild-type mice, accumulation of activity in the bones was observed indicating the release of [18F]fluoride from the tracer. In order to confirm the degradation of [18F]28 in vivo, an in vitro metabolism study was conducted with 28. The phenol 9 was detected as main metabolite, which was formed upon release of [18F]2-fluoroacetaldehyde. The release of [18F]2-fluoroacetaldehyde could explain the enrichment of [18F]fluoride in the bones. This finding is of general interest, since the [18F]2-fluoroethyl moiety is a well-known prosthetic group often used in PET tracers.45,46 The potential degradation to afford [18F]2-fluoroacetaldehyde should be generally taken into account, when designing novel PET tracers with the [18F]2-fluoroethyl moiety or using the popular prosthetic group [18F]fluoroethyl tosylate to alkylate phenols.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
We are grateful to Elke Naß, Institute of Physiology I, for performing the patch-clamp experiments with 28 and to Sandra Schimmelpfennig and Sarah Sargin, Institue of Physiology II, for cultivation and preparation of the A549-3R cells. This work was supported by the Deutsche Forschungsgemeinschaft (DFG), which is gratefully acknowledged. Moreover, we are grateful to Cells-in-Motion (CiM) Cluster of Excellence for supporting this project by a Pilot Project Fund. Furthermore, funding of the research training group “Chemical biology of ion channels (Chembion)” by the DFG is gratefully acknowledged. We acknowledge support from the Open Access Publication Fund of the University of Muenster. Furthermore, we are grateful to the Interdisciplinary Centre for Clinical Research (IZKF, core unit PIX), Münster, Germany for supporting this project.Notes and references
- Z. H. Yu, J. R. Xu, Y. X. Wang, G. N. Xu, Z. P. Xu, K. Yang, D. Z. Wu, Y. Y. Cui and H. Z. Chen, Am. J. Respir. Cell Mol. Biol., 2013, 48, 685–693 CrossRef CAS PubMed.
- P. Bradding and H. Wulff, Br. J. Pharmacol., 2009, 157, 1330–1339 CrossRef CAS PubMed.
- K. I. Ataga and J. Stocker, Expert Opin. Invest. Drugs, 2009, 18, 231–239 CrossRef CAS PubMed.
- K. I. Ataga, W. R. Smith, L. M. de Castro, P. Swerdlow, Y. Saunthararajah, O. Castro, E. Vichinsky, A. Kutlar, E. P. Orringer, G. C. Rigdon and J. W. Stacker, Blood, 2008, 111, 3991–3997 CrossRef CAS PubMed.
- K. I. Ataga, E. P. Orringer, L. Styles, E. P. Vichinsky, P. Swerdlow, G. A. Davis, P. A. DeSimone and J. W. Stocker, Pharmacotherapy, 2006, 26, 1557–1564 CrossRef CAS PubMed.
- D. L. Tharp, B. R. Wamhoff, H. Wulff, G. Raman, A. Cheong and D. K. Bowles, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 1084–1089 CrossRef CAS PubMed.
- I. Grgic, E. Kiss, B. P. Kaistha, C. Busch, M. Kloss, J. Sautter, A. Müller, A. Kaistha, C. Schmidt, G. Raman, H. Wulff, F. Strutz, H. J. Gröne, R. Köhler and J. Hoyer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14518–14523 CrossRef CAS PubMed.
- C. Huang, S. Shen, Q. Ma, J. Chen, A. Gill, C. A. Pollock and X. M. Chen, Diabetes, 2013, 62, 2923–2934 CrossRef CAS PubMed.
- R. Xu, C. Li, Y. Wu, L. Shen, J. Ma, J. Qian and J. Ge, Arterioscler., Thromb., Vasc. Biol., 2017, 37, 226–236 CrossRef CAS PubMed.
- Y. R. Zhu, X. X. Jiang and D. M. Zhang, J. Mol. Med., 2019, 97, 1219–1229 CrossRef PubMed.
- G. Anumanthan, S. Gupta, M. K. Fink, N. P. Hesemann, D. K. Bowles, L. M. McDaniel, M. Muhammad and R. R. Mohan, PLoS One, 2018, 13(3), e0192145 CrossRef PubMed.
- G. Anumanthan, P. J. Wilson, R. Tripathi, N. P. Hesemann and R. R. Mohan, Exp. Eye Res., 2018, 167, 140–144 CrossRef CAS PubMed.
- L. Jin, J. di Lucente, H. M. Nguyen, V. Singh, L. Singh, M. Chavez, T. Bushong, H. Wulff and I. Maezawa, Ann. Clin. Transl. Neurol., 2019, 6, 723–738 CrossRef CAS PubMed.
- I. Maezawa, D. P. Jenkins, B. E. Jin and H. Wulff, Int. J. Alzheimer's Dis., 2012, 2012 Search PubMed.
- M. Yi, P. Yu, Q. Lu, H. M. Geller, Z. Yu and H. Chen, Mol. Cell. Neurosci., 2016, 76, 21–32 CrossRef CAS PubMed.
- L. Sevelsted Møller, A. D. Fialla, R. Schierwagen, M. Biagini, C. Liedtke, W. Laleman, S. Klein, W. Reul, L. Koch Hansen, M. Rabjerg, V. Singh, J. Surra, J. Osada, R. Reinehr, O. B. S. de Muckadell, R. Köhler and J. Trebicka, Sci. Rep., 2016, 6, 28770 CrossRef PubMed.
- Z. Pethő, K. Najder, E. Bulk and A. Schwab, Cell Calcium, 2019, 80, 79–90 CrossRef PubMed.
- C. J. Mohr, D. Gross, E. C. Sezgin, F. A. Steudel, P. Ruth, S. M. Huber and R. Lukowski, Cancers, 2019, 11, 1285 CrossRef CAS PubMed.
- N. Prevarskaya, R. Skryma and Y. Shuba, Physiol. Rev., 2018, 98, 559–621 CrossRef CAS PubMed.
- K. L. Turner, A. Honasoge, S. M. Robert, M. M. Mcferrin and H. Sontheimer, Glia, 2014, 62, 971–981 CrossRef PubMed.
- M. Rabjerg, A. Oliván-Viguera, L. K. Hansen, L. Jensen, L. Sevelsted-Møller, S. Walter, B. L. Jensen, N. Marcussen and R. Köhler, PLoS One, 2015, 10, e0122992 CrossRef PubMed.
- M. Faouzi, F. Hague, D. Geerts, A. S. Ay, M. Potier-Cartereau, A. Ahidouch and H. Ouadid-Ahidouch, Oncotarget, 2016, 7, 36419–36435 CrossRef PubMed.
- N. Haren, H. Khorsi, M. Faouzi, A. Ahidouch, H. Sevestre and H. Ouadid-Ahidouch, Histol. Histopathol., 2010, 25, 1247–1255 CAS.
- E. Bulk, A.-S. Ay, M. Hammadi, H. Ouadid-Ahidouch, S. Schelhaas, A. Hascher, C. Rohde, N. H. Thoennissen, R. Wiewrodt, E. Schmidt, A. Marra, L. Hillejan, A. H. Jacobs, H.-U. Klein, M. Dugas, W. E. Berdel, C. Müller-Tidow and A. Schwab, Int. J. Cancer, 2015, 137, 1306–1317 CrossRef CAS PubMed.
- J. H. Ko, E. A. Ko, W. Gu, I. Lim, H. Bang and T. Zhou, Mol. Cancer, 2013, 12, 106 CrossRef PubMed.
- F. A. Steudel, C. J. Mohr, B. Stegen, H. Y. Nguyen, A. Barnert, M. Steinle, S. Beer-Hammer, P. Koch, W. Y. Lo, W. Schroth, R. Hoppe, H. Brauch, P. Ruth, S. M. Huber and R. Lukowski, Mol. Oncol., 2017, 11, 1172–1188 CrossRef CAS PubMed.
- H. Wulff and N. A. Castle, Expert Rev. Clin. Pharmacol., 2010, 3, 385–396 CrossRef CAS PubMed.
- C. Gross, Epilepsy Currents, 2020, 20, 211–213 CrossRef PubMed.
- S. Sugunan, S. Nampoothiri, T. Garg and R. Krishnamurthy, CNS Neurol. Disord.: Drug Targets, 2016, 15, 1299–1305 CrossRef CAS PubMed.
- V. Hofschröer, K. Najder, M. Rugi, R. Bouazzi, M. Cozzolino, A. Arcangeli, G. Panyi and A. Schwab, Front. Pharmacol., 2021, 11, 1979 Search PubMed.
- J. W. Stocker, L. de Franceschi, G. A. McNaughton-Smith, R. Corrocher, Y. Beuzard and C. Brugnara, Blood, 2003, 101, 2412–2418 CrossRef CAS PubMed.
- B. I. Mobele, S. Venkatraman, G. McNaughton-Smith, C. Gibb, L. G. Ulysse, C. A. Lindmark, S. Shaw, B. Marron, K. Spear and M. J. Suto, Org. Process Res. Dev., 2012, 16, 1385–1392 CrossRef CAS.
- C. N. Neumann, J. M. Hooker and T. Ritter, Nature, 2016, 534, 369–373 CrossRef CAS PubMed.
- T. Fujimoto, F. Becker and T. Ritter, Org. Process Res. Dev., 2014, 18, 1041–1044 CrossRef CAS PubMed.
- K. Brömmel, S. Maskri, I. Maisuls, C. P. Konken, M. Rieke, Z. Pethő, C. A. Strassert, O. Koch, A. Schwab and B. Wünsch, Angew. Chem., Int. Ed., 2020, 59, 8277–8284 CrossRef PubMed.
- M. Lasch, A. Caballero Martinez, K. Kumaraswami, H. Ishikawa-Ankerhold, S. Meister and E. Deindl, Cells, 2020, 9(4), 913 CrossRef CAS PubMed.
- S. A. Nicolaou, L. Neumeier, Y. Q. Peng, D. C. Devor and L. Conforti, Am. J. Physiol.: Cell Physiol., 2007, 292(4), 1431–1439 CrossRef PubMed.
- E. Cole, M. Stewart, R. Littich, R. Hoareau and P. Scott, Curr. Top. Med. Chem., 2014, 14, 875–900 CrossRef CAS PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- M. E. Sergeev, F. Morgia, M. Lazari, C. Wang and R. M. van Dam, J. Am. Chem. Soc., 2015, 137, 5686–5694 CrossRef CAS PubMed.
- M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751–7755 CrossRef CAS PubMed.
- F. Dollé, F. Hinnen, A. Damont, B. Kuhnast, C. Fookes, T. Pham, B. Tavitian and A. Katsifis, J. Labelled Compd. Radiopharm., 2008, 51, 435–439 CrossRef.
- S. Comagic and R. Schirrmacher, Synthesis, 2004, 2004, 885–888 Search PubMed.
- O. Prante, C. Hocke, S. Löber, H. Hübner, P. Gmeiner and T. Kuwert, Nuklearmedizin, 2006, 45, 41–48 CrossRef CAS PubMed.
- W. Mohnike, G. Hör, A. Hertel, H. Schelbert, PET/CT-Atlas, Springer, Berlin, Heidelberg, 2016 Search PubMed.
- Z. Li and P. S. Conti, Adv. Drug Delivery Rev., 2010, 62, 1031–1051 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03850h |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |