
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile method for Rh-catalyzed decarbonylative ortho-C–H alkylation of (hetero)arenes with alkyl carboxylic acids†
Yiqiang Tianab,
Xiaojie Liuab,
Bangyue Heb,
Yuxi Ren*b and
Weiping Su *b
*b
aCollege of Chemistry, Fuzhou University, Fuzhou 350116, China
bState Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, China. E-mail: wpsu@fjirsm.ac.cn; yxren@fjirsm.ac.cn
First published on 2nd June 2021
Abstract
A facile and effective method for Rh-catalyzed direct ortho-alkylation of C–H bonds in (hetero)arenes with commercially available carboxylic acids has been developed. This strategy was initiated by in situ conversion of carboxylic acids to anhydrides which, without isolation, underwent Rh-catalyzed direct decarbonylative cross-coupling of aryl carboxamides containing 8-aminoquinoline. The reaction proceeds with high regioselectivity and exhibits a broad substrate scope as well as functional group tolerance.
Alkylation of (hetero)arenes1 is one of the most fundamental reactions in synthetic chemistry, leading to ubiquitous alkylated scaffolds and revealing itself to be of great significance with widespread application in fine chemicals, pharmaceuticals, agrochemicals and so forth. One of the classical methods for the C–H alkylation of arenes is the Friedel–Crafts reaction,2 one of the oldest organic transformations and still a commonly used protocol nowadays which, however, suffers from severe limitations such as poor reactivity of electron-poor aromatic substrates, undesired cationic rearrangement, and low chemo- and/or regioselectivity. Recently, oxidative decarboxylative coupling of aliphatic carboxylic acids3,4 has provided complementary access to Friedel–Crafts reactions with opposite reactivity and selectivity. However, in this decarboxylative coupling, the substrate scopes of carboxylic acids were mainly restricted to arylacetic acids, secondary and tertiary alkyl acids, or alkyl acids with a stabilized atom (such as N, O, S) at the α-position of the carboxyl group. Additionally, regioselectivity in simple (hetero)arenes remains challenging.
These well-established limitations have encouraged the development of alternative metal-catalyzed directed alkylation of (hetero)arenes C–H bonds,5 one of the most accurate and effective tools, therein, highly regioselectivity mostly relies on the use of a directing group by allowing the metal center proximally close to the target C–H bonds in the starting (hetero)arenes. To date, this directed C–H alkylation of (hetero)arenes undergoes with diverse alkylating agents within which alkenes5j,l,p and alkyl halides5c are mostly used reagents. To avoid the multiple steps or limitations in synthesis of these agents from available starting materials, as well as to reduce the discharge of poisonous by-products, there is a need to explore novel and convenient alkyl donors beyond these commonly used reagents. Carboxylic anhydrides6 thereof have attracted considerable attention not only owing to their low cost and nontoxicity, but also the easy obtainment from commercially available carboxylic acids. Driven by their electron deficiency, the activated anhydrides may serve as potent alkylating sources in the metal-catalyzed direct decarbonylative coupling reaction of (hetero)arenes which is triggered by metal-catalyzed oxidative addition of a C(acyl)–O bond. Notably, this direct decarbonylative alkylation no longer confined to the use of ortho-substituted aromatic carboxylic acids which are required in conventional decarboxylative cross-coupling reactions.7 Following their first example of the decarbonylative methylation of arenes with benzoic acids via RhI/(tBuCO)2O catalytic system,6c Z.-J. Shi and co-workers further extended this concept to afford alkylated products, enabling to introduce methyl, ethyl, benzyl and phenethyl groups onto cyclic enamines,6d and later achieved methylation of indoles.6e In their research, the presence of a monodentate N-directing groups and the in situ generation of mixed anhydrides were crucial. Using a similar protocol, Z. Shi and co-workers developed Rh-catalyzed methylation of indoles with Ac2O in the presence of a PIII-directing groups.6g P. Walsh and co-workers demonstrated an analogous access to Rh-catalyzed C6-alkylated 2-pyridones with the assist of pyridine as the directing group, installing long chains and cyclic rings onto N-heteroarenes (Scheme 1a).6h Despite these significant progresses in recent years, there is still much room for improvement of this decarbonylative alkylation, particularly in terms of substrate scopes and functional group tolerance for both the starting (hetero)arenes and alkyl sources.
Encouraged by Daugulis's pioneering work and others' previous studies,5 herein we select 8-aminoquinoline (AQ), an excellent N,N-bidentate directing group in catalytic functionalization of C–H bonds,8,9 as the installed moiety on the starting (hetero)arenes, and expect to develop a general method for Rh-catalyzed decarbonylative C–H alkylation of (hetero)arenes with in situ generated alkyl carboxylic acid anhydrides (Scheme 1b).
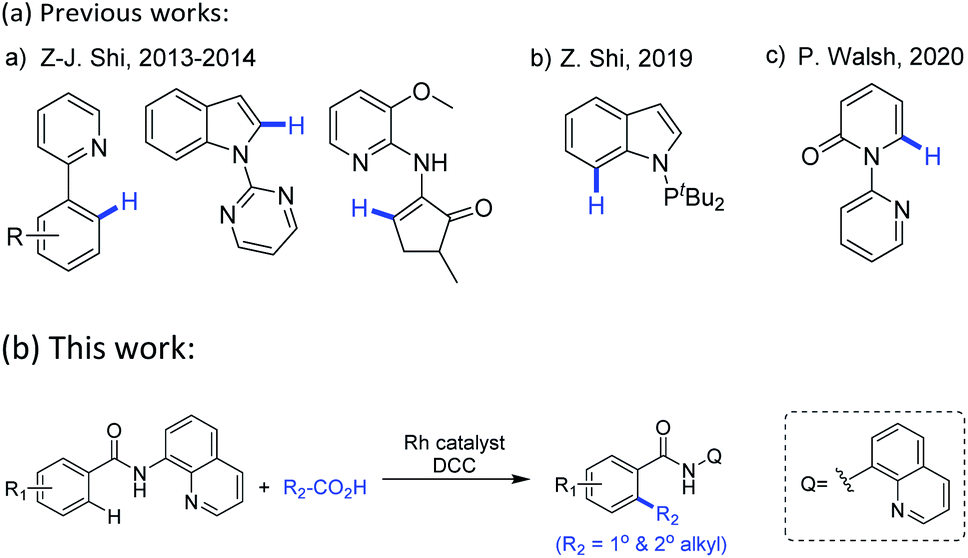 | ||
| Scheme 1 Transition-metal-catalyzed chelation-assisted decarbonylative alkylation reactions of (hetero)arenes with alkyl carboxylic acids or anhydrides. | ||
Based on our knowledge, we initially chose N,N′-dicyclohexylcarbodiimide (DCC) as the additive for the stoichiometric conversion of alkyl carboxylic acids into the corresponding anhydrides,10 and began our studies with a thorough optimization for this Rh-catalyzed C–H alkylation of AQ-substituted benzamide 1a with propionic acid 2a (Table 1). To our delight, the desired ortho-alkylated product 3a was delivered in the presence of 2.5 mol% [Rh(COD)Cl2] as the catalyst, DCC (3 equiv.) and Na2CO3 (3 equiv.) under N2 in toluene (1.5 ml) at 140 °C for 12 h (entry 1). The control experiments revealed that [Rh(COD)Cl2], DCC and Na2CO3 all were essential to this reaction (entries 2–4). Compared with other frequently employed RhI catalysts, [Rh(COD)Cl2] exhibited the most satisfied efficiency (see Table S1 in the ESI†). Na2CO3 was identified as potent base when compared with NaHCO3 and K2CO3 (entries 5 and 6). Using N,N′-diisopropylcarbodiimide (DIC) as an additive in the activation of carboxylic acid led to a drop in yield (entry 7). Lowing the reaction temperature hindered the reaction, probably because the high temperature was required for the decarbonylation step (entries 8 and 9). The use of polar solvent such as 1,4-dioxane gave a decreased yield (entry 10). Interestingly, this reaction still occurred in air, albeit with a lower yield, indicating its promising application in the practical synthesis (entry 11).
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol).b GC yield of 3a.c Yield of isolated 3a. DCC (0.6 mmol, N,N′-dicyclohexylcarbodiimide) DIC (0.6 mmol, N,N′-diisopropylcarbodiimide). | ||
| 1 | None | 75 (71c) |
| 2 | Without [Rh(COD)Cl]2 | NA |
| 3 | Without DCC | NA |
| 4 | Without Na2CO3 | NA |
| 5 | NaHCO3 instead of Na2CO3 | 60 |
| 6 | K2CO3 instead of Na2CO3 | 34 |
| 7 | DIC instead of DCC | 57c |
| 8 | 130 °C instead of 140 °C | 52 |
| 9 | 120 °C instead of 140 °C | 31 |
| 10 | 1,4-Dioxane as the solvent | 54 |
| 11 | Air instead of N2 | 59 |
With the optimized reaction condition in hand, we next examined the generality of this method by exploring the substrate scopes of alkyl carboxylic acids and 8-AQ-containing benzamides (Scheme 2). Gratifyingly, this protocol afforded expected alkylation and successfully introduced a vast set of primary and secondary alkyl chains on the ortho-position of the benzamide motif (3a–3r). Various functional groups on the scaffolds of linear aliphatic carboxylic acids, including chloro (3d), bromo (3e), ester (3f), alkenyl (3g, 3h) and alkyne (3i), were all compatible. It's worth noting that terminal C–Cl, C–Br and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in alkyl carboxylic acids hydrides remained intact (3d, 3e, 3g), indicating that hydrides might be applied as complementary alkylating agents to the commonly used alkyl halides or alkenes at present. Though metal-catalyzed cross-coupling with alkyl carboxylic acid derivatives bearing β-hydrogens are rather challenging,11 to our delight, no C–H alkenylation occurred in this Rh-catalyzed decarbonylative coupling of 1a with 3g, 3j, 3k and 3l which are inclined to form stable conjugated side products via β-hydride elimination process, ruling out the possible β-H elimination pathway in this catalytic cycle. The use of 8-AQ might be the key in this transformation which occupies the site of coordinative unsaturation on the metal cis to the alkyl group by flexible ligand association/dissociation and thus suppresses the possible β-H elimination.12 Besides, secondary alkyl carboxylic acids, including branched acids (3m) and cyclic acids with different ring size (3n–3r), delivered the desired alkylation products in moderate to good yield. Interestingly, this protocol was not limited to C–H alkylation of aryl benzamides, direct C–H arylation (3s) and alkenylation (3t) were also achieved under the standard conditions, demonstrating its promising utility in synthetic chemistry. Moreover, as shown in Scheme 3, this Rh-catalyzed method enabled ortho-C–H ethylation of diversely 8-AQ decorated amides containing electron-donating and electron-withdrawing substitutes on the arene rings (4b–4f), and tolerated C–Cl and C–Br bonds (4g, 4h). Polycyclic arene (4i) and heteroaryl arenes (4j–4l) also proved to be compatible with satisfactory yield. Thus, this protocol exhibits its broad substrate scope and implies its potential application.
C bonds in alkyl carboxylic acids hydrides remained intact (3d, 3e, 3g), indicating that hydrides might be applied as complementary alkylating agents to the commonly used alkyl halides or alkenes at present. Though metal-catalyzed cross-coupling with alkyl carboxylic acid derivatives bearing β-hydrogens are rather challenging,11 to our delight, no C–H alkenylation occurred in this Rh-catalyzed decarbonylative coupling of 1a with 3g, 3j, 3k and 3l which are inclined to form stable conjugated side products via β-hydride elimination process, ruling out the possible β-H elimination pathway in this catalytic cycle. The use of 8-AQ might be the key in this transformation which occupies the site of coordinative unsaturation on the metal cis to the alkyl group by flexible ligand association/dissociation and thus suppresses the possible β-H elimination.12 Besides, secondary alkyl carboxylic acids, including branched acids (3m) and cyclic acids with different ring size (3n–3r), delivered the desired alkylation products in moderate to good yield. Interestingly, this protocol was not limited to C–H alkylation of aryl benzamides, direct C–H arylation (3s) and alkenylation (3t) were also achieved under the standard conditions, demonstrating its promising utility in synthetic chemistry. Moreover, as shown in Scheme 3, this Rh-catalyzed method enabled ortho-C–H ethylation of diversely 8-AQ decorated amides containing electron-donating and electron-withdrawing substitutes on the arene rings (4b–4f), and tolerated C–Cl and C–Br bonds (4g, 4h). Polycyclic arene (4i) and heteroaryl arenes (4j–4l) also proved to be compatible with satisfactory yield. Thus, this protocol exhibits its broad substrate scope and implies its potential application.
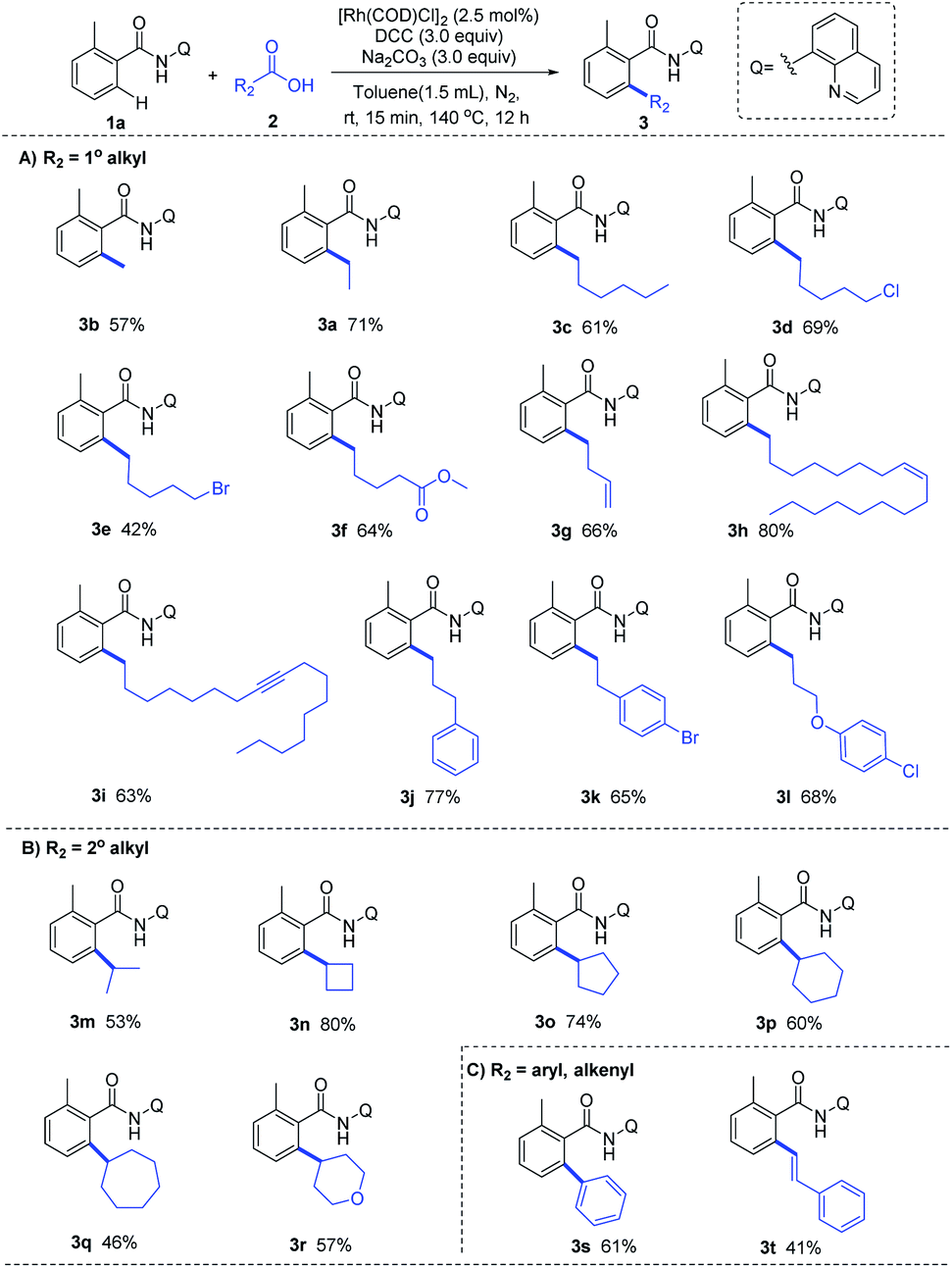 | ||
| Scheme 2 Substrate scope of carboxylic acids. | ||
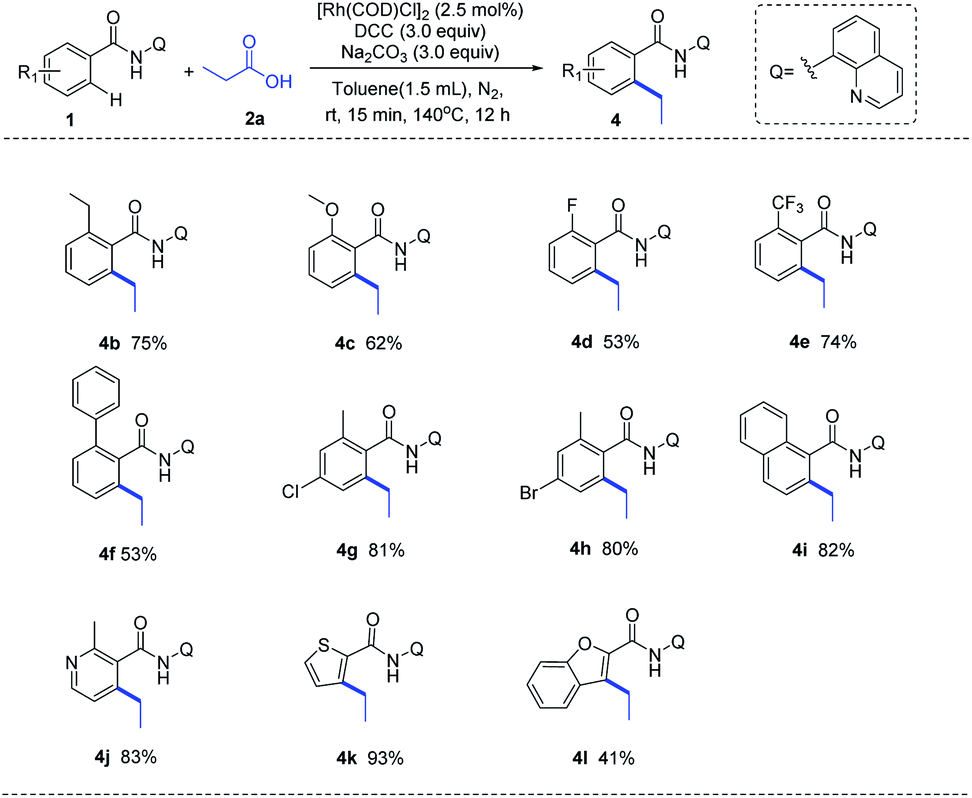 | ||
| Scheme 3 Substrate scope of aromatic amides. | ||
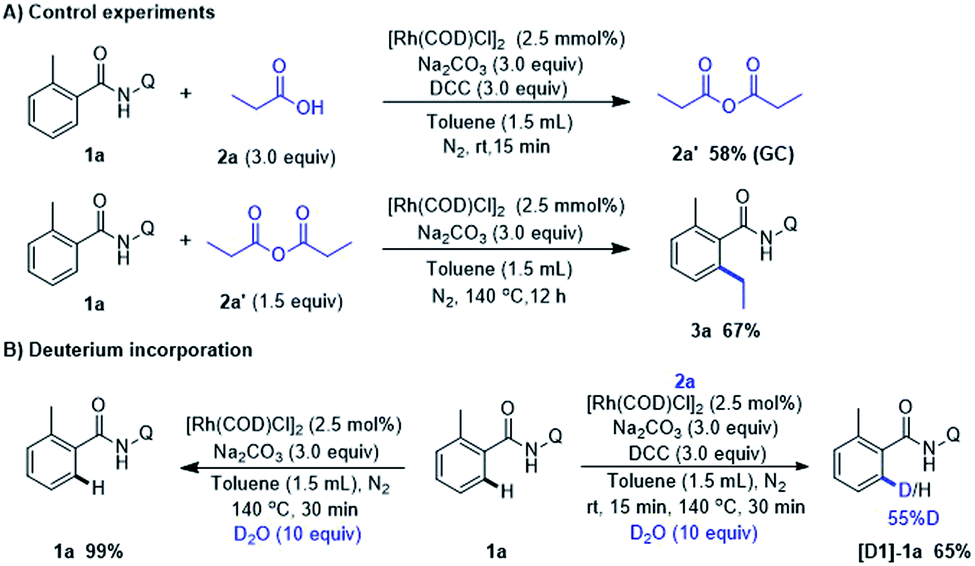
To gain insight of this Rh-catalyzed decarbonylative coupling reaction, a series of experiments was carried out (Scheme 4). In order to observe and verify the formation anhydride, the control experiment was conducted with amide (1a) and propionic acid (2a) under standard condition. The reaction was quenched after 15 min pre-stirring at room temperature and 58% yield of propionic anhydride (2a′) was detected. Then we performed experiment with 1a and possible intermediates 2a′ to confirm whether the alkylation product 3a could be formed. Notwithstanding a slightly drop in yield when compared with the output under the standard conditions (67% in Scheme 4A vs. 71% in Scheme 2, 3a). It consists with the hypothesis that in situ conversion of carboxylic acid into anhydride comprises the basic steps of this Rh-catalyzed alkylation reaction. When 1a was reacted with D2O for 30 min under otherwise standard conditions (Scheme 4B), we observed a significant difference of H/D exchange with or without 2a, which indicating the fact that RhI species did not react with ortho-C–H bond of 1a via catalyzed C–H activation even with the assistance of bidentate directing group. Instead, ortho-C–H bond of 1a was activated by RhIII complex which was formed by RhI oxidatively inserting into C(acyl)–O bond in anhydride.
 | ||
| Scheme 4 Mechanistic studies. | ||
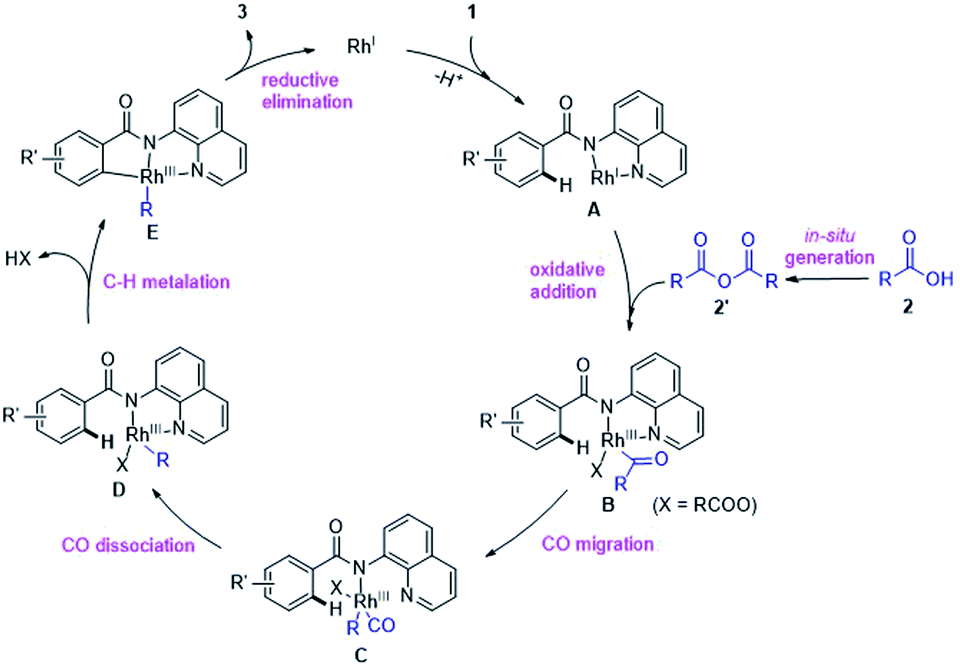
Thus, we propose a plausible catalytic pathway different from the previous researches,6c–e,g which likely involves: (i) in situ conversion of carboxylic acid 2 into anhydride 2′, (ii) oxidative addition of C(acyl)–O bond in 2′ by RhI species A, (iii) decarbonylation, (iv) chelation-assisted C–H cyclometalation and (v) C–C bond-forming reductive elimination to release the product 3 and regenerates RhI to propagate the reaction cycle (Scheme 5).
 | ||
| Scheme 5 Plausible mechanism. | ||
In conclusion, we have developed a facile method for Rh-catalyzed direct ortho-C–H alkylation of (hetero)arenes with readily available carboxylic acids, which involving an initial step of in situ conversion of carboxylic acids to the corresponding anhydrates and the subsequent Rh-catalyzed decarbonylative cross-coupling of the resultant anhydrates with (hetero)arenes. This reaction proceeds in highly regioselectivity by identifying 8-aminoquinoline as the efficient bidentate directing group embedded on the starting (hetero)arenes. Enabling a diversity of primary and secondary carboxylic acids as well as various benzamide derivatives as the cross-coupling substrates, our strategy reveals its promising utility in synthetic chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFA0704502), the National Natural Science Foundation of China (Grants No. 21931011, 22071241), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000 and XDB10040304), and the Key Research Program of Frontier Sciences, CAS (QYZDJ-SSW-SLH024).Notes and references
- For selective reviews on C–H alkyaltion, see: (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (c) E. J. E. Caro-Diaz, M. Urbano, D. J. Buzard and R. M. Jones, Bioorg. Med. Chem. Lett., 2016, 26, 5378 CrossRef CAS PubMed; (d) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7558 CrossRef CAS PubMed.
- (a) R. M. Roberts and A. A. Khalaf, Friedel-Crafts Alkylation Chemistry: A Century of Discovery, Marcel Dekker, New York, 1984 Search PubMed; (b) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS PubMed; (c) T. B. Poulsen and K. A. Jorgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS PubMed.
- For selective reviews on oxidative decarboxylative cross-coupling of alkyl carboxylic acids, see: (a) J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed; (b) M. O. Konev and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 11340 CrossRef CAS PubMed; (c) P. Liu, G. Zhang and P. Sun, Org. Biomol. Chem., 2016, 14, 10763 RSC; (d) H. Bao, Y. Li, L. Ge and M. Muhammad, Synthesis, 2017, 49, 5263 CrossRef; (e) T. Patra and D. Maiti, Chem, 2017, 23, 7382 CrossRef CAS PubMed.
- The selective examples of oxidative decarboxylative cross-coupling of alkyl carboxylic acids, see: (a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (b) M. Rueda-Becerril, O. Mahe, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J. F. Paquin, J. Am. Chem. Soc., 2014, 136, 2637 CrossRef CAS PubMed; (c) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (d) Q. Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef CAS PubMed; (e) Z. J. Liu, X. Lu, G. Wang, L. Li, W. T. Jiang, Y. D. Wang, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2016, 138, 9714 CrossRef CAS PubMed; (f) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed.
- The selective reviews on metal-catalyzed directed alkylation of (hetero)arenes, see: (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (d) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (e) M. Zhang, Y. F. Zhang, X. M. Jie, H. Q. Zhao, G. Li and W. P. Su, Org. Chem. Front., 2014, 1, 843 RSC; (f) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (g) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed; (h) G. Zhang, F. Jia and L. J. Goossen, Chem, 2018, 24, 4537 CrossRef CAS PubMed; (i) N. Chatani, Bull. Chem. Soc. Jpn., 2018, 91, 211 CrossRef CAS; (j) C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (k) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456 RSC; (l) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (m) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7202 CrossRef CAS PubMed; (n) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (o) S. B. Ankade, A. B. Shabade, V. Soni and B. Punji, ACS Catal., 2021, 11, 3268 CrossRef CAS; (p) T. Yamaguqi, S. Natsui, K. Shibata, K. Yamazaki, S. Rej, Y. Ano and N. Chatani, Chem.–Eur. J., 2019, 25, 6915 CrossRef PubMed.
- The selective examples of decarbonylative cross-coupling with anhydrides or carboxylic acids, see: (a) E. M. O'Brien, E. A. Bercot and T. Rovis, J. Am. Chem. Soc., 2003, 125, 10498 CrossRef PubMed; (b) W. Jin, Z. Yu, W. He, W. Ye and W. J. Xiao, Org. Lett., 2009, 11, 1317 CrossRef CAS PubMed; (c) F. Pan, Z. Q. Lei, H. Wang, H. Li, J. Sun and Z. J. Shi, Angew. Chem., Int. Ed., 2013, 52, 2063 CrossRef CAS PubMed; (d) Z.-Q. Lei, J.-H. Ye, J. Sun and Z.-J. Shi, Org. Chem. Front., 2014, 1, 634 RSC; (e) L. Zhang, X. Xue, C. Xu, Y. Pan, G. Zhang, L. Xu, H. Li and Z. Shi, ChemCatChem, 2014, 6, 3069 CrossRef CAS; (f) C. Liu, C. L. Ji, X. Hong and M. Szostak, Angew. Chem., Int. Ed., 2018, 57, 16721 CrossRef CAS PubMed; (g) X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504 CrossRef CAS PubMed; (h) H. Zhao, X. Xu, H. Yu, B. Li, X. Xu, H. Li, L. Xu, Q. Fan and P. J. Walsh, Org. Lett., 2020, 22, 4228 CrossRef CAS PubMed.
- The studies on ortho-substituent effect on metal-catalyzed decarboxylation, see: (a) J. Cornella, C. Sanchez, D. Banawa and I. Larrosa, Chem. Commun., 2009, 46, 7176 RSC; (b) L. Xue, W. Su and Z. Lin, Dalton Trans., 2011, 40, 11926 RSC; (c) R. Grainger, J. Cornella, D. C. Blakemore, I. Larrosa and J. M. Campanera, Chem.–Eur. J., 2014, 20, 16680 CrossRef CAS PubMed.
- The selective examples of 8-aminoquinoline-directed C–H functionalization, see: (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed; (c) S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed; (d) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477 CrossRef CAS PubMed; (e) E. R. Fruchey, B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 13130 CrossRef CAS PubMed; (f) L. Ilies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126 CrossRef CAS PubMed; (g) B. M. Monks, E. R. Fruchey and S. P. Cook, Angew. Chem., Int. Ed., 2014, 53, 11065 CrossRef CAS PubMed; (h) S. Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531 CrossRef CAS PubMed; (i) R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 7660 CrossRef CAS PubMed; (j) J. Tang, P. Liu and X. Zeng, Chem. Commun., 2018, 54, 9325 RSC; (k) R. C. Samanta, J. Struwe and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 14154 CrossRef CAS PubMed.
- The selective examples of directed C–H functionalization of (hetero)arenes, see: (a) Y. H. Cheng, S. J. Yu, Y. H. He, G. H. An, G. M. Li and Z. Y. Yang, Chem. Sci., 2021, 12, 3216 RSC; (b) C. Tian, Q. Wang, X. Wang, G. H. An and G. M. Li, J. Org. Chem., 2019, 84, 14241 CrossRef CAS PubMed; (c) K. Korvorapun, N. Kaplaneris, T. Rogge, S. Warratz, A. C. Stückl and L. Ackermann, ACS Catal., 2018, 8, 886 CrossRef CAS.
- D. G. Stark, L. C. Morrill, D. B. Cordes, A. M. Slawin, T. J. O'Riordan and A. D. Smith, Chem.–Asian J., 2016, 11, 395 CrossRef CAS PubMed.
- The selective examples of conversion of alkyl acid derivates into alkenes via β-H elimination, see: (a) L. J. Goossen and N. Rodriguez, Chem. Commun., 2004, 724 RSC; (b) Y. Liu, S. C. Virgil, R. H. Grubbs and B. M. Stoltz, Angew. Chem., Int. Ed., 2015, 54, 11800 CrossRef CAS PubMed; (c) A. John, M. O. Miranda, K. Ding, B. Dereli, M. A. Ortuño, A. M. LaPointe, G. W. Coates, C. J. Cramer and W. B. Tolman, Organometallics, 2016, 35, 2391 CrossRef CAS; (d) J. Hu, M. Wang, X. Pu and Z. Shi, Nat. Commun., 2017, 8, 14993 CrossRef PubMed; (e) X. Zhang, F. Jordan and M. Szostak, Org. Chem. Front., 2018, 5, 2515 RSC.
- The studies on ligands suppressing β-H elimination for cross-coupling of alkyl nucleophiles, see: (a) J. S. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726 CrossRef CAS PubMed; (b) J. Breitenfeld, O. Vechorkin, C. m. Corminboeuf, R. Scopelliti and X. Hu, Organometallics, 2010, 29, 3686 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03992j |
| This journal is © The Royal Society of Chemistry 2021 |