
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of indazoles from 2-formylphenylboronic acids†
Vitalii V. Solominab,
Alberts Seinsab and
Aigars Jirgensons *ab
*ab
aLatvian Institute of Organic Synthesis, Aizkraukles 21, Riga, LV-1006, Latvia. E-mail: aigars@osi.lv
bFaculty of Materials Science and Applied Chemistry, Riga Technical University, P. Valdena Str. 3, Riga, LV-1048, Latvia
First published on 28th June 2021
Abstract
A method for the synthesis of indazoles was developed which involves a copper(II) acetate catalysed reaction of 2-formylboronic acids with diazadicaboxylates followed by acid or base induced ring closure. Hydrazine dicarboxylates were also shown as competent reaction partners for the synthesis of indazoles, however, they required a stoichiometric amount of copper(II) acetate for the C–N bond formation step. The transformation can be efficiently performed as a two step-one pot procedure to give a range of 1N-alkoxycarbonyl indazoles.
Introduction
The indazole motif plays an important role in pharmaceutically relevant compounds including drugs and candidate drugs e.g. Lonidamine, Gamendazole, Bendazac, Pazopanib, Axitinib (Fig. 1).1–4 A number of approaches have been developed to assemble indazole from 2-aminotoluenes,5 2-acyl-halobenzenes,6–8 2-aminophenyloximes,9 and 2-nitrobenzaldehydes,10,11 and by [3 + 2] annulations of in situ generated arynes.12–14 Most of the above mentioned methods lead to the defined 1N or 2N indazole substitution pattern, however, they require harsh conditions or long routes to the key intermediates limiting their application. Selective N-functionalization of indazoles has been reported for alkylation reactions15–17 and few reports can be found on selective N-acylation of indazoles.18 | ||
| Fig. 1 Indazole containing drugs (Lonidamine, Gamendazole, Bendazac, Pazopanib) and candidate drug (Axitinib). | ||
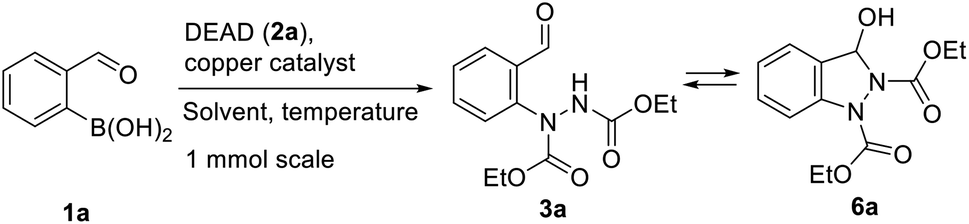
Previously, we demonstrated the construction of aminoquinazolines from 2-formylphenylboronic acids.19 This method involved the Chan–Evans–Lam reaction for the C–N bond formation. To extend this approach for the synthesis of indazoles, we turned our attention to copper catalysed addition of phenyl boronic acids to azodicarboxylates reported by Uemura and Chatani.20 Using 2-formylphenylboronic acids 1 as substrates, the addition to N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in azadicarboxylates 2 would give N-arylhydrazine intermediates 3 which could be further transformed to indazoles 4 and 5 (Scheme 1).
N bond in azadicarboxylates 2 would give N-arylhydrazine intermediates 3 which could be further transformed to indazoles 4 and 5 (Scheme 1).
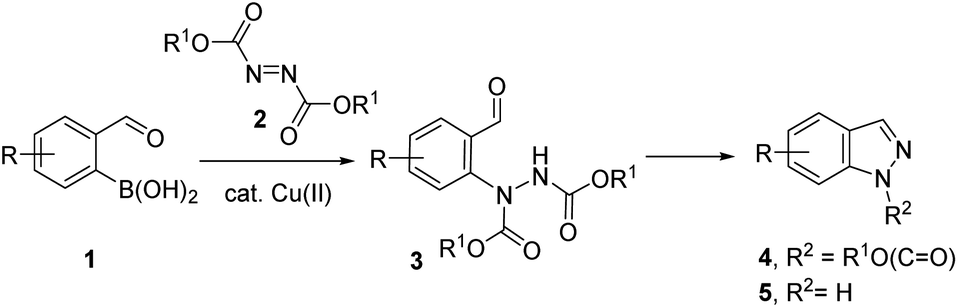 | ||
| Scheme 1 Indazole synthesis from 2-formylphenylboronic acids. | ||
Results and discussion
The initial investigation of the arylation conditions was performed for the reaction of 2-formylphenylboronic acid (1a) with diethylazodicarboxylate (DEAD, 2a) using Cu(OAc)2 as a catalyst in a range of solvents (Table 1, entries 1–9). Solvents such as MeCN, DMF and DMA were found to be appropriate to obtain the product 3a together with its cyclic tautomer 6a in a good yield (Table 1, entries 5 and 6). Decreased catalyst loading was also possible using DMA as a solvent without affecting the product 6a yield (Table 1, entries 7–9). Range of other copper sources was investigated (Table 1, entries 10–14). CuCl2 Cu(OTf)2 Cu(acac)2 performed as efficient catalysts for C–N bond formation giving the product 3a in high yield (Table 1, entries 10–12). Copper(I) source such as CuCl proved to be ineffective catalyst, while catalytic amount of CuI enabled product 3a formation in good yield (Table 1, entries 13 and 14).
|
|||
|---|---|---|---|
| Entry | Copper catalyst | Solvent | Isolated yield |
| a Violent DEAD decomposition observed.b N,N-Dimethylformamide.c N,N-Dimethylacetamide. | |||
| 1 | 20 mol% Cu(OAc)2 | MeOHa | 0% |
| 2 | 20 mol% Cu(OAc)2 | PhMe | 0% |
| 3 | 20 mol% Cu(OAc)2 | THF | 64% |
| 4 | 20 mol% Cu(OAc)2 | MeCN | 80% |
| 5 | 20 mol% Cu(OAc)2 | DMFb | 83% |
| 6 | 20 mol% Cu(OAc)2 | DMAc | 98% |
| 7 | 15 mol% Cu(OAc)2 | DMAc | 98% |
| 8 | 10 mol% Cu(OAc)2 | DMAc | 98% |
| 9 | 5 mol% Cu(OAc)2 | DMAc | 96% |
| 10 | 10 mol% CuCl2 | DMAc | 94% |
| 11 | 10 mol% Cu(OTf)2 | DMAc | 99% |
| 12 | 10 mol% Cu(acac)2 | DMAc | 97% |
| 13 | 10 mol% CuCl | DMAc | 25% |
| 14 | 10 mol% CuI | DMAc | 93% |
Next, the conditions were investigated for the indazole ring closure using arylhydrazine 3a (Table 2). Acidic reaction conditions enabled the condensation of arylhydrazine 3a to 1N-etoxycarbonyl indazole (4a) (Table 2, entries 1–5). TFA in DCM and in MeCN gave the expected product 4a in good yield (Table 2, entries 1 and 2). Neat AcOH at r. t. did not enable the cyclization of arylhydrazine 3a, while heating in a solution of MeCN induced formation of indazole 4a (Table 2, entries 3 and 4). Formic acid was strong enough to enable the formation of indazole 4a at room temperature in a solution of MeCN (Table 2, entry 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Temp., time | Product | Yield |
| 1 | 5 equiv. TFA | DCM | 25 °C, 12 h | 4a | 63% |
| 2 | 5 equiv. TFA | MeCN | 25 °C, 12 h | 4a | 64% |
| 3 | AcOH | Neat | r. t., 12 h | 4a | 0% |
| 4 | 30 equiv. AcOH | MeCN | 70 °C, 12 h | 4a | 56% |
| 5 | 30 equiv HCOOH | MeCN | r. t., 12 h | 4a | 56% |
| 6 | 3 equiv. K2CO3 | MeOH | 70 °C, 1 h | 5a | 67% |
| 7 | 3 equiv. K2CO3 | MeOH | 25 °C, 12 h | 5a | 67% |
| 8 | 4 equiv. KOH | EtOH | r. t., 12 h | 5a | 59% |

The use of a base in alcoholic solvent provided unprotected indazole 5a (Table 2, entries 6–8). Both K2CO3 and KOH could be efficiently used for the ring closure – deacylation reaction of arylhydrazine 3a.
Next, the one pot formation of 1N-etoxycarbonyl indazole (4a) from 2-formylphenylboronic acid (1a) was investigated (Table 3). Unfortunately, DMA which was the solvent of choice for high yielding arylation of DEAD was not suitable for the ring closure step in the presence of TFA (Table 3, entry 1). In this case, the arylhydrazine 3a intermediate was not transformed to product 4a, according to LC-MS. In turn, the addition of TFA in DCM in an amount to sufficiently dilute DMA, enabled the formation of expected product 4a in a good yield (Table 3, entry 2). The use of DCM as a solvent for both steps was less productive (Table 3, entry 3). However, MeCN was found as an appropriate solvent for both arylation and ring closure in the presence of TFA to give 1N-protected indazole 4a in a good overall yield (Table 3, entry 4).
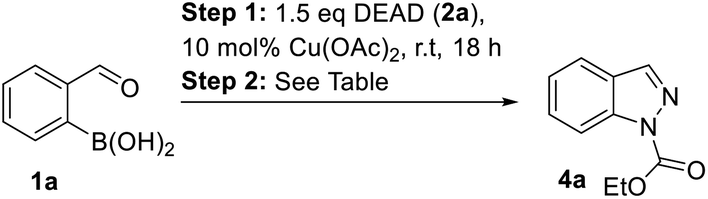
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:With one-pot conditions in hand, the synthesis of other alkoxycarbonylindazoles 4b–d was performed by the reaction of boronic acid 1a with azodicarboxylates 2b–d (Table 4). The best yield of product 4b was obtained with diisopropyl azodicarboxylate (DIAD, 2b, Table 4, entry 1).
|
||
|---|---|---|
| Entry | R | 4, isolated yield |
| 1 | i-Pr | 4b, 86% |
| 2 | Bn | 4c, 60% |
| 3 | i-Bu | 4d, 45% |

The scope of boronic acid substitution was investigated in the reaction of a range of formylboronic acids 1b–f with DIAD (2b) followed by cyclization (Scheme 2). Substrates 1b–d bearing methoxy and benzyloxy groups provided indazoles 4e–g in a good to moderate yield. In the case of substrates 1e,f bearing electron-withdrawing substituents, yields of products 4h, i were decreased.
 | ||
| Scheme 2 The reaction of substituted formylboronic acids with DIAD (2b). | ||
Thiophene boronic acid 8 was found a suitable substrate to obtain thienopyrazole derivative 9 in a good yield (Scheme 3).
 | ||
| Scheme 3 The reaction of substituted formylboronic acids with DIAD (2b). | ||
Hydrazine dicarboxylate 7a was also explored as a reagent for the synthesis of indazoles instead of azodicarboxylate 2a (Table 5). 2-Formylphenylboronic acid (1a) was subjected to the reaction with diethyl hydrazine dicarboxylate (7a) using the two-step one-pot procedure for the formation of indazole 4a. The catalytic amount of Cu(OAc)2 and excess of triethylamine was not sufficient to achieve good yield of product 4a formation (Table 5, entry 1). The use of equimolar amount of Cu(OAc)2 and an excess of triethylamine for the first step enabled good yield of product 4a over two steps, while increasing the amount of Cu(OAc)2 reduced the yield of product 4a (Table 5, entries 2 and 3). The transformation of 2-formylphenylboronic (1a) to indazole 4a was not efficient in the absence of base for the first step, however, TEA could be replaced by TMEDA and DIPEA without significantly reducing the product 4b yield (Table 5, entries 5 and 6). Several other Cu salts were tried for the first step of indazole 4b formation, however, were found to be ineffective (Table 5, entries 7 and 8). The need for an equimolar amount of Cu (OAc)2 for successful synthesis of indazole 4a using hydrazine dicarboxylate 7a implies in situ oxidation of reagent 7a to azodicarboxylate 2a (see also Scheme 5). However, C–N bond formation with hydrazine dicarboxylate 7a in the Chan–Evans–Lam reaction cannot be excluded.21
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Additive | NMR yielda |
| a NMR yield, using 1,3,5-trimethoxybenzene as internal standard. | ||||
| 1 | 20 mol% Cu(OAc)2 | MeCN | 3 equiv. TEA | 25% |
| 2 | 1 equiv. Cu(OAc)2 | MeCN | 3 equiv. TEA | 66% |
| 3 | 1.5 equiv. Cu(OAc)2 | MeCN | 3 equiv. TEA | 50% |
| 4 | 1 equiv. Cu(OAc)2 | MeCN | None | 26% |
| 5 | 1 equiv. Cu(OAc)2 | MeCN | 2 equiv. TMEDA | 67% |
| 6 | 1 equiv. Cu(OAc)2 | MeCN | 3 equiv. DIPEA | 60% |
| 7 | 1 equiv. CuCl | MeCN | 3 equiv. TEA | 35% |
| 8 | 1 equiv. CuCl2 | MeCN | 3 equiv. TEA | 25% |

 | ||
| Scheme 4 Scope of boronic acids 1 in the reaction with diazadicarboxylate 7g. | ||
 | ||
| Scheme 5 Proposed mechanism for the C–N bond forming step. | ||
Next, a range of hydrazine dicarboxylates 7a–g was explored as reaction components for a one-pot two-step synthesis of indazoles 4a–d, j–l (Table 6). TFA was a suitable acid for the cyclization step to give the corresponding products 4a–d, j, k from the reaction of boronic acid 1a with hydrazine dicarboxylates 7a–f (Table 6, entries 1–6). For the synthesis of product 4l bearing acid labile t-Bu group, acetic acid at elevated temperature was used instead of TFA (Table 6, entry 7). This approach successfully provided product 4l in a very good yield (Table 6, entry 8).
|
|||
|---|---|---|---|
| Entry | 7, R | Acid | 4, isolated yield |
| 1 | 7a, Et | TFA | 4a, 63% |
| 2 | 7b, i-Pr | TFA | 4b, 47% |
| 3 | 7c, Bn | TFA | 4c, 46% |
| 4 | 7d, i-Bu | TFA | 4d, 61% |
| 5 | 7e, Me | TFA | 4j, 63% |
| 6 | 7f, allyl | TFA | 4k, 40% |
| 7 | 7g, t-Bu | AcOH | 4l, 73% |

The scope of phenyl boronic acids 1b–i was explored with di-tert-butyl hydrazine dicarboxylate 7g as a reaction component for the synthesis of 1N-Boc indazoles 4m–t (Scheme 4). The major reason for reduction was formation of N-acetyl indazoles 10 as by-products (see ESI† for the characterization of 10a, R = H).
The mechanism for the C–N bond formation in the copper catalysed reaction of arylboronic acids with diazadicarboxylates has been proposed by Uemura and Chatani.20 According to this, the transmetalation reaction of arylboronic acid 1a with a copper catalyst would form an arylcopper species 10 (Scheme 5). Addition of intermediate 10 to NN double bond gives an arylhydrazine 11 which undergoes the transmetalation with boronic acid 1a to give intermediate 12 and return arylcopper species 10 into catalytic cycle. Work-up would produce arylhydrazine 3a. Noteworthy, it was shown by Uemura and Chatani that dialkoxycarbonyl hydrazines are not competent substrates for this reaction unless additional oxidant is added.20 This implies that hydrazine 7a is likely oxidised to diazadicarboxylate 2a by stoichiometric amount of copper source.
The proposed mechanism for the condensation of arylhydrazine intermediate into indazole is given in Scheme 6. In the presence of acid, N-acyliminium ion 13 is formed. Selective hydrolytic cleavage of one ethoxycarbonyl group in intermediate 13 gives 1N-ethoxycarbonyl indazole 4a. In turn, basic conditions would enable cleavage of both ethoxycarbonyl groups leading to intermediate 14 which eliminates water to give indazole 5a.
 | ||
| Scheme 6 Proposed mechanism for the condensation step. | ||
Conclusions
In summary, copper catalysed reaction of 2-formylboronic acids with diazadicaboxylates followed by acid or base induced ring closure is a convenient method for the synthesis of 1N-alkoxycarbonyl indazole derivatives. The indazole synthesis can also be performed using hydrazine dicarboxylates as reaction partners for the synthesis of indazoles, however, required a stoichiometric amount of copper(II) acetate for the C–N bond formation step. The method is based on readily available building blocks and can be performed at relatively mild reaction conditions which enables its application for the synthesis of indazole motif containing compounds.Author contributions
V. S. and A. S performed the synthesis. A. J. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from H2020 MSC-ITN project CARTNET “Combating Antimicrobial Resistance Training Network”, grant agreement ID: 765147 is acknowledged.Notes and references
- K. B. Goodman, H. Cui, S. E. Dowdell, D. E. Gaitanopoulos, R. L. Ivy, C. A. Sehon, R. A. Stavenger, G. Z. Wang, A. Q. Viet, W. Xu, G. Ye, S. F. Semus, C. Evans, H. E. Fries, L. J. Jolivette, R. B. Kirkpatrick, E. Dul, S. S. Khandekar, T. Yi, D. K. Jung, L. L. Wright, G. K. Smith, D. J. Behm, R. Bentley, C. P. Doe, E. Hu and D. Lee, J. Med. Chem., 2007, 50, 6–9 CrossRef CAS PubMed.
- Y. Hu, D. Cole, R. A. Denny, D. R. Anderson, M. Ipek, Y. Ni, X. Wang, S. Thaisrivongs, T. Chamberlain, J. P. Hall, J. Liu, M. Luong, L.-L. Lin, J.-B. Telliez and A. Gopalsamy, Bioorg. Med. Chem. Lett., 2011, 21, 4758–4761 CrossRef CAS PubMed.
- J. Schoene, T. Gazzi, P. Lindemann, M. Christmann, A. Volkamer and M. Nazaré, ChemMedChem, 2019, 14, 1514–1527 CrossRef CAS PubMed.
- S.-G. Zhang, C.-G. Liang and W.-H. Zhang, Molecules, 2018, 23, 2783 CrossRef PubMed.
- J.-H. Sun, C. A. Teleha, J.-S. Yan, J. D. Rodgers and D. A. Nugiel, J. Org. Chem., 1997, 62, 5627–5629 CrossRef CAS.
- X. Xiong, Y. Jiang and D. Ma, Org. Lett., 2012, 14, 2552–2555 CrossRef CAS PubMed.
- E. Dubost, S. Stiebing, T. Ferrary, T. Cailly, F. Fabis and V. Collot, Tetrahedron, 2014, 70, 8413–8418 CrossRef CAS.
- K. Lukin, M. C. Hsu, D. Fernando and M. R. Leanna, J. Org. Chem., 2006, 71, 8166–8172 CrossRef CAS.
- B. C. Wray and J. P. Stambuli, Org. Lett., 2010, 12, 4576–4579 CrossRef CAS PubMed.
- J. Schoene, H. Bel Abed, P. Schmieder, M. Christmann and M. Nazaré, Chem.–Eur. J., 2018, 24, 9090–9100 CrossRef CAS PubMed.
- H. Bel Abed, N. Weißing, J. Schoene, J. Paulus, N. Sewald and M. Nazaré, Tetrahedron Lett., 2018, 59, 1813–1815 CrossRef CAS.
- G. Chen, M. Hu and Y. Peng, J. Org. Chem., 2018, 83, 1591–1597 CrossRef CAS.
- Z. Liu, F. Shi, P. D. G. Martinez, C. Raminelli and R. C. Larock, J. Org. Chem., 2008, 73, 219–226 CrossRef CAS PubMed.
- P. Li, C. Wu, J. Zhao, D. C. Rogness and F. Shi, J. Org. Chem., 2012, 77, 3149–3158 CrossRef CAS.
- K. W. Hunt, D. A. Moreno, N. Suiter, C. T. Clark and G. Kim, Org. Lett., 2009, 11, 5054–5057 CrossRef CAS PubMed.
- M. Cheung, A. Boloor and J. A. Stafford, J. Org. Chem., 2003, 68, 4093–4095 CrossRef CAS.
- G. Luo, L. Chen and G. Dubowchik, J. Org. Chem., 2006, 71, 5392–5395 CrossRef CAS PubMed.
- D. M. M. M. Dissanayake and A. K. Vannucci, Org. Lett., 2019, 21, 457–460 CrossRef CAS PubMed.
- V. Solomin, A. Seins and A. Jirgensons, Synlett, 2020, 31 Search PubMed.
- T. Uemura and N. Chatani, J. Org. Chem., 2005, 70, 8631–8634 CrossRef CAS PubMed.
- L. Raus, O. Tsubrik and U. Maeorg, Proc. Est. Acad. Sci. Chem., 2005, 54, 12 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04056a |
| This journal is © The Royal Society of Chemistry 2021 |