
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructurally diverse diterpenoid alkaloids from the lateral roots of Aconitum carmichaelii Debx. and their anti-tumor activities based on in vitro systematic evaluation and network pharmacology analysis†
Yang Yuab,
Shifei Wua,
Jianqing Zhanga,
Jiayuan Lia,
Changliang Yaoa,
Wenyong Wuac,
Yingying Wangac,
Hongjian Jiac,
Wenlong Weia,
Min Gaoab,
Yun Liad,
Shuai Yaoa,
Yong Huanga,
Qirui Bia,
Hua Quac and
De-an Guo *ab
*ab
aShanghai Research Center for Modernization of Traditional Chinese Medicine, National Engineering Laboratory for TCM Standardization Technology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: daguo@simm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China
dSchool of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 210009, China
First published on 4th August 2021
Abstract
Thirty-seven diterpenoid alkaloids (DAs) with diverse structures were isolated and identified from the lateral roots of Aconitum carmichaelii Debx., comprising eight C20-DAs and twenty-nine C19-DAs. Besides the 31 known DAs identified by comparing the 1H NMR and 13C NMR data with those reported in the literature, the structures of four new compounds (1, 14, 17, and 25), and two other compounds (26 and 37) which were reported to be synthesized previously, were also elucidated based on the comprehensive analysis of their HR-ESI-MS, 1D and 2D NMR spectra, including 1H–1H COSY, HSQC and HMBC and NOESY/ROESY. Among them, compound 1 represents the first example of a C20-DA glucoside. Besides, the anti-tumor activities of all the isolated compounds against human non-small-cell lung cancer A549 and H460 cells were systematically evaluated by MTT methods. The results revealed that all of the C19-DAs possessed moderate activities against both of the two cell lines with IC50 values ranging from 7.97 to 28.42 μM, and their structure–activity relationships indicated the active sites of C-8, C-10, and C-14 positions and the nitrogen atom in the C19-DA skeleton. In addition, all of the isolated DAs, with chemical structures confirmed, were further applied for network pharmacology analysis, in order to give an insight into the possible mechanisms of their anti-tumor activities. As a result, 173 potential targets and three most important pathways related to non-small-cell lung carcinoma were finally unearthed.
Introduction
Malignant tumor is one of the major diseases threatening human health with high morbidity and mortality. With the continuous development of tumor pharmacology, a deeper understanding of the process of tumorigenesis and development was achieved, which in turn facilitated the development of new therapeutic drugs. Natural products and their derivatives are always considered as one of the most important sources of drug entities for cancer treatment,1,2 such as taxol, camptothecin, and vincristine, which have already been applied in clinics, and also provided the leading structures for different classes of conventional anti-tumor drugs. Take camptothecin as an example, which has been proved to possess the inhibitory effect of topoisomerase I; researchers have been devoted to developing more high-efficiency and low-toxic camptothecin derivatives for treating cancer, including hydroxycamptothecin, irinotecan, topotecan, etc.3 In a word, the discovery, development and further pharmacological activity investigations of new natural products and their derivatives are undoubtedly important for further development of anti-tumor agents with new structures and new mechanisms of action.Diterpenoid alkaloids (DAs) are a kind of nitrogen-containing compounds with complex structures and significant bioactivities and toxicities, which have always aroused the scientists' interests all over the world.4 Some DAs such as 13-acetylaconitine, lappaconitine, crassicauline A, and guanfu-base A have already been applied in the clinic as analgesic and anti-arrhythmia agents. Some DAs, including hypaconine, mesaconine, and beiwutinine, demonstrated cardiac effects,5 while other DAs including fuziline and neoline were found to possess cardiomyocyte protective activities,6 etc. Recent research also showed that DAs could induce apoptosis of tumor cells, thus possessing potential in the development of anti-tumor agents.7–10 DAs are mainly derived from plants of the genera Aconitum and Delphinium, and usually classified into four subtypes according to their carbon skeletons, namely C18-, C19-, C20-, and bis-DAs.4 More than one hundred DAs (mainly C19 and C20 subtypes) have ever been isolated from Aconitum carmichaelii Debx., which is a well-known traditional Chinese medicine (TCM) dubbed “fuzi” in China, and possesses significant bioactivities, including cardioactive effects, anti-arrhythmic , anti-inflammatory, and analgesic activities, etc.11 It is required to be processed before oral administration to prevent poisoning, as recorded in Chinese Pharmacopoeia (2020), since the diester C19-DAs would transform into monoester C19-DAs and aminoalcohol C19-DAs which were less toxic.12 The structural differences between these three subtypes lie in whether an acetyl or benzoyl group substituted at the C-8 or C-14 positions, respectively, which suggested the variety of substituents also play an important part in their bioactivities and toxicities. Despite the extensive studies about DAs, there are still multitudes of DAs varied in skeletons and substituents needing to be further investigated for their activities and mechanisms of action.
This study aims at exploring more DAs with anti-tumor activities from the lateral roots of Aconitum carmichaelii Debx., and systematically evaluate their anti-tumor effects and possible mechanisms for future drug development. As a result, a total of thirty-seven DAs were isolated by integration of different chromatographic methods, including eight aminoalcohol-C19 DAs, sixteen monoester-C19 DAs, three diester-C19 DAs, one 8, 15-seco C19 DA, one pyro-type C19 DA, and eight C20 DAs. Among them, the structures of four new compounds, hetisane-15β-O-β-D-glucoside (1), 6-demethoxyhypaconine (14), carmichaeline K (17), and 8-O-ethyl-benzoyldeoxyaconine (25), were determined by a combination of HR-ESI-MS, 1D NMR and 2D NMR spectra, and the NMR data of other two DAs, 8-O-ethyl-benzoylhypaconine (26)13 and 3-deoxypyromesaconine (37),14 which were reported to be synthesized previously, were also elucidated and listed in Tables S1 and S2.† The remaining 31 known compounds were all identified based on the 1H NMR and 13C NMR data reported in literatures, including nominine (2),15 hetisine (3),16 13β-acetoxy-15β,19α-dihydroxyhetisane (4),17 songorine (5),18 napelline (6),19 dihydroatisine (7),20 trifoliolasine E (8),21 karacoline (9),22 isotalatizidine (10),23 neoline (11),24 8-dehydroxyl-bikhaconine (12),25 fuziline (13),26 hypaconine (15),27 mesaconine (16),27 14-benzoyltalatisamine (18),28 14-benzoylchasmanine (19),29 benzoylhypaconine (20),30 benzoyldeoxyaconine (21),31 benzoylmesaconine (22),30 benzoylaconine (23),30 14-benzoylbeiwutinine (24),32 8-O-ethyl-14-benzoylmesaconine (27),24 8-O-ethyl-14-benzoylaconine (28),33 beiwucine (29),34 condelphine (30),35 14-O-acetyltalatisamine (31),36 14-O-acetylneoline (32),37 hypaconitine (33),38 deoxyaconitine (34),39 mesaconitine (35),39 and nagarine A (36).40 The structures of all the isolated C20- and C19-DAs were presented in Fig. 1 and 2, respectively. And in vitro systematic evaluation of their anti-tumor activities against human non-small-cell lung cancer A549 and H460 cells and non-cancerous HBE cells were conducted, their structure–activity relationships were also discussed, rendering a reference for the development of new anti-tumor agents. To further interpret the underlying mechanisms of DAs in anti-tumor activities, network pharmacology analysis was subsequently conducted for predicting the possible targets and pathways, laying the theoretical foundations for future mechanism research of DAs.
 | ||
| Fig. 1 The structures of C20-DAs (compounds 1–8). | ||
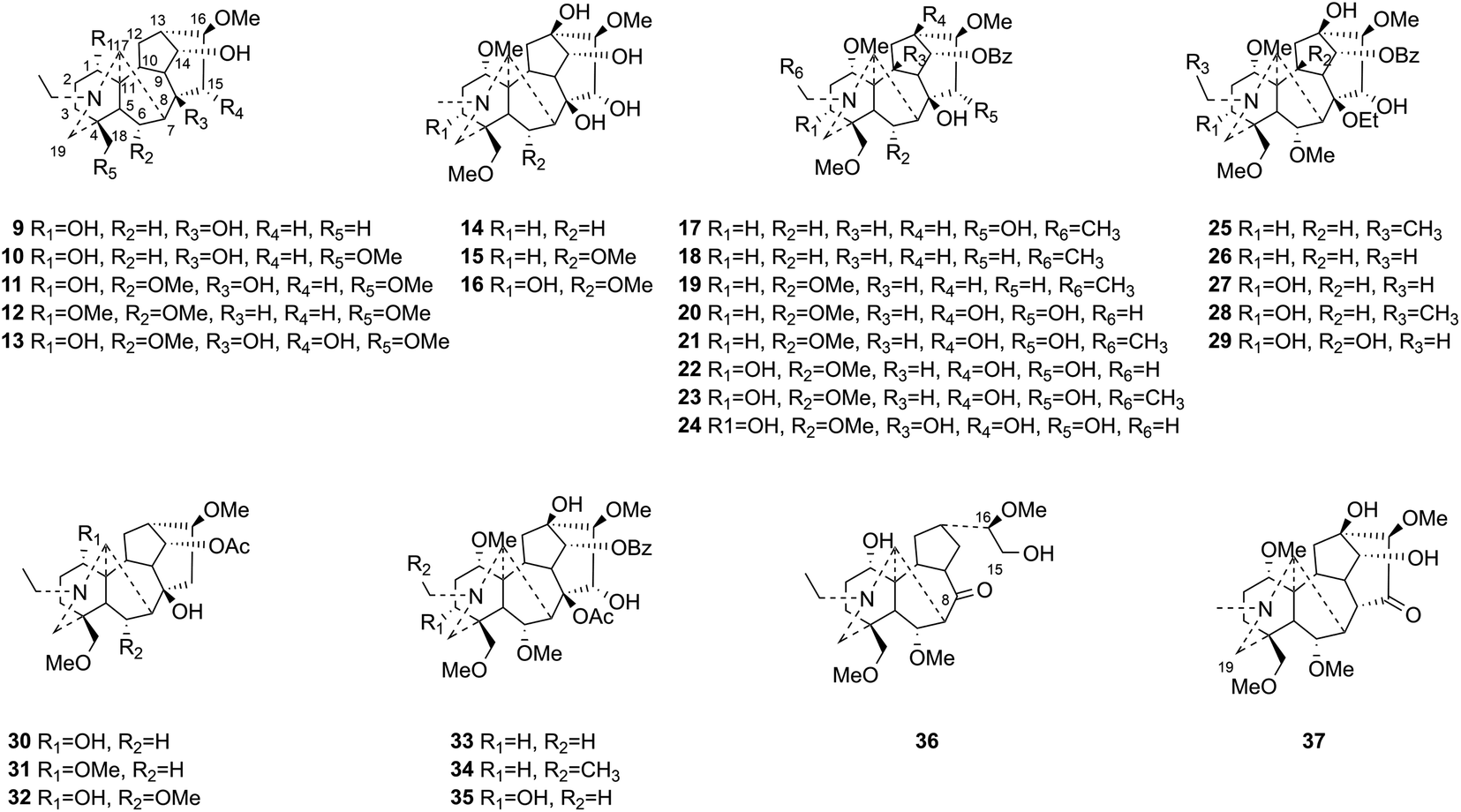 | ||
| Fig. 2 The structures of C19-DAs (compounds 9–37). | ||
Results and discussion
Structure elucidation of new compounds
Compound 1 was purified as a white, amorphous powder, and showed a protonated molecular ion at m/z 460.2702 ([M + H]+, calcd for C26H38NO6, 460.2699) in the HR-ESI-MS analysis, corresponding to the molecular formula C26H37NO6. The NMR data (Table 1) of compound 1 demonstrated 26 carbon signals. Among them, 20 carbons including one methyl carbon (δC 28.59), seven methylenes, seven methines, three non-oxygenated quaternary carbons (δC 48.6, 44.8, 37.4), and an exocyclic double bond unit (δC 149.3, 111.2), constituted a typical C20-DA skeleton.41 And correlation of H-19 [δH 2.22–2.35 (2H, m)] and C-6 (δC 64.6) in HMBC spectrum (Fig. 3) further supported the hetisine-type C20-DA skeleton with (C-19)–N–(C-6) connection. The other 6 carbons represented a hexose moiety consisting of five oxygenated methines [δH 4.36 (1H, d, J = 7.8 Hz), 2.97 (1H, t, J = 8.3 Hz), 3.09–3.14 (1H, m), 3.00–3.03 (1H, m), 3.00–3.03 (1H, m); δC 97.7, 73.5, 77.1, 76.8, 74.4] and an oxygenated methylene [δH 3.67 (1H, d, J = 11.6 Hz), 3.43–3.46 (1H, m); δC 61.3]. All evidences mentioned above indicated it was a hetisine-type C20-DA with one hexose substituent. The only one oxygenated carbon signal (δC 74.3) in the skeleton was assigned to C-15 based on the HMBC correlation from H-15 [δH 4.13 (1H, s)] to C-1′ (δC 97.75) and C-17 (δC 111.2). Besides, correlation of H-17 [δH 4.86 (1H, s)] with H-15, and H-1′ [δH 4.36 (1H, d, J = 7.8 Hz)] in NOESY spectrum implied the β-configuration of 15-glucoside. And the vicinal coupling constant value of H-1′ and H-2′ [δH 2.97 (1H, t, J = 8.3 Hz)], and NOESY correlation between H-4′ [δH 3.00–3.03 (1H, m)] and H-6′ [δH 3.67 (1H, d, J = 11.6 Hz)] indicated the hexose was a β-glucoside. In addition, the D,L-configuration of the glucoside was determined by acid hydrolysis and then derivatization, as reported in literature.42 By comparing its UHPLC-MS chromatogram (Fig. S9†) with glucoside derivatives prepared in the same way, structure of compound 1 was fully established and named as hetisane-15β-O-β-D-glucoside. Since the previously reported glycosidic DAs all bear a C19-skeleton,43,44 this is the first C20-DA glycoside isolated from Aconitum carmichaelii Debx.| Position | δH (ppm) | δC (ppm) |
|---|---|---|
| 1 | 1.91–1.98 (1H, m); 1.12–1.26 (1H, m) | 26.8 |
| 2 | 1.48–1.55 (2H, m) | 19.3 |
| 3 | 1.36–1.40 (1H, m); 1.12–1.26 (1H, m) | 33.9 |
| 4 | — | 37.4 |
| 5 | 2.44 (1H, s) | 74.4 |
| 6 | 3.07 (1H, s) | 64.6 |
| 7 | 1.97 (1H, dd, J = 13.3, 2.6 Hz); 1.41–1.46 (1H, m) | 32.6 |
| 8 | — | 48.6 |
| 9 | 1.66–1.74 (1H, m) | 42.1 |
| 10 | — | 44.8 |
| 11 | 1.58–1.66 (1H, m); 1.05–1.10 (1H, m) | 35.0 |
| 12 | 2.07–2.13 (1H, m) | 33.2 |
| 13 | 1.66–1.74 (1H, m); 1.55–1.58 (1H, m) | 24.3 |
| 14 | 1.75–1.83 (1H, m) | 44.1 |
| 15 | 4.13 (1H, s) | 74.3 |
| 16 | — | 149.3 |
| 17 | 4.99 (1H, s); 4.86 (1H, s) | 111.2 |
| 18 | 0.91 (3H, s) | 28.6 |
| 19 | 2.22–2.35 (2H, m) | 62.1 |
| 20 | 1.36 (1H, s) | 60.9 |
| 1′ | 4.36 (1H, d, J = 7.8 Hz) | 97.7 |
| 2′ | 2.97 (1H, t, J = 8.3 Hz) | 73.5 |
| 3′ | 3.09–3.14 (1H, m) | 77.1 |
| 4′ | 3.00–3.03 (1H, m) | 76.8 |
| 5′ | 3.00–3.03 (1H, m) | 74.4 |
| 6′ | 3.67 (1H, d, J = 11.6 Hz); 3.43–3.46 (1H, m) | 61.3 |
| 2′-OH | 4.64–4.69 (1H, m) | — |
| 3′-OH | 4.88–4.91 (1H, m) | — |
| 4′-OH | 4.88–4.91 (1H, m) | — |
| 6′-OH | 4.44–4.50 (1H, m) | — |
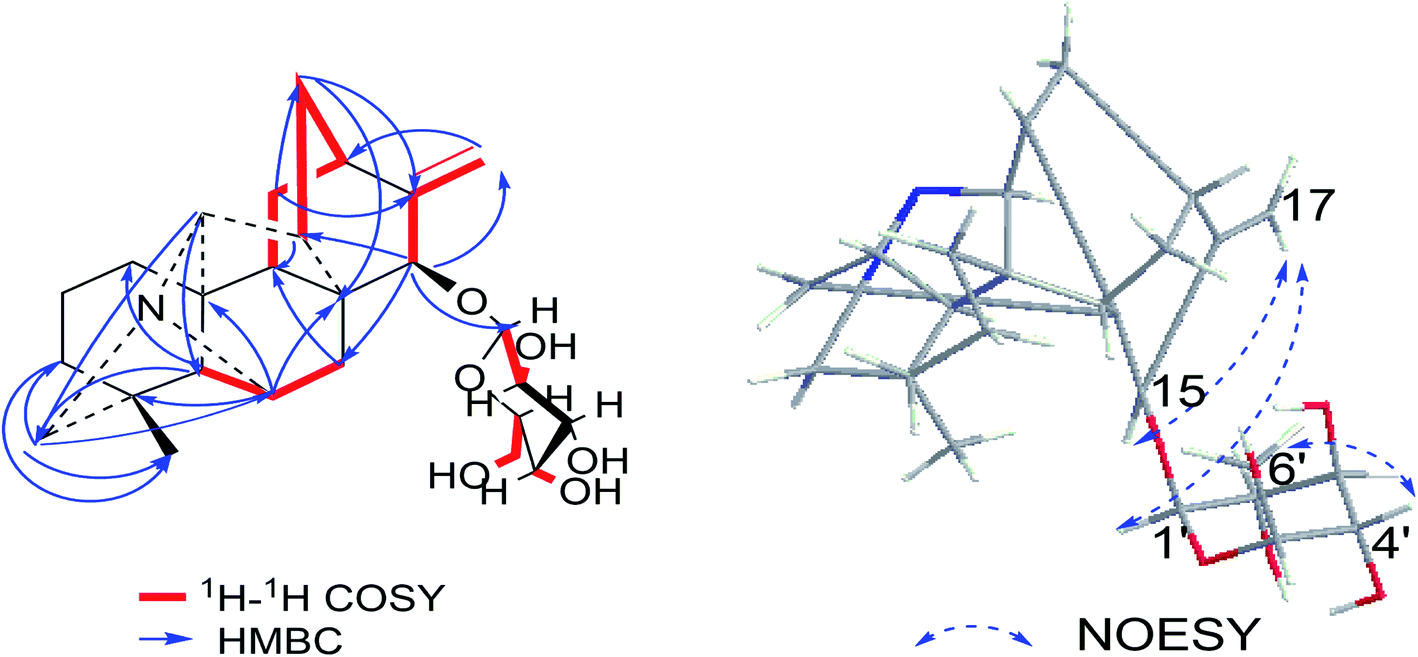 | ||
| Fig. 3 Key HMBC, 1H–1H COSY and NOESY correlations of compound 1. | ||
Compound 14a (the acetate salt of compound 14) was obtained as a white, amorphous powder, and its molecular formula was determined to be C23H37NO7 by HR-ESI-MS data at m/z 440.2636 ([M-CH3COO−]+, calcd for C23H38NO7, 440.2648), indicative of six degrees of unsaturation. The 13C NMR spectra displayed 25 carbon signals, including the characteristic signals attributed to one N-methyl group [δH 2.29 (3H, s), δC 42.9], three methoxy groups [δH 3.27(3H, s), δC 56.8; δH 3.62 (3H, s), δC 61.1; δH 3.28(3H, s), δC 59.6], and one acetyl group [δC 172.8; δH 2.01 (3H, s), δC 22.8]. Except for the above signals, the remaining 19 carbon signals, constituting six methylenes, nine methines, and four quaternary carbons, revealed that compound 14a was a C19-DA.45 Considering that DAs usually display protonated ions in HR-ESI-MS spectrum, the number of carbons calculated from HR-ESI-MS data are supposed to be consistent with those displayed in 13C NMR spectrum, compound 14a might be a salt due to the acidic conditions used in the isolation process. And because the acetyl unit had no correlations with other signals in either HMBC or ROESY spectrum (Fig. 4), it might be an acetate. Besides, the HMBC correlations between 18-OMe [δH 3.28 (3H, s)] and the methylene carbon C-18 (δC 79.7), 1-OMe [δH 3.27 (3H, s)] and C-1 (δC 85.8), 16-OMe [δH 3.62 (3H, s)] and C-16 (δC 91.3) indicated the three methoxy groups were substituted at C-18, C-1, and C-16, respectively. It was also speculated to contain four hydroxy groups according to the seven oxygenated carbon signals observed in the 13C NMR spectrum. And the doublet peak of H-14 [δH 3.93 (1H, d, J = 5.1 Hz)], the 1H–1H COSY cross-peaks of H-14/H-10 [δH 1.87–1.95 (1H, m)]/H-9 [δH 2.29–2.34 (1H, m)], H-15 [δH 4.38–4.49 (1H, m)]/H-16 [δH 3.16 (1H, d, J = 5.8 Hz)], and the HMBC correlations of H-6a [δH 1.79–1.87 (1H, m)], H-14, H-15, H-16 with C-8 (δC 79.0), and H-9, H-10, H-12b [δH 1.96–1.98 (1H, m)], H-14, H-16 with C-13 (δC 74.71) implied the four hydroxy groups were located at the oxygenated quaternary carbon C-8, C-13, and the oxygenated methine carbon C-14 (δC 79.0) and C-15 (δC 82.4), respectively. After the planar structure of compound 14a was constructed, its relative configuration was also deduced from the ROESY spectrum (Fig. 5). ROE correlations of H-1 [δH 3.06 (1H, dd, J = 10.5, 6.7 Hz)] with H-10β, and H-14 with H-9β indicated the α-configurations of 1-OMe and 14-OH, respectively, while correlations of one proton of H-12 [δH 1.96–1.98 (1H, m)] with H-14, and another proton of H-12 [δH 2.72 (1H, dd, J = 13.6, 4.1 Hz)] with H-16, and H-15 with 16-OMe implied the β-configuration of 16-OMe and α-configuration of 15-OH. At last, compound 14 (5 mg) was obtained as a free base by further alkaline treatment of compound 14a using preparative TLC (n-hexane–ethyl acetate–diethylamine, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1). By comparing their 1H NMR and 13C NMR data (Table 2), compound 14a was confirmed to be an acetate, and named as 6-demethoxyhypaconine acetate, while compound 14 was named as 6-demethoxyhypaconine, and its structure was presented in Fig. 1.
:2:1). By comparing their 1H NMR and 13C NMR data (Table 2), compound 14a was confirmed to be an acetate, and named as 6-demethoxyhypaconine acetate, while compound 14 was named as 6-demethoxyhypaconine, and its structure was presented in Fig. 1.
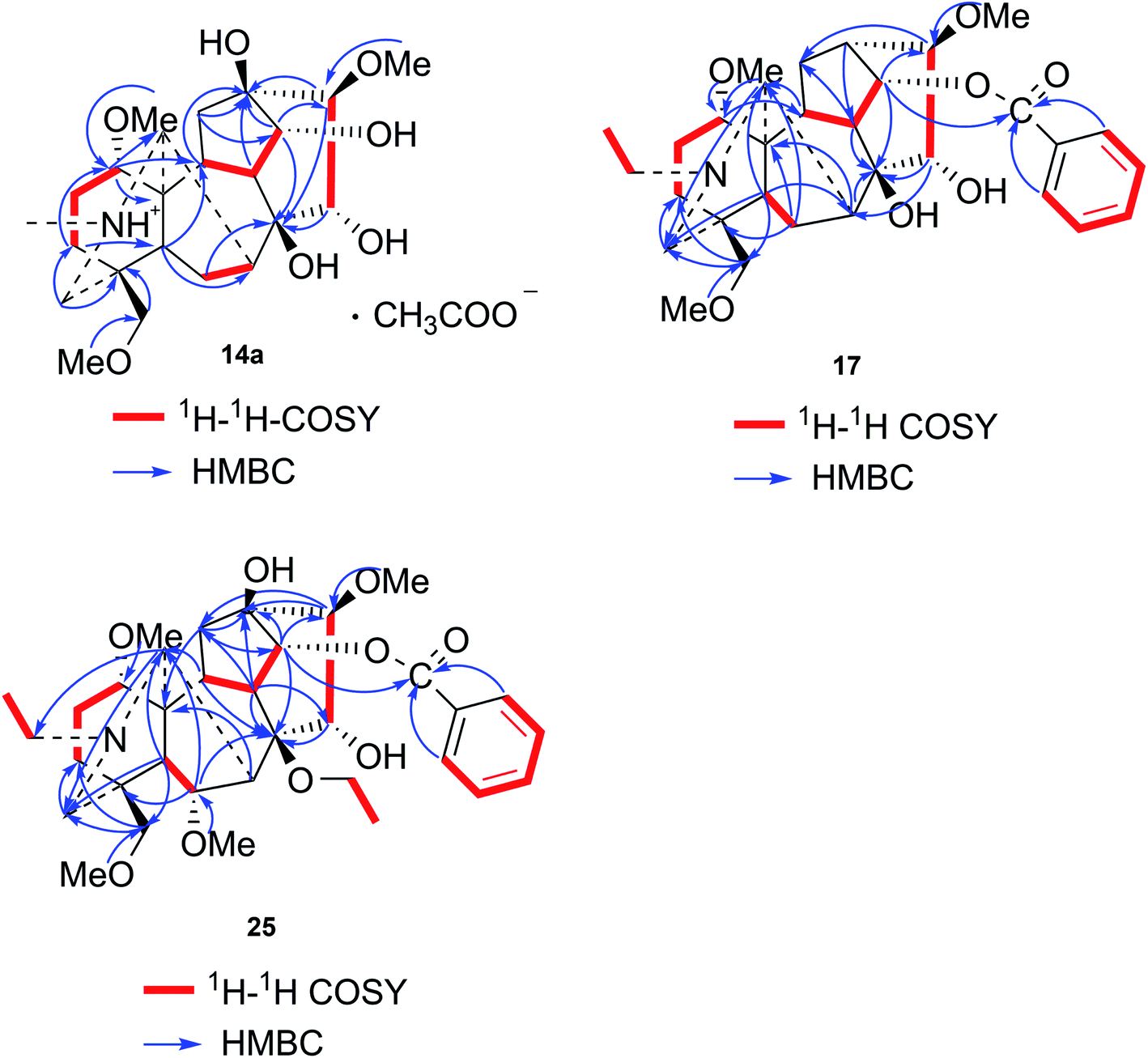 | ||
| Fig. 4 Key 1H–1H COSY and HMBC correlations of compounds 14a, 17 and 25. | ||
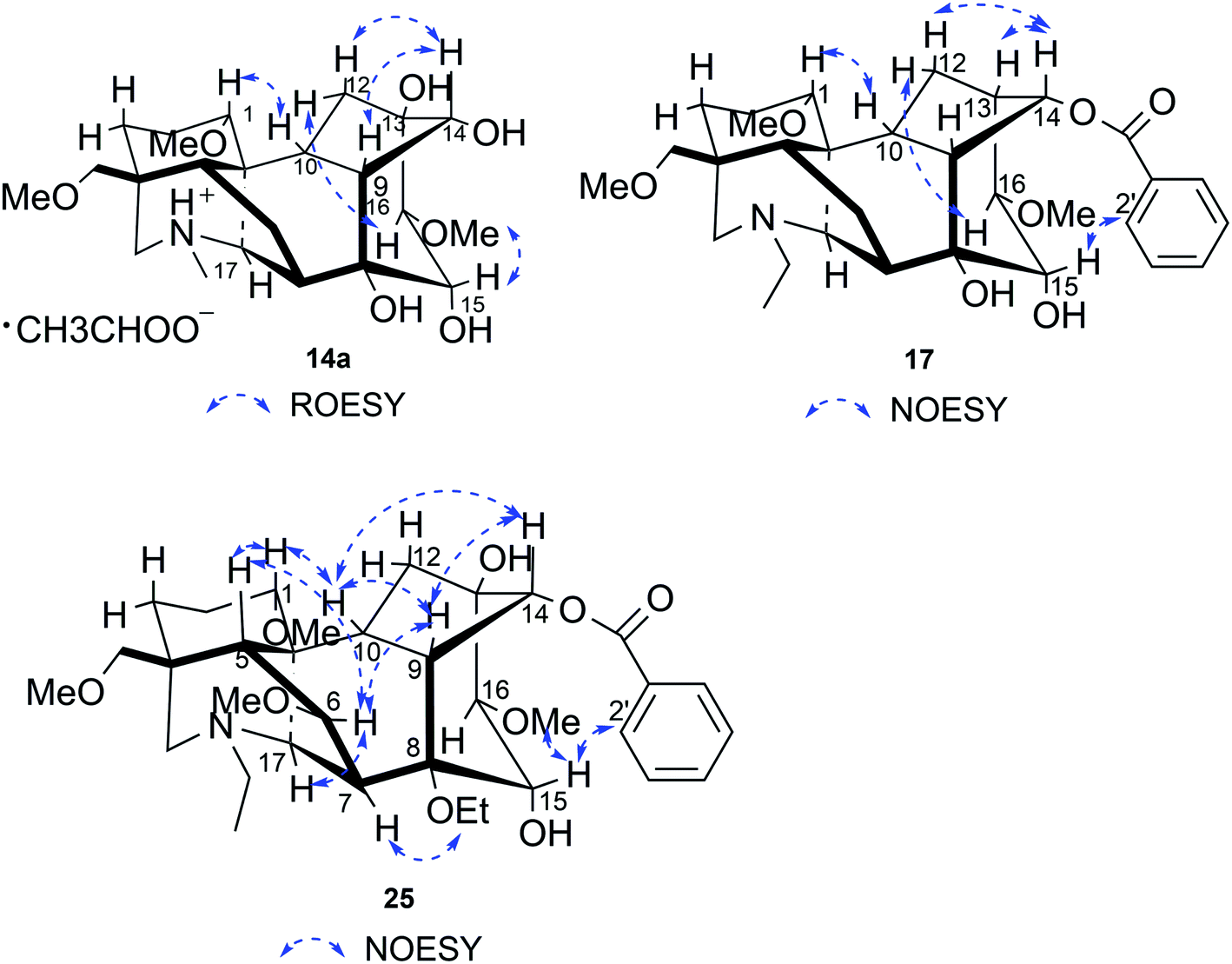 | ||
| Fig. 5 Key NOESY/ROESY correlations of compounds 14a, 17 and 25. | ||
| Position | 14a | 14ab | 17a | 25b | ||||
|---|---|---|---|---|---|---|---|---|
| δH (ppm) | δC (ppm) | δH (ppm) | δC (ppm) | δH (ppm) | δC (ppm) | δH (ppm) | δC (ppm) | |
| a In 1H (600 MHz) and 13C (150 MHz) NMR.b In 1H (500 MHz) and 13C (125 MHz) NMR. | ||||||||
| 1 | 3.05–3.10 (1H, m) | 85.7 | 3.06 (1H, dd, J = 10.5, 6.7 Hz) | 85.8 | 3.17–3.23 (1H, m) | 85.3 | 3.28–3.31 (1H, m) | 83.4 |
| 2 | 2.16–2.20 (1H, m); 2.04–2.09 (1H, m) | 26.6 | 2.12–2.26 (1H, m); 2.03–2.06 (1H, m) | 26.7 | 2.23–2.34 (1H, m); 1.91–1.98 (1H, m) | 26.1 | 2.09–2.19 (1H, m); 1.53–1.65 (1H, m) | 24.2 |
| 3 | 1.72–1.80 (1H, m); 1.38–1.49 (1H, m) | 32.7 | 1.71–1.79 (1H, m); 1.35–1.48 (1H, m) | 32.7 | 1.71–1.78 (1H, m); 1.48–1.56 (1H, m) | 32.0 | 1.82–1.95 (1H, m); 1.65–1.76 (1H, m) | 31.2 |
| 4 | — | 38.5 | — | 38.7 | — | 38.4 | — | 38.7 |
| 5 | 1.50–1.64 (1H, m) | 45.0 | 1.58 (1H, d, J = 8.1 Hz) | 45.0 | 1.68 (1H, d, J = 7.5 Hz) | 45.0 | 2.23–2.25 (1H, m) | 44.8 |
| 6 | 1.82–1.89 (1H, m); 1.54–1.64 (1H, m) | 24.8 | 1.79–1.87 (1H, m); 1.52–1.56 (1H, m) | 24.8 | 1.87 (1H, dd, J = 15.0, 7.6 Hz); 1.62 (1H, dd, J = 15.0, 8.4 Hz) | 25.0 | 4.09 (1H, d, J = 6.4 Hz) | 82.6 |
| 7 | 2.36–2.47 (1H, m) | 38.7 | 2.44 (1H, d, J = 8.1 Hz) | 38.7 | 2.58 (1H, s) | 40.4 | 2.88 (1H, s) | 43.7 |
| 8 | — | 79.0 | — | 79.0 | — | 78.3 | — | 82.6 |
| 9 | 2.31–2.34 (1H, m) | 47.9 | 2.29–2.34 (1H, m) | 47.8 | 2.47–2.49 (1H, m) | 45.5 | 2.60–2.68 (1H, m) | 44.8 |
| 10 | 1.91–1.96 (1H, m) | 42.1 | 1.87–1.95 (1H, m) | 42.3 | 1.98–2.07 (1H, m) | 45.4 | 2.09–2.19 (1H, m) | 40.9 |
| 11 | — | 48.7 | — | 48.7 | — | 49.1 | — | 50.6 |
| 12 | 2.66–2.73 (1H, m); 1.97–2.01 (1H, m) | 37.1 | 2.72 (1H, dd, J = 13.6, 4.1 Hz); 1.96–1.98 (1H, m) | 37.2 | 2.47–2.49 (1H, m); 1.98–2.07 (1H, m) | 29.7 | 2.20–2.23 (2H, m) | 36.7 |
| 13 | — | 76.7 | — | 76.8 | 2.52–2.56 (1H, m) | 37.9 | — | 74.8 |
| 14 | 3.96 (1H, d, J = 5.1 Hz) | 79.3 | 3.93 (1H, d, J = 5.1 Hz) | 79.0 | 5.07 (1H, t, J = 4.8 Hz) | 76.9 | 4.83 (1H, d, J = 5.1 Hz) | 79.3 |
| 15 | 4.39–4.50 (1H, m) | 83.1 | 4.38–4.49 (1H, m) | 82.4 | 4.39 (1H, d, J = 6.7 Hz) | 79.9 | 4.59 (1H, d, J = 5.5 Hz) | 77.7 |
| 16 | 3.17 (1H, d, J = 5.6 Hz) | 90.8 | 3.16 (1H, d, J = 5.8 Hz) | 91.3 | 3.09 (1H, d, J = 6.7 Hz) | 90.7 | 3.34–3.38 (1H, m) | 93.1 |
| 17 | 3.02 (1H, s) | 62.9 | 2.99 (1H, s) | 62.8 | 3.07 (1H, s) | 62.3 | 3.29 (1H, s) | 63.3 |
| 18 | 3.11 (1H, d, J = 9.0 Hz); 2.97 (1H, d, J = 9.0 Hz) | 79.6 | 3.11 (1H, d, J = 9.0 Hz); 2.96 (1H, d, J = 9.0 Hz) | 79.7 | 3.12 (1H, d, J = 9.2 Hz); 3.01 (1H, d, J = 9.2 Hz) | 79.6 | 3.58 (1H, d, J = 8.2 Hz); 3.18 (1H, d, J = 8.2 Hz) | 79.2 |
| 19 | 2.34–2.36 (1H, m); 2.01–2.04 (1H, m) | 55.9 | 2.38 (1H, d, J = 11.6 Hz); 1.99–2.02 (1H, m) | 55.8 | 2.56–2.57 (1H, m); 2.07–2.14 (1H, m) | 53.6 | 3.02–3.10 (1H, m); 2.85–2.91 (1H, m) | 55.3 |
| NCH3 | 2.31 (3H, s) | 42.9 | 2.29 (3H, s) | 42.9 | — | — | — | — |
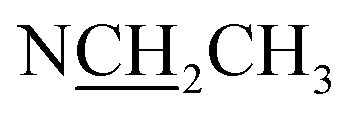 |
— | — | — | — | 2.68–2.78 (1H, m); 2.40–2.47 (1H, m) | 49.7 | 3.34–3.38 (1H, m); 2.92–3.01 (1H, m) | 50.1 |
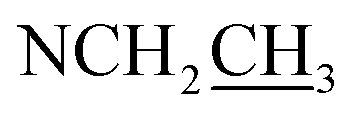 |
— | — | — | — | 1.09 (3H, t, J = 7.2 Hz) | 13.4 | 1.29 (3H, t, J = 5.5 Hz) | 12.3 |
| 1-OMe | 3.28 (3H, s) | 56.9 | 3.27 (3H, s) | 56.8 | 3.31 (3H, s) | 56.4 | 3.34 (3H, s) | 56.3 |
| 6-OMe | — | — | — | — | — | — | 3.27 (3H, s) | 58.8 |
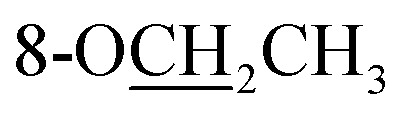 |
— | — | — | — | — | — | 3.46–3.50 (1H, m); 3.38–3.44 (1H, m) | 57.5 |
 |
— | — | — | — | — | — | 0.56 (3H, t, J = 6.9 Hz) | 15.3 |
| 16-OMe | 3.62 (3H, s) | 60.8 | 3.62 (3H, s) | 61.1 | 3.37 (3H, s) | 57.1 | 3.79 (3H, s) | 62.0 |
| 18-OMe | 3.29 (3H, s) | 59.6 | 3.28 (3H, s) | 59.6 | 3.29 (3H, s) | 59.6 | 3.31 (3H, s) | 59.3 |
| 14-OCOC6H5 | — | — | — | — | — | 166.4 | — | 166.4 |
| 1′ | — | — | — | — | — | 130.3 | — | 130.3 |
| 2′ | — | — | — | — | 7.98–8.03 (1H, m) | 129.8 | 8.02–8.07 (1H, m) | 129.9 |
| 3′ | — | — | — | — | 7.37–7.44 (1H, m) | 128.6 | 7.45 (1H, t, J = 7.7 Hz) | 128.5 |
| 4′ | — | — | — | — | 7.49–7.55 (1H, m) | 133.0 | 7.53–7.59 (1H, m) | 133.1 |
| 5′ | — | — | — | — | 7.37–7.44 (1H, m) | 128.6 | 7.45 (1H, t, J = 7.7 Hz) | 128.5 |
| 6′ | — | — | — | — | 7.98–8.03 (1H, m) | 129.8 | 8.02–8.07 (1H, m) | 129.9 |
CH3![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) OO− OO− |
— | — | — | 172.8 | — | — | — | — |
 |
— | — | 2.01 (3H, s) | 22.8 | — | — | — | — |
Compound 17 was isolated as a white, amorphous powder with a molecular formula of C31H43NO7, as deduced from the HR-ESI-MS data at m/z 542.3129 ([M + H]+, calcd for C31H44NO7, 542.3118). The NMR data (Table 2) verified compound 17 was substituted with one N-ethyl group [δH 2.68–2.78 (1H, m), 2.40–2.47 (1H, m), 1.09 (3H, t, J = 7.2 Hz); δC 49.7, 13.4], three methoxy groups [δH 3.29 (3H, s), δC 59.6; δH 3.37 (3H, s), δC 57.1; 3.31 (3H, s), δC 56.4], one benzoyl group [δH 7.49–7.55 (1H, m), 7.98–8.03 (2H, m), 7.37–7.44 (2H, m); δC 166.4, 133.0, 130.3, 129.8 × 2, 128.6 × 2]. The remaining 19 carbons, comprising six methylenes, ten methines, and three quaternary carbons, suggested it was a typical C19-DA.41 Besides, the 6 oxygenated carbons, indicated the substitution of another two hydroxy groups, except for the three methoxy groups and one benzoyl group. HMBC cross-peaks of the three methoxy groups (δH 3.31, 3.37, 3.29) with correlated carbons attested their substitutions at C-1 (δC 85.3), C-16 (δC 90.7), and the methylene carbon C-18 (δC 79.6), respectively. And correlations between signals of H-14 [δH 5.07 (1H, t, J = 4.8 Hz)] and the ester carbonyl carbon (δC 166.4) obtained from HMBC spectrum revealed the substitution of benzoyl group at C-14 (δC 76.9). Moreover, 1H–1H COSY cross-peak between H-15 [δH 4.39 (1H, d, J = 6.7 Hz)] and H-16 [δH 3.09 (1H, d, J = 6.7 Hz)] indicated a hydroxyl group located at C-15 (δC 79.9). And another hydroxyl group was speculated to be placed at C-8 (δC 78.3) based on the long-range correlations of the oxygenated quaternary carbon C-8 with H-7 [δH 2.58 (1H, s)], H-9 [δH 2.47–2.49 (1H, m)], H-14, and H-15 in HMBC spectrum. In addition, the relative configuration of compound 17 was established by NOE correlations between H-1 [δH 3.20 (1H, m)] and H-10β [δH 1.98–2.07 (1H, m)], H-14 and H-13β [δH 2.52–2.56 (1H, m)], and between H-15 and H-2′, 6′ [δH 7.98–8.03 (2H, m)], indicating the α-configurations of 1-OMe, 14-OH, and 15-OH, respectively. And correlations of one proton at C-12 [δH 1.98–2.07 (1H, m)] with H-14, and another proton at C-12 [δH 2.47–2.49 (1H, m)] with H-16 implied the β-configuration of 16-OMe. Accordingly, the structure of compound 17 was determined as shown in Fig. 2, and was named as carmichaeline K, which was isolated as a free base for the first time in this study, compared with the previously reported carmichaeline K trifluoroacetate.32,46–48 And the main differences of their NMR spectra lie in the chemical shifts of 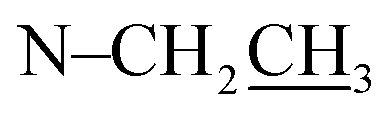 group, which were δH 1.09 (3H, t, J = 7.2 Hz) and δC 13.4 for compound 17, while δH 1.36–1.58 and δC 10.0–11.4 for the corresponding salts.
group, which were δH 1.09 (3H, t, J = 7.2 Hz) and δC 13.4 for compound 17, while δH 1.36–1.58 and δC 10.0–11.4 for the corresponding salts.
Compound 25 was isolated as a white, amorphous powder, and its molecular formula was established as C34H49NO9 based on its HR-ESI-MS data at m/z 616.3509 ([M + H]+, calcd for C34H50NO9, 616.3486). The NMR signals (Table 2) implied the existence of one N-ethyl group [δH 3.34–3.38 (1H, m), 2.92–3.01 (1H, m), 1.29 (3H, t, J = 5.5 Hz); δC 50.1, 12.3], four methoxy groups [δH 3.34 (3H, s), δC 56.3; δH 3.27 (3H, s), δC 58.8; δH 3.79 (3H, s), δC 62.0; δH 3.31 (3H, s), δC 59.3], one benzoyl group [δH 8.02–8.07 (2H, m), 7.45 (2H, t, J = 7.7 Hz), 7.53–7.59 (1H, m); δC 166.4, 130.3, 129.9 × 2, 128.5 × 2, 133.1] and one ethoxy group [δH 3.46–3.50 (1H, m), 3.38–3.44 (1H, m), 0.56 (3H, t, J = 6.9 Hz); δC 57.5, 15.3]. The remaining 19 carbons, including 8 oxygenated carbons, constituted the skeleton of C19-DA,45 and indicated the substitution of two hydroxy groups. The locations of four methoxy groups and one benzoyl group were confirmed according to the correlation signals of H-1/H-2/H-3, H-5/H-6, and H-14/H-9/H-10 in 1H–1H COSY spectrum, as well as the HMBC correlations between signals of 1-OMe [δH 3.34 (3H, s)], 6-OMe [δH 3.27 (3H, s)], 16-OMe [δH 3.79 (3H, s)], 18-OMe (δH 3.31), H-14 [δH 4.83 (1H, d, J = 5.1 Hz)] and C-1 (δC 83.4), C-6 (δC 82.6), C-16 (δC 93.1), C-18 (δC 79.2), 14-OBz (δC 166.4), respectively. One hydroxy group was attached to C-15 (δC 77.7) based on the H-15/H-16 correlation in 1H–1H COSY spectrum, and the other one attached to C-13 (δC 74.8) according to the doublet peak of H-14, and HMBC correlations of H-9 [δH 2.60–2.68 (1H, m)], H-12 [δH 2.20–2.23 (2H, m)], H-14, and H-16 [3.34–3.38 (1H, m)] with C-13 (δC 74.8). Besides, the ethoxy group was placed at the oxygenated quaternary carbon C-8 (δC 82.6) according to the HMBC correlations from H-6 [δH 4.09 (1H, d, J = 6.4 Hz)], H-9, H-10 [2.15 (1H, m)], H-14, and H-15 [δH 4.59 (1H, d, J = 5.5 Hz)] to C-8 (δC 82.6), and the NOE correlation of H-7 [δH 2.88 (1H, s)] to 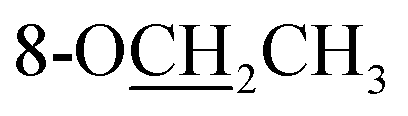 [δH 3.48 (1H, m)]. In addition, NOESY cross-peaks of H-1/H-5β, and H-10β; H-6/H-5β, H-9β, and H-17β; H-14/H-9β, and H-10β; H-15/H-2′, 6′, and 16-OMe indicated that 1-OMe, 6-OMe, 14-OBz, and 15-OH took α-configurations, while 16-OMe was β-configuration. Consequently, as exhibited in Fig. 2, compound 25 was assigned as 8-O-ethyl-benzoyldeoxyaconine.
[δH 3.48 (1H, m)]. In addition, NOESY cross-peaks of H-1/H-5β, and H-10β; H-6/H-5β, H-9β, and H-17β; H-14/H-9β, and H-10β; H-15/H-2′, 6′, and 16-OMe indicated that 1-OMe, 6-OMe, 14-OBz, and 15-OH took α-configurations, while 16-OMe was β-configuration. Consequently, as exhibited in Fig. 2, compound 25 was assigned as 8-O-ethyl-benzoyldeoxyaconine.
In order to verify whether these compounds were artifacts, the powdered dried lateral roots of Aconitum carmichaelii Debx. were also extracted by 1% hydrochloric acid (HCl) containing water, and the HR-ESI-MS spectrum showed compounds 25–29 with an oxyethyl group substituted at C-8 position were artificial products, which could be produced in the presence of ethanol according to previous reports.32
Anti-tumor activity assay
Anti-tumor activities of all the isolated compounds and the positive drug 5-fluorouracil (5-FU) against human non-small-cell lung cancer A549 and H460 cells and non-cancerous HBE cells were examined, the IC50 values expressed as means ± s.d. from three independent experiments were listed in Table 3, and representative IC50 curves for each compound were displayed in Fig. S53.† It could be concluded that all the C19 DAs showed moderate inhibitory activities against both of the two cancerous cell lines with IC50 values in the range of 7.97–28.42 μM, while all the C20-DAs only exhibited moderate activities against H460 cells with IC50 values from 13.56 to 27.44 μM, but several C20-DAs seemed to possess little inhibition against A549 cells, displaying IC50 values of more than 30 μM. Besides, most of the DAs displayed selectivity towards one or both of the two cancerous cell lines over non-cancerous cell line to some extent, and several compounds such as compounds 9, 25, etc. exhibited higher selectivity. Furthermore, the predicted properties for compound 9 also fall within the Lipinski's rule of five, with molecular weight 377.52, 3 hydrogen bond donors, 5 hydrogen bond acceptors, 5 freely rotatable bonds and log P value 1.278 ± 0.521. And its ADME parameters predicted by a web-based application PreADMET (https://preadmet.bmdrc.kr/) also indicated its moderate absorption and bioavailability, including the permeability of Caco-2 cell (value of 15.54), percentage of human intestinal absorption (HIA, value of 88.24), and skin permeability (value of −5.58) to estimate absorption, blood brain barrier (BBB, value of 0.31) and plasma protein binding (PPB, value of 29.33) for drug penetration, etc. On basis of these results, DAs and their derivatives, especially those with the C19-skeletons, may have the potentials for future development of anti-tumor agents. Besides, their physicochemical properties, biochemical properties, pharmacokinetic and pharmacodynamic parameters including ADMET and so on also need to be further evaluated.| Compound | HBE | A549 | H460 | Compound | HBE | A549 | H460 |
|---|---|---|---|---|---|---|---|
| 1 | 37.86 ± 2.16 | 48.10 ± 11.65 | 27.44 ± 0.55 | 20 | 30.66 ± 10.14 | 21.54 ± 2.86 | 21.79 ± 2.11 |
| 2 | 39.91 ± 6.20 | 75.30 ± 12.48 | 24.33 ± 1.66 | 21 | 25.66 ± 3.72 | 16.07 ± 0.73 | 24.06 ± 4.74 |
| 3 | 21.77 ± 2.24 | 17.65 ± 1.35 | 14.14 ± 0.80 | 22 | 32.80 ± 3.66 | 25.95 ± 10.25 | 22.31 ± 1.04 |
| 4 | 36.47 ± 2.49 | 16.49 ± 0.29 | 24.14 ± 1.71 | 23 | 28.74 ± 1.05 | 20.60 ± 0.25 | 18.90 ± 4.45 |
| 5 | 32.41 ± 2.62 | 21.24 ± 0.84 | 25.10 ± 0.59 | 24 | 21.56 ± 0.99 | 17.09 ± 2.08 | 13.48 ± 1.00 |
| 6 | 28.76 ± 2.36 | 25.24 ± 0.25 | 20.94 ± 0.68 | 25 | 50.07 ± 9.82 | 12.58 ± 1.82 | 12.76 ± 2.10 |
| 7 | 21.59 ± 8.19 | 13.67 ± 0.47 | 13.56 ± 0.23 | 26 | 25.51 ± 2.22 | 14.49 ± 0.71 | 13.87 ± 0.67 |
| 8 | 43.72 ± 4.39 | 64.16 ± 1.70 | 26.46 ± 3.08 | 27 | 30.21 ± 1.28 | 17.61 ± 1.17 | 18.81 ± 1.57 |
| 9 | 49.74 ± 8.11 | 8.28 ± 0.41 | 9.69 ± 0.40 | 28 | 31.35 ± 2.62 | 20.13 ± 0.73 | 18.31 ± 1.07 |
| 10 | 21.38 ± 1.18 | 8.33 ± 0.20 | 12.23 ± 0.97 | 29 | 32.09 ± 1.85 | 17.82 ± 1.00 | 19.38 ± 4.82 |
| 11 | 24.30 ± 2.18 | 17.09 ± 3.69 | 12.72 ± 3.68 | 30 | 31.43 ± 6.00 | 16.36 ± 2.38 | 16.00 ± 1.15 |
| 12 | 21.15 ± 0.72 | 8.36 ± 0.82 | 9.92 ± 0.71 | 31 | 27.73 ± 0.98 | 18.46 ± 2.03 | 22.22 ± 2.58 |
| 13 | 26.21 ± 3.78 | 12.25 ± 1.05 | 10.70 ± 1.60 | 32 | 29.63 ± 1.73 | 24.56 ± 3.24 | 18.50 ± 1.58 |
| 14 | 27.31 ± 3.65 | 18.16 ± 1.32 | 22.80 ± 0.81 | 33 | 32.70 ± 4.56 | 14.31 ± 1.79 | 16.52 ± 2.22 |
| 15 | 24.84 ± 2.50 | 12.33 ± 1.59 | 13.69 ± 0.77 | 34 | 30.99 ± 4.80 | 14.87 ± 4.02 | 14.64 ± 1.32 |
| 16 | 20.20 ± 1.83 | 8.92 ± 0.86 | 7.97 ± 0.31 | 35 | 26.31 ± 0.98 | 13.01 ± 5.69 | 15.55 ± 4.84 |
| 17 | 31.03 ± 10.73 | 21.30 ± 3.27 | 22.30 ± 2.16 | 36 | 26.85 ± 0.62 | 26.46 ± 0.86 | 28.42 ± 8.89 |
| 18 | 44.53 ± 13.56 | 25.43 ± 9.64 | 23.36 ± 2.35 | 37 | 32.59 ± 5.94 | 19.43 ± 1.57 | 14.41 ± 0.64 |
| 19 | 34.11 ± 2.88 | 13.88 ± 0.52 | 15.69 ± 0.18 | 5-FU | 8.93 ± 6.05 | 12.52 ± 8.81 | 6.16 ± 1.80 |
Structure–activity relationship
The structure–activity relationships of the isolated C19-DAs were discussed based on the comprehensive analysis of different types and positions of their substituents, as well as the IC50 values against A549 and H460 cells. First of all, it is obvious that substituents at C-8 position exerted an influence on the anti-tumor activities of C19-DAs. The diester C19-DAs with an acetyl group attached to the C-8 position showed stronger activities than monoester C19-DAs with the hydroxy group, as indicated by comparison of compounds 33, 34 and 35 with compounds 20, 21, and 22, respectively, which is similar with those reported in literatures.8 Furthermore, when the hydroxy group at C-8 position was further replaced by an ethoxy group, such as in compounds 25–27, the inhibitory activities were also strengthened. Besides, the hydroxy group at C-10 position displayed a slight increase in anti-tumor activities of C19-DAs, as illustrated by compounds 24 and 29, compared with compounds 22 and 27. On top of that, the replacement of the 14-acetyl or benzoyl with the hydroxy group also leads to the improvement in inhibitory activities on A549 and H460 cells, which was attested by pairs of compounds 10 and 30, 11 and 32, 15 and 20, 16 and 22. Additionally, slight decrease was observed in the anti-tumor activities of N-methyl substituted compounds 20, 22, 26, and 27, than N-ethyl substituted compounds 21, 23, 25, and 28, suggesting that substituents at nitrogen atom of C19-DAs have slight effects on their anti-tumor activities against A549 and H460 cells. However, hydroxy group substituted at C-3 position seemed to exert no significant effects on the anti-tumor activities of C19-DAs, by comparing the IC50 values of compounds 20 with 22, and 33 with 35. Based on all of the above evidences, it is concluded that C-8 and C-14 positions of C19-DAs are active sites which could be replaced by different substituents for development of potential anti-tumor agents. Besides, substituents at C-10 position and nitrogen atom may also slightly alter the anti-tumor activities of C19-DAs.Network pharmacology analysis
All of the isolated DAs, with chemical structures confirmed, were further applied for network pharmacology analysis to explore the possible mechanisms of their anti-tumor activities. Compound-associated-target prediction was firstly performed by screening the metaTarFisher (https://metatarget.scbdd.com/) search server which integrated more than 10 popular target fishing tools, resulting in discovery of 362 target genes. Meanwhile, DisGeNET platform was also applied for discovery of 3926 genes closely related with non-small-cell lung carcinoma. The intersection of these compound-associated genes and disease-associated genes generated 173 potential target genes, which was further used for protein–protein interaction (PPI) analysis by STRING database to search for the proteins that may jointly contribute to a function as well as the core proteins which may play a pivotal role in the entire network, according to the PPI network node degree value, as depicted in Fig. 6 and listed in Table S3.† Furthermore, GO/KEGG enrichment analysis was performed on Metascape platform to reveal the most important KEGG pathways, reactome gene sets and GO biological processes involved in their bioactivities. According to the Qvalues (Table S4† and Fig. 7), “pathways in cancer”, “signaling by interleukins” and “regulation of MAPK cascade” were the three most important pathways related to their anti-tumor effects. Moreover, the whole “compound-target-disease” network was constructed in Cytoscape software to demonstrate the potential anti-tumor mechanisms of DAs in treating cancer, as represented in Fig. 8.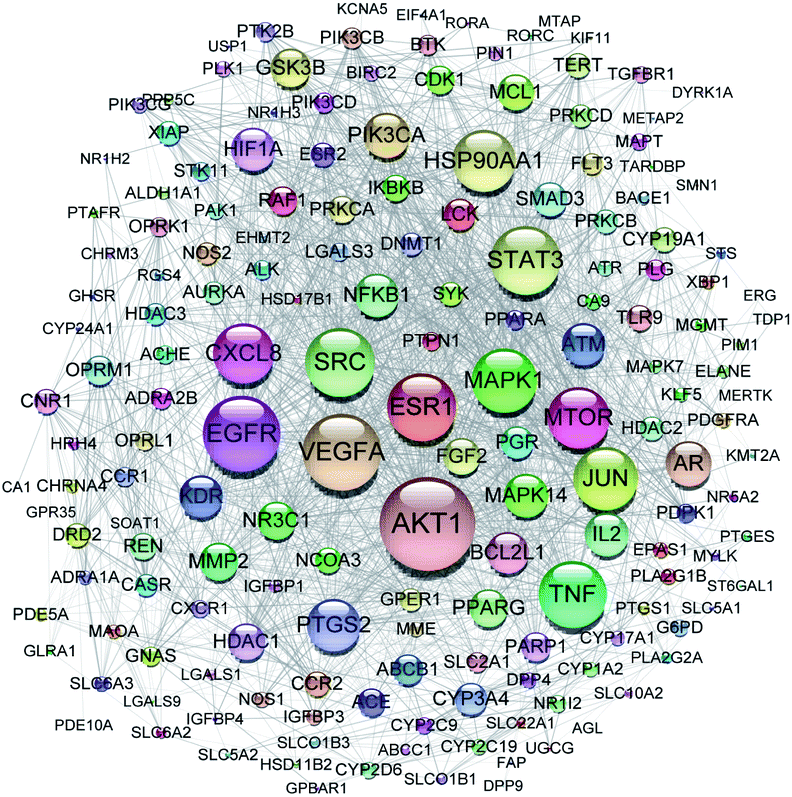 | ||
| Fig. 6 PPI network of 173 target genes. The size of the target genes represented the node degree value. | ||
 | ||
| Fig. 7 Bubble graph demonstrating GO/KEGG enrichment analysis results. | ||
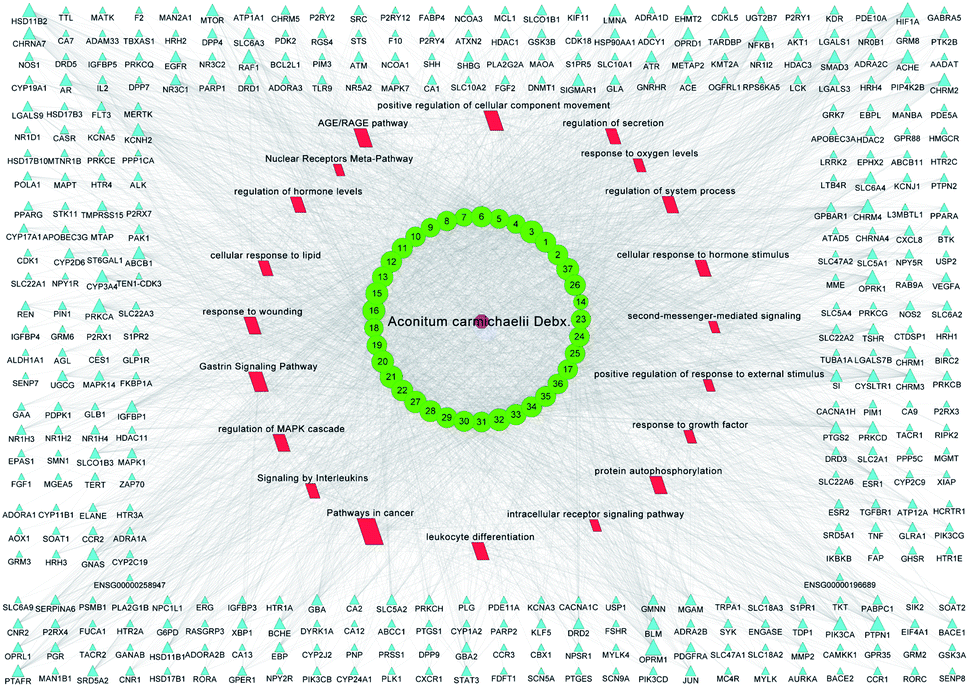 | ||
| Fig. 8 Visualization of “compound-target-pathway” network. The purple octagon-shaped node represented Aconitum carmichaelii Debx., the green ellipse-shaped node represented the DA compounds 1–37, the blue triangle-shaped node represented the target genes, the red parallelogram-shaped node represented the pathways, while the size of the node represented the degree value. | ||
Based on the above target genes and related pathways, the underlying anti-tumor mechanisms of DAs might be explained in the following aspects. Firstly, pathways in cancer, especially the PI3K-AKT signaling pathway, according to the target genes (AKT1, PIK3CA, PIK3CB, PIK3CD, mTOR, etc.) involved, as listed in Table S4,† which plays a crucial part in tumor progression.49 PI3K was activated when stimulated by upstream signals, such as TGF, EGF, PDGF, FLT3 and so on, and further activated AKT and various downstream signaling molecules, including NF-κB, mTOR, eNOS, VEGF, GSK3β, etc. to regulate the proliferation, invasion and metastasis, apoptosis, angiogenesis and carbohydrate metabolism of tumor cells.50 By inhibiting the expression of various molecules in the pathway, the growth and metastasis of tumors can be inhibited, and various therapeutic drugs targeting this pathway have been continuously developed and used in pre-clinical research and clinical trials for tumor treatment.51 DAs and their derivatives may also have the anti-tumor effects by blocking the PI3K-AKT signaling pathway.
Besides, DAs were also associated with signaling by interleukins. Interleukins were first identified as cytokines interacting between leukocytes during immune responses, further studies showed they could also take part in regulations of many other cells throughout the body, and play an important role in tumor immunobiology.52 For example, IL-2 exerted an influence on cell growth and activation, and showed clinical efficacy in both solid tumors and hematologic malignancies.53 Therefore, DAs may have potentials in treating cancer by influencing interleukins signals.
Moreover, DAs may also influence cell proliferation, differentiation, apoptosis, metastasis, and invasion by regulation of MAPK cascade, which mainly contains four main families, ERKs, p38MAPKs, JNKs, and ERK5.54 Among them, ERK1/2 were activated abnormally in a variety of tumors after phosphorylation, and acted on certain regulatory factors to promote cell proliferation, inhibit cell apoptosis, and regulate cell cycle.55 Many chemotherapeutic drugs inhibit the growth of tumors by inhibiting the activation of ERK. Some research also found the activation of ERK-MAPK transduction pathway could activate downstream tumor suppressor genes and initiate the p53-dependent apoptosis pathway to inhibit tumor growth.56 Besides, the mechanism of DAs in regulation of MAP kinase activity was also supported by previous report that Aconitum species played an anti-tumor role by inducing apoptosis of A549 cells through the activation of p38 MAPK-pathway.7
In summary, DAs may inhibit tumor cell growth and promote apoptosis by regulating different signal transduction pathways including the PI3K-AKT signaling pathway, interleukins signaling pathways, MAPK signaling pathway, etc. And the whole mechanism network formed by their intricate relationships ultimately determines the survival and apoptosis of tumor cells, which deserves further investigation.
Experimental section
General experimental procedures
The optical rotation data were determined on an Autopol VI Automatic Polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). The IR spectra in the range of 4000–650 cm−1 were recorded on a Nicolet iS50 FTIR spectrometer using a Diamond ATR module and processed by OMNIC v9.7.7 software (Thermo Fisher Scientific, Waltham, MA, USA). HR-ESI-MS analysis were conducted on Waters UPLC HClass system and Xevo QTOF mass spectrometer (Waters Corporation, Milford, MA, USA) using a Waters CSH C18 column (100 mm × 2.1 mm, 1.8 μm). All of the 1D and 2D NMR spectra (1H NMR, 13C NMR, DEPT-135 NMR, 1H–1H COSY, HSQC, HMBC, and NOESY) were obtained on a Bruker Avance III 500 MHz spectrometer (Bruker BioSpin AG, Fällanden, Switzerland) or a Bruker Avance III HD 600 MHz spectrometer (Bruker BioSpin AG, Fällanden, Switzerland) using tetramethylsilane as an internal standard with chemical shifts reported in parts per million (ppm). Preparative high-performance liquid chromatography (HPLC) separations were carried out on Agilent 1200 HPLC system with an ODS column (50 cm × 34 mm, 50 μm; YMC Co., Ltd., Kyoto, Japan), BUCHI Pure C-815 Flash system (BÜCHI Labortechnik, Flawil, Switzerland) with a YMC Triart Prep C18 column (40 × 160 mm, 20 μm; YMC Co., Ltd., Kyoto, Japan), Hanbon NP7001C HPLC system (Hanbon Sci. & Tech, Jiangsu, China) with an Acchrom XCharge C18 column (20 × 250 mm, 5 μm), and a TBE-300B high-speed counter-current chromatography equipped with a Model TBP-1002 constant-flow pump and a Model TBD-2000UV detector (Tauto Biotech Co. Ltd, Shanghai, China). Semi-preparative HPLC purifications were carried out on an Agilent 1100 HPLC system with a VWD detector using an Agilent Eclipse XDB-C18 column (9.4 × 250 mm, 5 μm; Agilent Technologies, Palo Alto, CA, USA), or a Waters XSelect CSH C18 column (10 × 250 mm, 5 μm; Waters, Milford, MA, USA) for acidic mobile phase, and a Phenomenex Gemini C18 column (10 × 250 mm, 5 μm; Phenomenex, Torrance, CA, USA), or a Waters Xbridge Prep C18 column (10 × 250 mm, 5 μm; Waters, Milford, MA, USA) for alkaline mobile phase. Column chromatography (CC) was carried out on silica gel (200–300 mesh; Qingdao Marine Chemical, Inc., Qingdao, China), MCI gel (75–100 μm, Mitsubishi Chemical Co., Ltd., Japan), and macroporous resin D101 (Shanghai Sinopharm Chemical Reagent Co. Ltd., Shanghai, China). And preparative thin layer chromatography (TLC) was performed on PLC silica gel 60 F254 plates (Merk Chemicals Co. Ltd., Shanghai, China).The organic solvents for sample extraction, column chromatography, preparative HPLC, and preparative TLC were of analytical grade and supplied by Shanghai Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Acetonitrile (MeCN; Shanghai Sinopharm Chemical Reagent Co. Ltd., Shanghai, China), diethylamine (DEA; TCI, Tokyo, Japan), and formic acid (FA; Shanghai Aladdin Bio-Chem Technology Co. Ltd, Shanghai, China) applied in semi-preparative HPLC were of HPLC-grade, and ammonium acetate (NH4OAc; ≥98%) was purchased from Sigma-Aldrich (St. Louis, MO, US). And the ultra-pure water was prepared by a Millipore Alpha-Q water purification system (Millipore, Bedford, USA).
Plant material
The dried lateral roots of Aconitum carmichaelii Debx. were purchased from Jiangyou, Sichuan province, China, in January 2019 and identified by Prof. De-an Guo. A voucher specimen was deposited at the National Engineering Laboratory for Traditional Chinese Medicine Standardization Technology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China.Extraction and isolation
The powdered dried lateral roots of Aconitum carmichaelii Debx. (10.0 kg) were extracted by heating at reflux with 75% ethanol (EtOH) for three times. After filtration, the extracting solvents were combined and evaporated under reduced pressure using a rotary evaporation equipment (Buchi, Switzerland) at the temperature of 40 °C. Subsequently, the obtained extracts (1.27 kg) were dissolved in distilled water, and successively applied for liquid–liquid partition using petroleum ether (PET; 60–90 °C), dichloromethane (CH2Cl2), ethyl acetate (EtOAc), and n-butanol (BuOH) saturated with water, and then evaporated under reduced pressure to obtain 38.49 g of the PET extracts, 76.71 g of the CH2Cl2 extracts, 7.66 g of the EtOAc extracts, and 151.28 g of the BuOH extracts. Integration of various chromatographic separation methods were carried out for further isolation and purification of DAs.The PET extracts were separated on a silica gel column eluted with a gradient of PET–acetone containing 1% DEA (50:1–1:4, v/v) to afford 14 fractions (PET-Fr.1 to Fr.14). Among them, Fr.4 was separated using silica gel column with a gradient elution of cyclohexane–acetone containing 1% DEA (30:1–6:1), and then preparative TLC (n-hexane–EtOAc–DEA, 5:2:1, v/v) to give compound 33 (31 mg). Fr.5 and Fr.7 were directly purified by semi-preparative HPLC (55% MeCN–10 mM NH4OAc containing H2O, and 74% MeCN–5 mM NH4OAc containing H2O), and basified with aqueous saturated sodium bicarbonate to obtain compound 21 (2 mg) and 24 (4 mg), respectively. And preparative HPLC with a gradient of MeCN–5 mM NH4OAc containing H2O was applied to separate Fr.12, followed by semi-preparative HPLC (52% MeCN–5 mM NH4OAc containing H2O) and alkaline treatment to yield compound 22 (5 mg).
For CH2Cl2 extracts, an MCI gel column (500 g) was applied to obtain 14 fractions (Fr.1–Fr.14) with a gradient elution of 5–95% EtOH containing 0.5% ammonium hydroxide. Fr.2 (4.41 g) and Fr.11 (3.03 g) were subsequently loaded onto a Triart Prep C18 column on Flash system with the gradient elution of MeCN–H2O (containing 0.015% DEA) to obtain Fr.2-1 to Fr.2-9, and Fr.11-1 to Fr.11-7, respectively. Semi-preparative HPLC (37% MeCN–0.015% DEA containing H2O, and 33% MeCN–0.015% DEA containing H2O) was further applied in purification of Fr.2-5 (113.9 mg) and Fr.2-6 (39.8 mg) to obtain compound 10 (19 mg) and compound 9 (7 mg), respectively. Fr.11-6 (384.6 mg) was separated on Flash system using an ODS column and MeCN–0.1% FA containing H2O as mobile phase, followed by preparative TLC (n-hexane–EtOAc–DEA, 5:2:1, v/v) to yield compound 28 (15 mg). And Fr.11-7 was purified by semi-preparative HPLC (67% MeCN–0.015% DEA containing H2O) to give compound 2 (3 mg). Other fractions including fraction 3 (2.77 g), 6 (5.72 g), 10 (5.55 g), 13 (5.97 g), and 14 (8.39 g) were all separated on an ODS column on MHPLC system with a gradient elution of MeCN–0.1% FA containing H2O to obtain Fr.3-1 to Fr.3-8, Fr.6-1 to Fr.6-11, Fr.10-1 to Fr.10-13, Fr.13-1 to Fr.13-7, and Fr.14-1 to Fr.14-9, respectively. And semi-preparative HPLC was further applied to obtain compound 4 (2 mg), 11 (77 mg), 29 (2 mg), 30 (14 mg), 32 (5 mg), 36 (5 mg), and 37 (17 mg) from the main subfractions of Fr.6, compound 5 (19 mg), 8 (14 mg), 12 (5 mg), 14a (9 mg), 15 (47 mg), 16(35 mg), 27 (20 mg), and 31 (21 mg) from subfractions of Fr.10, compound 20 (3 mg) and 26 (9 mg) from Fr.13, and compound 17 (3 mg), 18 (2 mg), 19 (15 mg), and 34(2 mg) from Fr.14.
Besides, the BuOH extracts were chromatographed over a macroporous resin D101 column (1.5 kg) eluted with H2O–EtOH (50:1 to 3:5) gradient system to give five fractions (Fr.1–Fr.5). Then an MCI column was applied to separate Fr.2 into 13 fractions (Fr.2-1 to Fr.2-13), among which Fr.2-8 was purified by semi-preparative HPLC (30% MeCN–0.015% DEA containing H2O) to obtain compound 13 (25 mg), Fr.2-9, Fr.2-13 and Fr.2-12 were all successively separated by preparative HPLC (5–8% MeCN–0.1% FA containing H2O), and semi-preparative HPLC (5% MeCN–0.1% FA containing H2O, 7% MeCN–0.1% FA containing H2O, and 40% MeCN–0.015% DEA containing H2O) to afford compound 3 (7 mg), 7 (8 mg), 6 (68 mg), and 1 (12 mg), respectively.
In addition, HSCCC equipment was applied for rapid preparation of the major DAs in Aconitum carmichaelii Debx. As reported previously,57 CH2Cl2–EtOAc–MeOH–0.3% HCl containing H2O (2.75:1:1.5:2) and CH2Cl2–BuOH–MeOH–0.4% HCl containing H2O were adopted as the solvent systems for separation of Aconitum carmichaelii Debx. and its processed products, respectively. And then preparative TLC was further used for purification of compound 23 (2 mg) and compound 35 (5 mg).
Cell culture and cell viability assays
Human lung cancer cell lines A549, H460 and human bronchial epithelial cell line HBE from the American Type Culture Collection (Manassas, VA34) were maintained in Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY) supplemented with 100 units per μL penicillin, 100 μg mL−1 streptomycin and 10% fetal bovine serum at 37 °C in a 5% CO2 incubator. Cells were seeded in 96-well plates at a concentration of 5 × 103 cells per well with complete culture medium and allowed to adhere to the plate overnight. Cell proliferation assays were performed using the MTT Cell Proliferation Kit (BSD Applied Science, Wuhan, China) according to the manufacturer's instructions. Each combination of cell line and drug concentration was set up in five replicates and repeated at least three times. Data were visualized using GraphPad Prism v.8.01 and IC50 curves were fitted using a non-linear regression model with a sigmoidal dose response characteristic.Network pharmacology analysis
All of the isolated DAs in this study were used to explore the “compound-target-pathway” network of DAs. Firstly, their chemical structures were converted into SMILES formats, and uploaded onto metaTarFisher (https://metatarget.scbdd.com/) search server for prediction of compound-associated targets. MetaTarFisher can be applied to obtain the possible targets of a compound based on its comprehensive information by integrating more than 10 widespread target fishing tools, including SwissTargetPrediction,58 Similarity ensemble approach (SEA),59 Polypharmacology Browser,60,61 PASSonline,62 TargetHunter,63 HitPickV2,64 TargetNet,65 DNNTargetPridict, SimRanking, ChemMapper,66 and ChEMBL.67 Meanwhile, genes associated with non-small-cell lung carcinoma were also explored using DisGeNET (https://www.disgenet.org/) platform. Intersection of the compound-associated targets and disease-associated targets were obtained, and Uniprot ID of these obtained targets were further transformed into preferred name (gene symbol) by ID mapping in STRING68 database (https://string-db.org/) for the convenience of following gene enrichment analysis. And protein–protein interaction (PPI) analysis was also conducted using STRING database, choosing the parameters of organisms as Homo sapiens, and others default (medium confidence 0.400). Moreover, GO/KEGG enrichment analysis was performed on MetaScape69 platform (https://metascape.org/) to interpret the possible molecular functions and pathways of these target genes, which was depicted by a bubble graph drawn in imageGP platform (http://www.ehbio.com/ImageGP/). At last, “compound-target-pathway” network was constructed using Cytoscape v3.8.2 software70 to demonstrate the bioactive mechanisms of DAs, which may provide possible explanations to their anti-tumor activities, rendering a theoretical reference for future mechanism research.Conclusions
A total of thirty-seven structurally diverse DAs were obtained from the lateral roots of Aconitum carmichaelii Debx., including three new natural compounds, one new DA derivative. Among them, compound 1 represented the first example of C20-DA glucoside. The structures of all the isolated DAs were identified by a combination of HR-ESI-MS and NMR spectra, and their anti-tumor activities were systematically evaluated against human non-small-cell lung cancer cell lines A549 and H460 for discussion of their structure–activity relationships. Results showed that all the C19-DAs exhibited moderate inhibitory activities with IC50 values in the range of 7.97–28.42 μM, and positions C-8, C-10, C-14 and nitrogen atom in the C19 skeleton were reckoned as the active sites, which may affect their anti-tumor activities. In addition, all of the isolated DAs were applied for network pharmacology analysis to interpret the possible mechanisms of DAs in treating cancer, laying the theoretical foundations for future research.Author contributions
D. G. conceived the experiment. Y. Y. carried out the isolation experiment, analyzed the data, and wrote the manuscript. S. W. completed the cell culture and cell viability assays investigation. J. Z., J. L., C. Y. and W. W. (Wenlong Wei) offered technical support of the experiment and revised the manuscript. W. W. (Wenyong Wu) helped with the network pharmacology analysis. Y. W., H. J., M. G. and Y. L. took part in the experiment. S. Y., Y. H., Q. B., and H. Q. provided resources of the experiment. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2019YFC1711000), and Qi-Huang Chief Scientist Program of National Administration of Traditional Chinese Medicine (2020).References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- C. L. Yao, J. Q. Zhang, J. Y. Li, W. L. Wei, S. F. Wu and D. A. Guo, Nat. Prod. Rep., 2021 10.1039/d0np00057d.
- W. J. Slichenmyer, E. K. Rowinsky, R. C. Donehower and S. H. Kaufmann, J. Natl. Cancer Inst., 1993, 85, 271–291 CrossRef CAS PubMed.
- F. P. Wang, Q. H. Chen and X. Y. Liu, Nat. Prod. Rep., 2010, 27, 529–570 RSC.
- X. X. Liu, X. X. Jian, X. F. Cai, R. B. Chao, Q. H. Chen, D. L. Chen, X. L. Wang and F. P. Wang, Chem. Pharm. Bull., 2012, 60, 144–149 CrossRef CAS PubMed.
- L. Xiong, C. Peng, X. F. Xie, L. Guo, C. J. He, Z. Geng, F. Wan, O. Dai and Q. M. Zhou, Molecules, 2012, 17, 9939–9946 CrossRef CAS.
- Y. P. Fan, Y. D. Jiang, J. J. Liu, Y. X. Kang, R. Q. Li and J. Y. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 380–387 CrossRef CAS PubMed.
- F. Gao, Y. Y. Li, D. Wang, X. Huang and Q. Liu, Molecules, 2012, 17, 5187–5194 CrossRef CAS.
- M. Y. Ren, Q. T. Yu, C. Y. Shi and J. B. Luo, Molecules, 2017, 22, 267 CrossRef PubMed.
- K. Wada and H. Yamashita, Molecules, 2019, 24, 2317 CrossRef CAS.
- G. H. Zhou, L. Y. Tang, X. D. Zhou, T. Wang, Z. Z. Kou and Z. J. Wang, J. Ethnopharmacol., 2015, 160, 173–193 CrossRef CAS.
- S. Liu, F. Li, Y. Li, W. Li, J. Xu and H. Du, J. Ethnopharmacol., 2017, 207, 237–250 CrossRef PubMed.
- M. Murayama, Jpn. Pat., JP06016553A, 1994 Search PubMed.
- M. Murayama, Jpn. Pat., JP07112971A, 1995 Search PubMed.
- L. V. Beshitaishvili and M. N. Sultankhodzhaev, Chem. Nat. Compd., 1992, 28, 206–208 CrossRef.
- A. G. González, G. de La Fuente, M. Reina and R. D. Timón, Phytochemistry, 1986, 25, 1971–1973 CrossRef.
- X. X. Zong, X. J. Yan, J. L. Wu, Z. Q. Liu, H. Zhou, N. Li and L. Liu, J. Nat. Prod., 2019, 82, 980–989 CrossRef CAS.
- D. Csupor, P. Forgo, K. Csedo and J. Hohmann, Helv. Chim. Acta, 2006, 89, 2981–2986 CrossRef CAS.
- T. Kiss, P. Orvos, S. Bánsághi, P. Forgo, N. Jedlinszki, L. Tálosi, J. Hohmann and D. Csupor, Fitoterapia, 2013, 90, 85–93 CrossRef CAS.
- J. G. Díaz, J. G. Ruiz and G. de La Fuente, J. Nat. Prod., 2000, 63, 1136–1139 CrossRef PubMed.
- Y. Liang, J. L. Wu, X. Li, M. Q. Guo, E. L. H. Leung, H. Zhou, L. Liu and N. Li, Tetrahedron Lett., 2016, 57, 5881–5884 CrossRef CAS.
- A. Ulubelen, J. Nat. Prod., 1996, 59, 360–366 CrossRef CAS PubMed.
- P. Forgo, B. Borcsa, D. Csupor, L. Fodor, R. Berkecz, V. A. Molnar and J. Hohmann, Planta Med., 2011, 77, 368–373 CrossRef CAS PubMed.
- Q. Li, S. D. Sun, M. Y. Wang, C. F. Li, D. Yuan and H. Z. Fu, J. Chin. Pharm. Sci., 2018, 27, 855–863 CrossRef.
- W. Y. Liu, D. He, D. K. Zhao, Y. P. Chen and Y. Shen, J. Asian Nat. Prod. Res., 2019, 21, 9–16 CrossRef CAS PubMed.
- F. Zhang, S. L. Peng, X. Liao, K. B. Yu and L. S. Ding, Planta Med., 2005, 71, 1073–1076 CrossRef CAS PubMed.
- W. Wei, X. W. Li, H. Y. Zhou, R. J. Yang, X. L. Shi, J. Shan, L. Meng and Y. R. Jin, J. Jilin Univ., Sci. Ed., 2010, 48, 127–132 CAS.
- C. Konno, M. Shirasaka and H. Hikino, J. Nat. Prod., 2004, 45, 128–133 CrossRef.
- F. P. Wang and S. W. Pelletier, J. Nat. Prod., 2004, 50, 55–62 CrossRef.
- W. Wei, X. W. Li, Y. R. Jie, X. L. Shi and H. F. Sun, Chin. J. Magn. Reson., 2010, 27, 238–248 CAS.
- H. C. Wang and A. N. Lao, Heterocycles, 1988, 27, 1615–1621 CrossRef CAS.
- Z. T. Zhang, L. Wang, Q. F. Chen, Q. H. Chen, D. L. Chen, X. Y. Liu and F. P. Wang, Tetrahedron, 2013, 69, 5859–5866 CrossRef CAS.
- L. M. Gao, H. Y. Yan, Y. Q. He and X. M. Wei, J. Integr. Plant Biol., 2006, 48, 364–369 CrossRef CAS.
- H. L. Yu and S. S. Jia, Acta Pharmacol. Sin., 2000, 35, 232–234 CAS.
- H. Hikino, Y. Kuroiwa and C. Konno, J. Nat. Prod., 2004, 46, 178–182 CrossRef.
- H. M. Liu and A. Katz, J. Nat. Prod., 1996, 59, 135–138 CrossRef CAS.
- J. M. Yue, X. Jun and Y. Z. Chen, Phytochemistry, 1994, 35, 829–831 CrossRef CAS.
- S. H. Shim, J. S. Kim, S. S. Kang, K. H. Son and K. Bae, Arch. Pharmacal Res., 2003, 26, 709–715 CrossRef CAS PubMed.
- S. W. Pelletier and Z. Djarmati, J. Am. Chem. Soc., 1976, 98, 2626–2636 CrossRef CAS.
- T. P. Yin, Y. Shu, H. Zhou, L. Cai and Z. T. Ding, Fitoterapia, 2019, 135, 1–4 CrossRef CAS PubMed.
- F. P. Wang, and X. T. Liang, in Alkaloids Chem. Biol., ed. G. A. Cordell, Academic Press, Pittsburgh, 2002, vol. 59, pp. 1–280 Search PubMed.
- T. Tanaka, T. Nakashima, T. Ueda, K. Tomii and I. Kouno, Chem. Pharm. Bull., 2007, 55, 899–901 CrossRef CAS PubMed.
- X. H. Meng, Q. L. Guo, C. G. Zhu and J. G. Shi, Chin. Chem. Lett., 2017, 28, 1705–1710 CrossRef CAS.
- Q. Guo, H. Xia, X. Meng, G. Shi, C. Xu, C. Zhu, T. Zhang and J. Shi, Acta Pharm. Sin. B, 2018, 8, 409–419 CrossRef.
- F. P. Wang, and Q. H. Chen, in Alkaloids Chem. Biol., ed. G. A. Cordell, Academic Press, Pittsburgh, 2010, vol. 69, pp. 1–577 Search PubMed.
- F. P. Wang, D. L. Chen, H. Y. Deng, Q. H. Chen, X. Y. Liu and X. X. Jian, Tetrahedron, 2014, 70, 2582–2590 CrossRef CAS.
- B. Jiang, S. Lin, C. Zhu, S. Wang, Y. Wang, M. Chen, J. Zhang, J. Hu, N. Chen, Y. Yang and J. Shi, J. Nat. Prod., 2012, 75, 1145–1159 CrossRef CAS PubMed.
- B. Y. Jiang, S. Lin, C. G. Zhu, S. J. Wang, Y. N. Wang, M. H. Chen, J. J. Zhang, J. F. Hu, N. H. Chen, Y. C. Yang and J. G. Shi, J. Nat. Prod., 2013, 76, 2008 CrossRef CAS.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489–501 CrossRef CAS.
- S. Revathidevi and A. K. Munirajan, Semin. Cancer Biol., 2019, 59, 80–91 CrossRef CAS PubMed.
- I. Pal and M. Mandal, Acta Pharmacol. Sin., 2012, 33, 1441–1458 CrossRef CAS PubMed.
- S. Lee and K. Margolin, Cancers, 2011, 3, 3856–3893 CrossRef CAS.
- M. B. Atkins, Semin. Oncol., 2002, 29, 12–17 CrossRef CAS PubMed.
- M. Cargnello and P. P. Roux, Microbiol. Mol. Biol. Rev., 2011, 75, 50–83 CrossRef CAS PubMed.
- Y. J. Guo, W. W. Pan, S. B. Liu, Z. F. Shen, Y. Xu and L. L. Hu, Exp. Ther. Med., 2020, 19, 1997–2007 CAS.
- C. Lin, D. R. Crawford, S. Lin, J. Hwang, A. Sebuyira, R. Meng, J. E. Westfall, H. Y. Tang, S. Lin, P. Y. Yu, P. J. Davis and H. Y. Lin, Carcinogenesis, 2011, 32, 19–26 CrossRef CAS.
- Q. B. Han, W. L. Tang, C. X. Dong, H. X. Xu and Z. H. Jiang, J. Sep. Sci., 2013, 36, 1304–1310 CrossRef CAS PubMed.
- D. Gfeller, A. Grosdidier, M. Wirth, A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2014, 42, W32–W38 CrossRef CAS.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Nat. Biotechnol., 2007, 25, 197–206 CrossRef CAS.
- M. Awale and J. L. Reymond, J. Cheminf., 2017, 9, 11 Search PubMed.
- M. Awale and J. L. Reymond, J. Chem. Inf. Model., 2019, 59, 10–17 CrossRef CAS.
- P. V. Pogodin, A. A. Lagunin, D. A. Filimonov and V. V. Poroikov, SAR QSAR Environ. Res., 2015, 26, 783–793 CrossRef CAS.
- L. R. Wang, C. Ma, P. Wipf, H. B. Liu, W. W. Su and X. Q. Xie, AAPS J., 2013, 15, 395–406 CrossRef CAS.
- S. Hamad, G. Adornetto, J. J. Naveja, A. Chavan Ravindranath, J. Raffler and M. Campillos, Bioinformatics, 2019, 35, 1239–1240 CrossRef CAS.
- Z. J. Yao, J. Dong, Y. J. Che, M. F. Zhu, M. Wen, N. N. Wang, S. Wang, A. P. Lu and D. S. Cao, J. Comput.-Aided Mol. Des., 2016, 30, 413–424 CrossRef CAS.
- J. Y. Gong, C. Q. Cai, X. F. Liu, X. Ku, H. L. Jiang, D. Q. Gao and H. L. Li, Bioinformatics, 2013, 29, 1827–1829 CrossRef CAS PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrian-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magarinos, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- D. Szklarczyk, A. L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, M. Simonovic, N. T. Doncheva, J. H. Morris, P. Bork, L. J. Jensen and C. V. Mering, Nucleic Acids Res., 2019, 47, D607–D613 CrossRef CAS.
- Y. Zhou, B. Zhou, L. Pache, M. Chang, A. H. Khodabakhshi, O. Tanaseichuk, C. Benner and S. K. Chanda, Nat. Commun., 2019, 10, 1523 CrossRef PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04223h |
| This journal is © The Royal Society of Chemistry 2021 |