
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization and mechanism study of aqueous cationic polymerization of p-methylstyrene†
Shichao You ab,
Jiwen Ren*c,
Jinghan Zhangab,
Zhaopeng Yuab,
Chenqi Zhaoab,
Yibo Wuab and
Ruofan Liu*ab
ab,
Jiwen Ren*c,
Jinghan Zhangab,
Zhaopeng Yuab,
Chenqi Zhaoab,
Yibo Wuab and
Ruofan Liu*ab
aBeijing Key Lab of Special Elastomer Composite Materials, Beijing Institute of Petrochemical Technology, Beijing, 102617, China. E-mail: 0020200006@bipt.edu.cn
bCollege of New Materials and Chemical Engineering, Beijing Institute of Petrochemical Technology, Beijing, 102617, China
cZhejiang Cenway Materials Co Ltd, Zhejiang, 314201, China. E-mail: jiwen.Ren@cenwaymaterials.com
First published on 8th August 2021
Abstract
Aqueous cationic polymerization has attracted considerable interest as a novel polymerization technique, because it conforms to the “green chemistry” trend and challenges the concept of traditional cationic polymerization. In this paper, a CumOH/B(C6F5)3/Et2O system was used to initiate the aqueous polymerization of p-methylstyrene through suspension and emulsion methods. Several types of surfactants were used, including the cationic surfactant CTAB, non-ionic surfactant NP-40, and anionic surfactant SDBS, and the influences of initiator concentration and temperature on polymerization were investigated. Consistent with previous literature, initiator activity was positively correlated with temperature unlike in traditional cationic polymerization. Gaussian 09W simulation software was used to calculate and optimize changes in the bond lengths and angles of B(C6F5)3 after ether was added to the system. The addition of ether increased the polarity of B(C6F5)3, rendering it soluble in water. 1H-NMR was used in identifying the main chain and terminal structures of the polymer, and the mechanism of p-methylstyrene aqueous phase cationic polymerization was proposed.
1 Introduction
Water is a cheap and eco-friendly solvent. Thus, replacing organic solvents with water for polymer synthesis has become a major topic in the field of cationic polymerization. Aqueous synthesis systems for cationic polymerization are rarely explored due to the high sensitivity of the traditional co-initiator Lewis acid to water. Even an extremely small amount of water leads to the loss of initiator activity in these systems. However, in 1999, Sawamoto1 have achieved a great breakthrough in aqueous cationic polymerization with the highly active monomer vinyl ether and p-methoxystyrene, with ytterbium triflate [Yb(OTF)3] as the co-initiator, which has strong water resistance. The traditional concept “the cationic polymerization can only be carried out under the condition of no oxygen nor water” was broken.The development of water-resistant initiator systems has led to breakthroughs in the field of cationic polymerization. Water-resistant initiators include INIURF2,3 (acts as an initiator and surfactant), lanthanum trifluoraldehyde,1,4–10 Lewis acid surfactant combined catalyst,7,11,12 BF3·OEt2,13–16 aromatic borane-based co-initiators,17–24 tris(pentafluorophenyl)gallium [Ga(C6F5)3] and tris(pentafluorophenyl)aluminum [Al(C6F5)3] systems,25 and heteropoly acid and its salts.26,27 Except the first and seventh systems, these initiators are all co-initiators. Owing to the development of these water-resistant initiating systems, progress has been achieved in cationic polymerization, including the development of systems with high water contents and aqueous dispersion. Aqueous cationic polymerization has shown great potential, not only because it facilitates the synthesis of specific polymer structures, but also because it has invigorated the prospects of traditional suspension and emulsion polymerization methods.
Borane-based co-initiators have shown considerable potential in the synthesis of various well-structured polymers. They can be applied to many important monomers, such as isobutene,23,25,28 styrene and its derivatives,4,13–16 isoprene,21 and cyclopentadiene.22,29 Tris (pentafluorophenyl) boron [B(C6F5)3] is not only a strong Lewis acid with water resistance but also has higher acidity than other water-resistant co-initiators, such as Ln(OTF)3 and BF3·Et2O.
In 2006, Ganachaud17 used B(C6F5)3 as a co-initiator to initiate the aqueous cationic suspension polymerization of styrene, a low-activity monomer, under mild conditions (room temperature and air atmosphere). The molecular weight of the product reached 2000 g mol−1. In 2019, our group used CumOH/B(C6F5)3 to initiate the aqueous cationic polymerization of isobutyl vinyl ether, 2-chloroethyl vinyl ether, and n-butyl vinyl ether.30
The cationic initiation of p-methylstyrene with BF3·Et2O as an initiator has been achieved previously in the presence of a small amount of water.13 In this paper, the aqueous cationic polymerization of p-methylstyrene was achieved. To further improve the solubility of B(C6F5)3 in water, diethyl ether was used as the third component to form a complex with B(C6F5)3. The complex improved initiating efficiency. The suspension and emulsion polymerization of p-methylstyrene were characterized. A polymerization mechanism based on the analyzed microstructure of the polymer was proposed.
2 Materials and methods
2.1 Materials
Cumyl alcohol (CumOH, 97%), tris(pentafluorophenyl)boron (B(C6F5)3, 97%), lithium chloride (LiCl, 99%), cetyltrimethylammonium bromide (CTAB, 99%), sodium dodecyl benzene sulfonate (SDBS, 95%), polyoxyethylene mono-4-octylphenyl ether (NP-40) and tetrahydrofuran (THF, 99.8%) were purchased from J&K Scientific Ltd., Beijing, China, and used as received. Sodium chloride (NaCl, 99.5%) and diethyl ether (Et2O, 99.7%) were purchased from Beijing Chemical Works. Methanol (MeOH, 99.5%) and ethanol (EtOH, 99.5%) were purchased from Tianjin Guangfu Technology Development Co., Ltd, Tianjin, China, and used as received. Calcium hydride (CaH2, 97%) was from Shanghai Yien Chemical Technology Co., Ltd. p-Methylstyrene (p-MSt, J&K Scientific Ltd., Beijing, China, 98%) was vacuum distillation at 75 °C with CaH2 before use.2.2 Suspension polymerization of p-MSt
The temperature of the system was controlled by a temperature controller (Huber D77656) between 0 °C and 30 °C. The cooling circulating medium was the mixture of water and ethanol (v/v = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). To prevent the crystallization of water, LiCl/NaCl/H2O (wt% = 23%/1.2%/75.8%) solution was prepared as brine bath. High-purity water (3 mL), p-methylstyrene (1.75 M) and CumOH were poured into the reaction vessel in sequence. Then, in order to prepare the co-initiator system, B(C6F5)3 (0.128 g), high-purity water (2 mL) and diethyl ether (0.2 g) were added into a beaker in sequence. Slightly shake the beaker until the solid is completely dissolved. The temperature of the monomer-initiator system and co-initiator system should be carefully kept at the controlled temperature. Then pour the co-initiator into the reaction vessel to initiate the reaction. After a set period of time, the reaction was terminated by adding excessive methanol, the polymer was precipitated by centrifugation, and dried to constant weight in vacuum drying oven at 35 °C. The conversion of the reaction was determined by gravimetric method.
:50). To prevent the crystallization of water, LiCl/NaCl/H2O (wt% = 23%/1.2%/75.8%) solution was prepared as brine bath. High-purity water (3 mL), p-methylstyrene (1.75 M) and CumOH were poured into the reaction vessel in sequence. Then, in order to prepare the co-initiator system, B(C6F5)3 (0.128 g), high-purity water (2 mL) and diethyl ether (0.2 g) were added into a beaker in sequence. Slightly shake the beaker until the solid is completely dissolved. The temperature of the monomer-initiator system and co-initiator system should be carefully kept at the controlled temperature. Then pour the co-initiator into the reaction vessel to initiate the reaction. After a set period of time, the reaction was terminated by adding excessive methanol, the polymer was precipitated by centrifugation, and dried to constant weight in vacuum drying oven at 35 °C. The conversion of the reaction was determined by gravimetric method.
2.3 Emulsion polymerization of p-MSt
In the air atmosphere, the above-mentioned temperature control method was used to explore the emulsion polymerization of methylstyrene at 20 degrees celsius. The high-purity water (3 mL) was mixed with three different kinds of surfactants (0.02 g, CTAB, NP-40, SDBS). When the surfactants are totally dissolved, p-methylstyrene (1.75 M) and cumyl alcohol were added. The solution was then mechanically stirred at 150 rpm. To prepare the co-initiator system, B(C6F5)3 (0.128 g), high-purity water (2 mL) and ether (0.2 g) were added into a beaker in sequence. Shake the beaker until the solid is completely dissolved, and then put the beaker into the brine bath for temperature control. When it reached the set temperature, pour the solution into the reactor to initiate the reaction. When the predetermined reaction time is reached, add excessive methanol to terminate the reaction, and centrifuge to precipitate the polymer. The conversion was determined by gravimetric method.2.4 Characterization
The molecular weight and molecular weight distribution of the polymer were determined by GPC at 35 °C, which is manufactured by Waters Co., which has automatic sampler, chromatographic column incubator (three cross-linked polystyrene columns contact), RI differential detector and UV detector. The standard molecular weight curve was plotted by using 500, 103, 104 and 105 g mol−1 molecular weight polystyrene standard samples which were dissolved in THF. The concentration of the polymer was 1.5–2 mg mL−1. The structure of the polymer was characterized by Bruker-500 MHz 1H-NMR at 25 °C, using CDCl3 as solvent, tetramethylsilane (TMS) as internal standard. Nanosight LM20 was used to analyze the size of latex particles at room temperature. All the simulations were carried out with Gaussian 09W software package, by using the 6-31G calculation method, mode B3LYP. A hybrid model of density functional theory (DFT), was used to optimize the molecular structure and calculate the molecular fluctuating frequency. In this paper, the unit of all bond lengths is Å.3 Results
3.1 Effect of diethyl ether on the initiation system
The solvability of B(C6F5)3 in high-purity water was extremely low (Fig. 1a). After ether was added to the system, B(C6F5)3 was completely dissolved in water within 1 min, forming into a milky solution (Fig. 1b). Gaussian 09W simulation software was used in exploring this phenomenon. The results are provided in Table 1 and Table 2. The state of complexation is illustrated in Fig. 2. In the B(C6F5)3 structure, three covalent bonds (B1–C2, B1–C8, and B1–C14) and three bond angles (C2–B1–C8, C8–B1–C14, and C14–B1–C2) were changed when it complexed with diethyl ether. Notably, the fluorine atoms in B(C6F5)3 formed hydrogen bonds with the hydrogen atoms of diethyl ether, and the oxygen atoms formed coordination bonds with boron atoms, which the bond length was 5.266 Å (Fig. 2). The bond angle of C2–B1–C8 decreased from 120.06° to 119.17°. The polarity of B(C6F5)3 increased after changes in bond length and angle, and thus the solubility of B(C6F5)3 in water improved. The largest change was observed in the bond angle of C8–B1–C14, implying that the OH− of CumOH was deprived off. The created cation served as an active center to initiate polymerization. | ||
| Fig. 1 Solution state before and after addition of ether. | ||
| Bond name | Bond length (Å) | Triangle name | Triangle (°) |
|---|---|---|---|
| B1–C2 | 1.569 | C2–B1–C8 | 120.06 |
| B1–C8 | 1.569 | C8–B1–C14 | 119.98 |
| B1–C14 | 1.569 | C14–B1–C2 | 119.96 |
| Bond name | Bond length (Å) | Triangle name | Triangle (°) |
|---|---|---|---|
| B1–C2 | 1.571 | C2–B1–C8 | 119.17 |
| B1–C8 | 1.573 | C8–B1–C14 | 120.74 |
| B1–C14 | 1.567 | C14–B1–C2 | 120.08 |
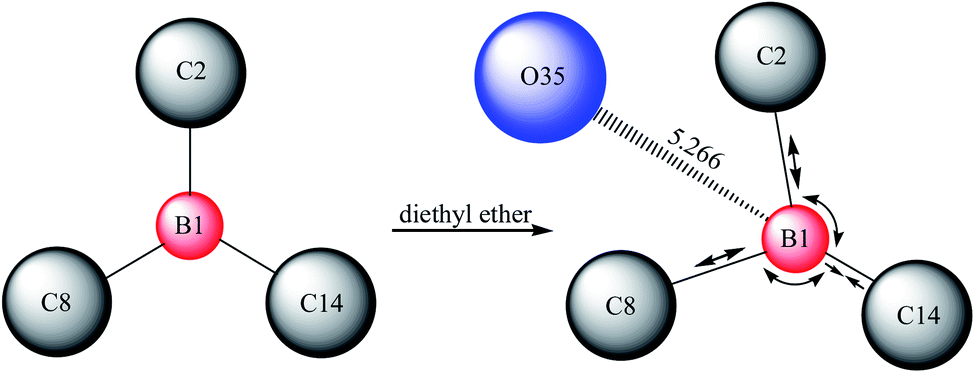 | ||
| Fig. 2 Schematic diagram of the structure changes of B(C6F5)3 after adding diethyl ether. | ||
3.2 Suspension of p-methylstyrene polymerization
The suspension of p-methylstyrene polymerization was initiated by B(C6F5)3/CumOH/Et2O system. The influence of CumOH concentration on polymerization was investigated (Fig. 3a). Since p-methylstyrene is a high-activity monomer, the polymerization completed within 2 h, and the conversion rates kept at steady levels. The conversion trends for different concentrations of CumOH were similar. The highest conversion rate was 26.04% for the blank control group and 49.74% for the 0.15 M CumOH concentration group. The comparison of the curves indicated that H2O was deprived off OH− by B(C6F5)3·Et2O and formed the active center H+ −HOB(C6F5)3·Et2O, thereby initiating polymerization. This phenomenon implied that H2O and CumOH, when both present in the system, undergo complexation competition reaction to form an active center. CumOH forms a stable tertiary carbocation cation, and benzene increases the steric hindrance of the active center and thus renders the CumOH active center stable and took advantages in the complexation competition.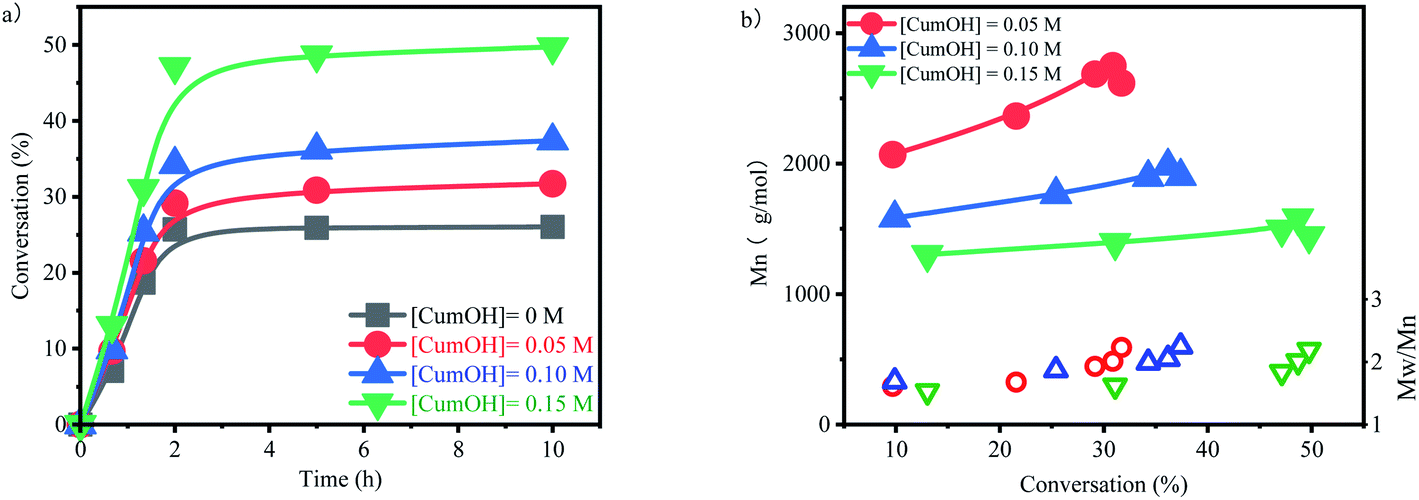 | ||
| Fig. 3 Influence of the concentration of cumyl alcohol on the polymerization of p-methylstyrene aqueous cationic suspension polymerization at 20 °C.(a) con. vs. time, (b) Mn and PDI vs. con.[p-MSt] = 1.75 M; [B(C6F5)3] = 0.05 M; Et2O = 0.2 g. | ||
The molecular weight of the polymer and its range were tested using GPC (Fig. 3b). Compared with the calculated value, the molecular weight is basically the same at the high CumOH concentration and much lower at the low CumOH concentration (Table S1†). The molecular weight decreased with increasing initiator concentration, indicating that excessive CumOH participated in the chain transfer process.31 The average molecular weight decreased at the end of the polymerization during CumOH conversion, showing that the macromolecular active centers transferred to new primary active centers continuously. The low monomer concentration of the system resulted in the low molecular weight at the end of polymerization, which reduced the average molecular weight at full scale. The molecular weight ranged from 1.6 to 2.3, showing the synthesis process was not a controlled polymerization process. In addition, the molecular weight range at the end of polymerization stage markedly changed with variable CumOH concentrations. This change indicated that polymers with low molecular weights formed at this stage and active centers transferred in the system.
3.3 Influence of temperature on polymerization reaction
The effect of temperature on active center activity was studied (Fig. 4). The conversion rate increased with temperature. These results indicated that the activity of B(C6F5)3·Et2O and its ability to derive OH− from CumOH is positively correlated to temperature. This result is consistent with the results reported in the literature.32–35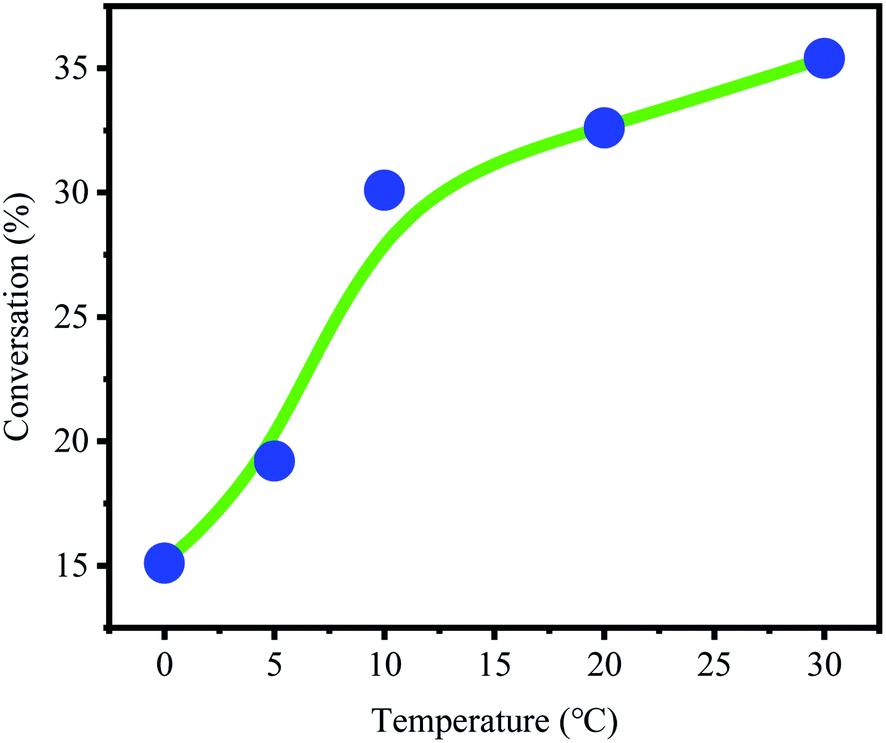 | ||
| Fig. 4 Conversion rate of p-methylstyrene at different temperatures [CumOH] = 0.05 M, [p-MSt] = 1.75 M, [B(C6F5)3] = 0.05 M; Et2O = 0.2 g. | ||
3.4 Emulsion polymerization of p-methylstyrene
The particle sizes of p-methylstyrene with different surfactants measured by dynamic light scattering (DLS) (Table 3). The average particle sizes of the emulsions, which had different kinds but the same amounts of surfactant, were roughly the same. The DLS images of monomers dispersed with different surfactants are shown in Fig. 5.| Surfactant types | Add amount (g) | Mean (nm) | Mode (nm) | SD (nm) |
|---|---|---|---|---|
| CTAB | 0.02 | 52 | 43 | 18 |
| NP-40 | 0.02 | 55 | 43 | 26 |
| SDBS | 0.02 | 52 | 44 | 20 |
 | ||
| Fig. 5 Emulsion DLS diagram of p-methylstyrene in CTAB, NP-40, SDBS. | ||
The influences of three different types of surfactants on p-methylstyrene emulsion polymerization were investigated (Fig. 6a). The surfactants reduced the conversion rates of the monomers. Given that the active centers were located on water–oil interfaces, adding surfactants increased the steric hindrance of the interfaces and reduced the collision probability of the active centers and monomers. This is one of the differences between emulsion and suspension polymerization (Fig. 7).
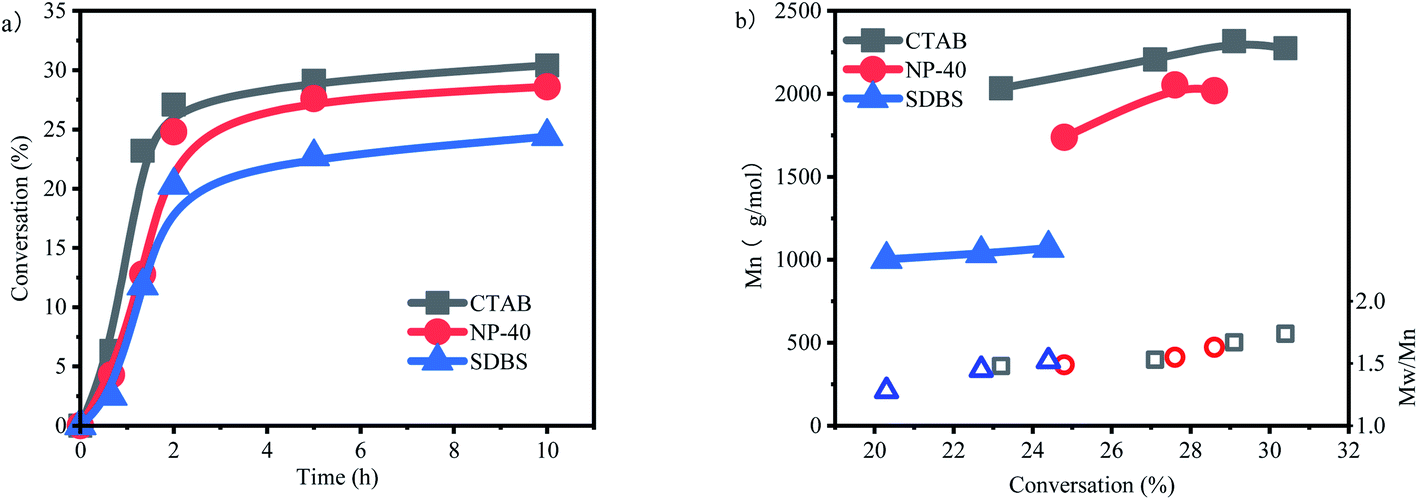 | ||
| Fig. 6 Effect of different surfactants on the polymerization of p-methylstyrene aqueous cationic emulsion polymerization. (a) con. vs. time, (b) Mn and PDI vs. con.[CumOH] = 0.05 M; [p-MSt] = 1.75 M; [B(C6F5)3] = 0.05 M; Et2O = 0.2 g; CTAB NP-40 SDBS = 0.02 g. | ||
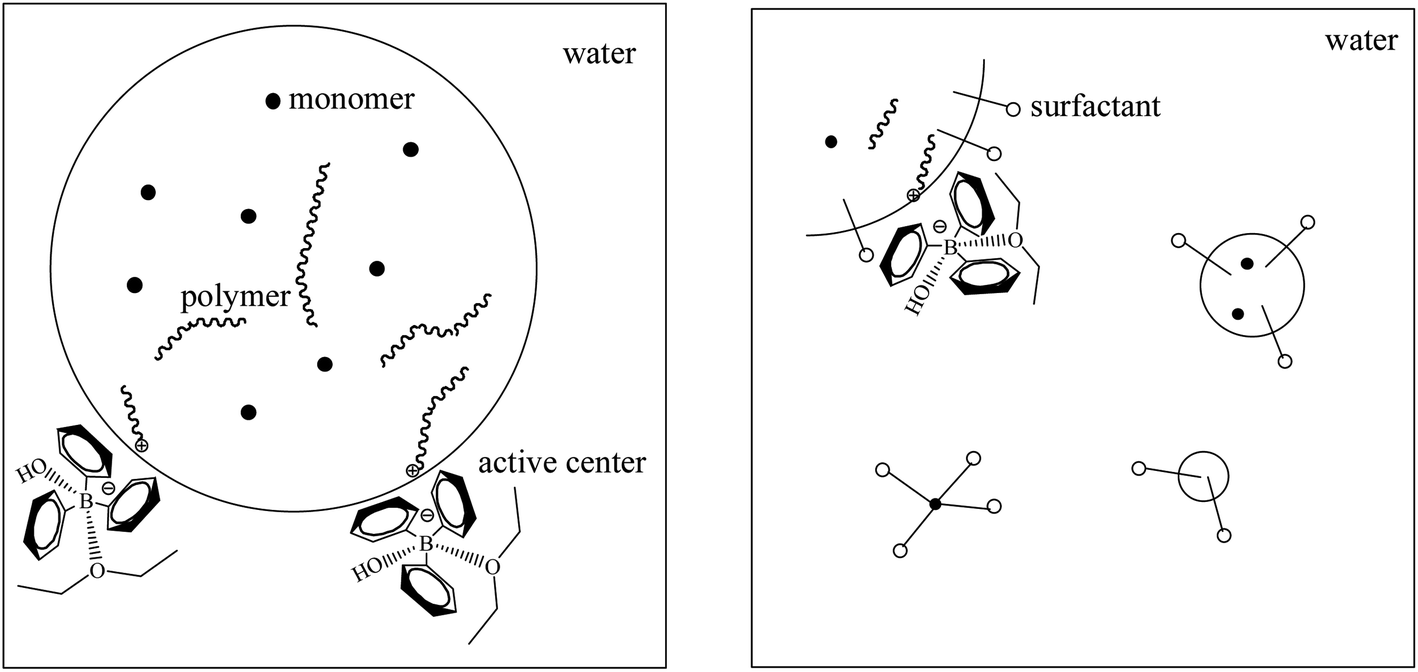 | ||
| Fig. 7 Illusion of basic reaction unit of suspension polymerization (left) and emulsion polymerization (right). | ||
The inhibition effect on polymerization of the cationic surfactant CTAB was weaker than that of NP-40 or SDBS. The reason is that CTAB forms a cationic layer on the interface. The resulting charge effect repulses carbon cations, which are undergoing chain growth. Active centers “open doors” on interfaces, thereby weakening steric hindrance. Therefore, the highest conversion rate of CTAB surfactant reached 30.4%. However, this charge effect is not the determining factor of surfactants. The conversion rate of the nonionic surfactant NP-40 was 28.6%, which was slightly lower than that of CTAB.
SDBS is an anionic surfactant that forms an anionic layer at a water–oil interface. The influence of anionic layers is threefold. First, the anions of an anionic layer compete with counter anions for carbon cations, breaking the equilibrium between the anions and cations of active centers. Second, the layer partially binds with carbon cations, weakening the activities of carbon cations and reducing initiation activity. Finally, opposite charges attract each other and cause the steric hindrance among other surfactants. Thus, the highest conversion rate of SDBS is only 24.4%.
Molecular weight is significantly influenced by surfactant type (Fig. 6b). The molecular weights of CTAB and NP-40 were 2000–2300 and 1700–2000 g mol−1, respectively, which were not greatly different from those in suspension polymerization. The average molecular weight decreased at the end of the polymerization stage, and the molecular weight distribution widened. These results indicated that the macromolecular active centers transferred to new primary active centers continuously. By contrast, molecular weight was reduced to 1000 g mol−1 after SDBS was applied. Moreover, no substantial change in molecular weight was observed after conversion rate increased, indicating that the anionic surfactant inhibited the activities of carbon cations. The active center activity was impeded by the charge effect.
3.5 Structural analysis of poly(p-methylstyrene)
Fig. 8 and 9 show the structure and 1H-NMR spectrum of poly(p-methylstyrene). As shown in Fig. 8, δ = 1.1–2.1 ppm (peak C, peak D) and δ = 6.2–7.2 ppm (peak B) represents the main chain structures of poly(p-methylstyrene) and phenyl side groups, respectively; δ = 2.3 ppm (peak E) represents the methyl group; and δ = 1.0 ppm (peak α) represents the first end methyl group. The methyl groups originated from two separate sources. The first source was CumOH, and the other was the chain transfer from the active center to water. δ = 2.9–3.0 ppm (peak ω1) represents the methoxy structure at the end of the chain, which was introduced by termination using methanol.22,36 The chemical shift of 4.1–4.3 ppm (peaks F and G) was attributed to end indene and hydroxyl structures, which also originated from two sources. The first was Friedel–Crafts alkylation chain transfer reaction to the monomers, and the second originated from chain transfer to water. The δ value ranging from 5.9 ppm to 6.1 ppm (peaks ω2, ω3) represents the carbon double bonds at the end of the polymers. They were formed by the β-H elimination.3,17,21 The end group ratios of the suspension and emulsion polymerization of poly(p-methylstyrene) are shown in Table 4. In suspension or emulsion polymerization, the proportion of end methoxy groups decreases over time, indicating that B(C6F5)3·Et2O is gradually deactivated in the reaction process. The proportion of terminal hydroxyl group in suspension polymerization was higher than that in emulsion polymerization, indicating that the steric hindrance effect impeded the active center to water chain transfer.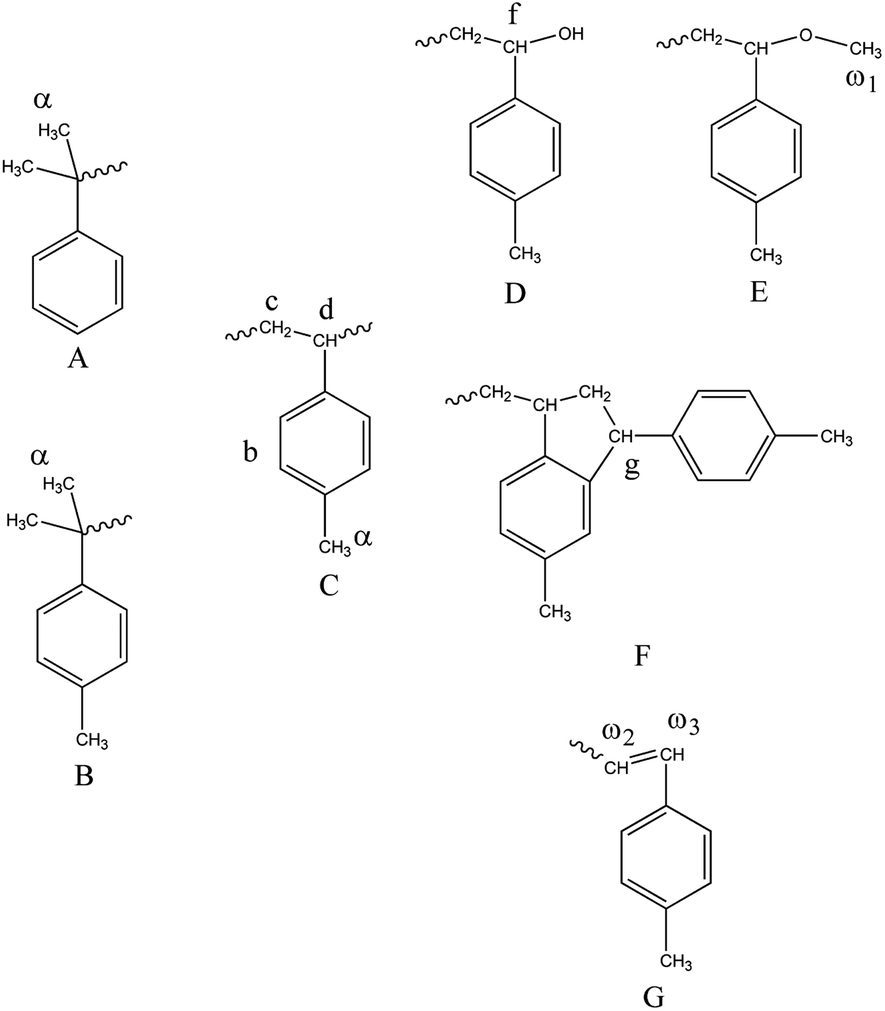 | ||
| Fig. 8 Molecular structure of poly(p-methylstyrene). | ||
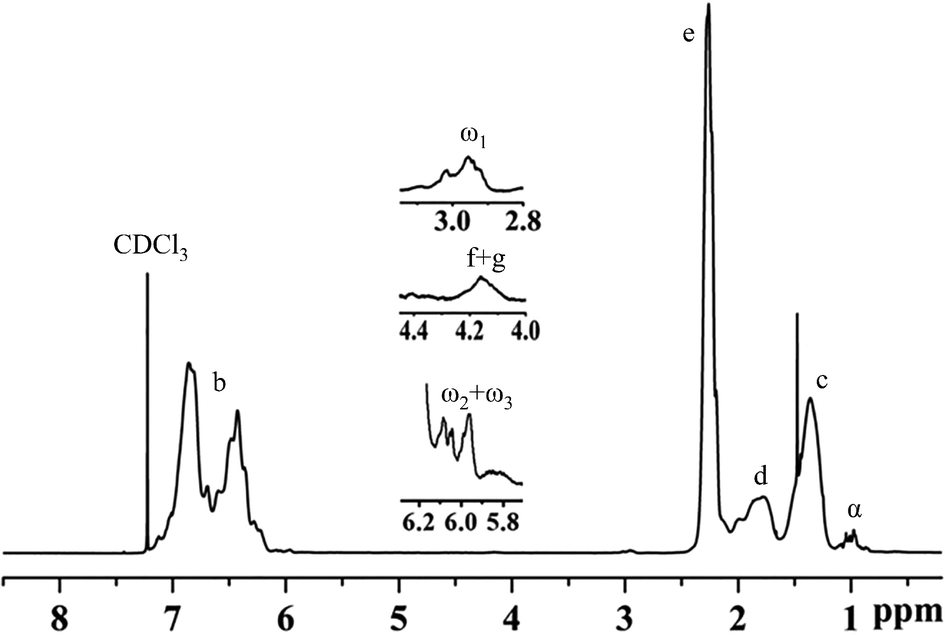 | ||
| Fig. 9 1H-NMR spectrum of poly(p-methylstyrene). | ||
| Reaction time (h) | Surfactant | End group | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Methoxy (%) | Hydroxyl (%) | Indene (%) | Double bond (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Suspension.b Emulsion. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1a | — | 63.4 | 8.4 | 13.0 | 15.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2a | — | 58.7 | 15.8 | 10.8 | 14.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5a | — | 20.1 | 22.5 | 30.4 | 26.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10a | — | 8.9 | 20.3 | 32.4 | 38.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2b | CTAB | 30.8 | 15.9 | 26.1 | 27.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5b | CTAB | 28.0 | 11.4 | 15.3 | 45.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10b | CTAB | 6.3 | 13.7 | 61.2 | 18.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5b | NP-40 | 24.0 | 21.1 | 16.6 | 38.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10b | NP-40 | 10.1 | 24.7 | 46.3 | 18.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5b | SDBS | 15.8 | 20.3 | 35.9 | 28.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10b | SDBS | 8.5 | 17.7 | 39.6 | 34.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.6 Polymerization mechanism of p-methylstyrene
We determined the possible mechanism of aqueous cationic polymerization in p-methylstyrene. The mechanism is illustrated in Fig. 10. In the chain initiation stage, B(C6F5)3·Et2O formed the active center H+ −HOB(C6F5)3·Et2O with water. When the monomer (mixed with the CumOH) was mixed with the co-initiator solution, hydrophobic groups, such as phenyl in the CumOH, were transferred to the monomer micelles. The hydrophilic groups (hydroxyls) were exposed outside the monomer micelles. CumOH underwent a complexation competition reaction with H+ −HOB(C6F5)3·Et2O and formed new active centers, ph(CH3)2C+ −HOB(C6F5)3·Et2O. Ph(CH3)2C+ was more stable in initiating monomer polymerization than H+. The chain growth reaction is shown in Fig. 10.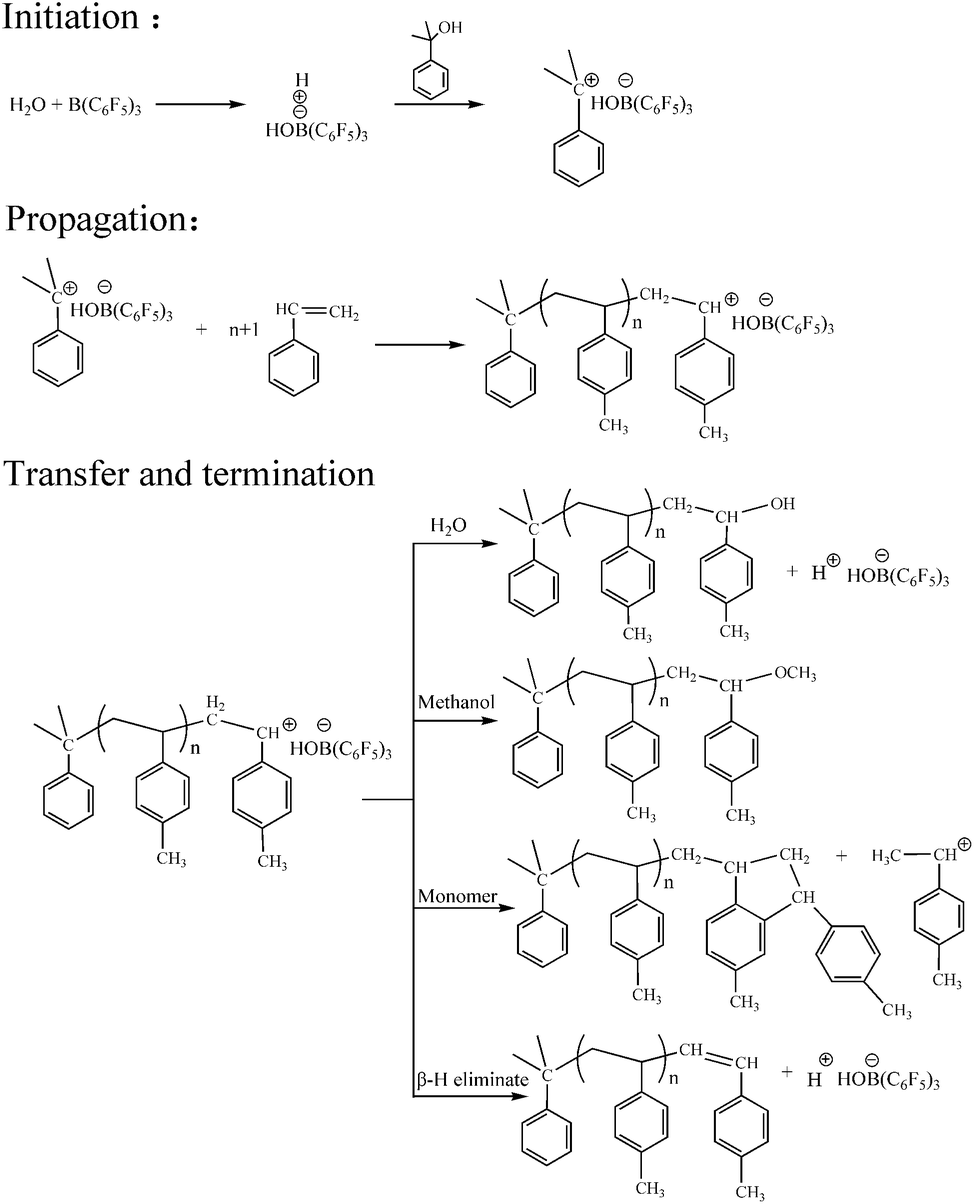 | ||
| Fig. 10 Reaction mechanism of p-methylstyrene aqueous cationic polymerization. | ||
The elementary reactions (chain initiation, growth, and transfer) sites for suspension and emulsion polymerization are at water–oil interfaces. The initiators were all “locked” on the interface between micelles and water. In suspension polymerization, there was no steric hindrance at the water–oil interface, thus the chain growth rate was high. When the polymer reached the “critical degree of polymerization”, which is the degree of polymerization when active center loses surface activity, chain transfer reaction occurred. Active centers transferred to water or monomers in the micelles or initiated β-H elimination themselves. By contrast, emulsion polymerization is stable. The heat release degree is not serious in comparison to suspension polymerization. Three typical surfactants have different steric hindrance and charge effects that impede chain initiation and propagation. The surfactants impeded chain growth and transfer. Thus, the final molecular weights were generally lower than that of suspension polymerization.
Although the suspension and emulsion polymerization of p-methylstyrene had been investigated, the molecular weights are still low. The “critical degree of polymerization” is the biggest factor restricting the growth of molecular weight.3 Depending on our mechanism, there are two possible methods might raise the molecular weight. The first is moving the reaction place from the water–oil surface into the monomer micelles. Once the active center enters the monomer droplet, it will breakthrough the restriction at the water–oil interface. It could reduce the possibility of the activity centers to lose activity. Another one is improving co-initiator's Lewis acidity. According to the experience of traditional cationic polymerization, the central atom of the co-initiator determines the molecular weight of the polymer. With boron atoms, the molecular weight can reach 103 magnitude (e.g., BF3);14,37,38 titanium atoms, 104 magnitude (e.g., TiCl4);39 and aluminum atoms, 105 magnitude (e.g., AlEt2Cl and AlEtCl2).40 With the increase of the Lewis acidity of the co-initiator, the strong nucleophilicity of the counter anion formed, which may help enhance the surface activity of the active center and reduce the possibility of the active center falling off from the water–oil interface.
4 Conclusions
Aqueous cationic polymerization of p-methylstyrene was successfully achieved, and diethyl ether-induced change in the polarity of B(C6F5)3 was investigated using the Gaussian 09W software package. Owing to the change in B–C bond length and C–B–C bond angle, B(C6F5)3 changed from a nonpolar molecule to a polar molecule when diethyl ether was added to the system. Changes in polarity increased the solubility of B(C6F5)3 in water.The suspension and emulsion polymerization of p-methylstyrene was successfully initiated by the CumOH/B(C6F5)3/Et2O system. The active center underwent chain transfer reaction and was transferred to water and monomer to form a new active center or occurred β-H elimination to deactivate the active center. The steric hindrance and charger effects on polymerization using CTAB, NP-40, and SDBS were investigated. The system added SDBS, forming an anionic layer, had the highest degree of suppression effect on polymerization.
The polymerization mechanism of p-methylstyrene aqueous cationic polymerization was determined through 1H-NMR analysis. Although we successfully initiated the aqueous cationic polymerization of p-methylstyrene, several questions need to be answered in future works. For instance, how to drive initiators from water–oil interface into the monomer micelles. And how to improve co-initiate Lewis acid to hold active center activity. These processes may increase the molecular weight and are potential topics of the next stage of our study.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 52073033), Beijing Excellent Talents Training Fund (No. Z2019-042) and Petrochina Company Limited Lanzhou Chemical Research Center Research Fund (No. KYWX-20-006).References
- K. Satoh, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 3827–3832 CrossRef CAS.
- S. Cauvin, A. Sadoun, R. Dos Santos, J. Belleney, F. Ganachaud and P. Hemery, Macromolecules, 2002, 35, 7919–7927 CrossRef CAS.
- S. V. Kostjuk and F. Ganachaud, Acc. Chem. Res., 2010, 43, 357–367 CrossRef CAS PubMed.
- K. Satoh, M. Kamigaito and M. Sawamoto, Macromolecules, 2000, 33, 4660–4666 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2728–2733 CrossRef CAS.
- R. F. Storey and A. D. Scheuer, Journal of Macromolecular Science, Part A, 2004, 41, 257–266 CrossRef.
- V. Touchard, C. Graillat, C. Boisson, F. d'Agosto and R. Spitz, Macromolecules, 2004, 37, 3136–3142 CrossRef CAS.
- P. De and R. Faust, Macromolecules, 2004, 37, 7930–7937 CrossRef CAS.
- S. Cauvin, F. Ganachaud, V. Touchard, P. Hémery and F. Leising, Macromolecules, 2004, 37, 3214–3221 CrossRef CAS.
- S. Cauvin and F. Ganachaud, Macromol. Symp., 2004, 215, 179–190 CrossRef CAS.
- S. Cauvin, F. Ganachaud, M. Moreau and P. Hemery, Chem. Commun., 2005, 2713–2715, 10.1039/b501489a.
- I. V. Vasilenko, H. Y. Yeong, M. Delgado, S. Ouardad, F. Peruch, B. Voit, F. Ganachaud and S. V. Kostjuk, Angew. Chem., Int. Ed. Engl., 2015, 54, 12728–12732 CrossRef CAS.
- K. Satoh, J. Nakashima, M. Kamigaito and M. Sawamoto, Macromolecules, 2001, 34, 396–401 CrossRef CAS.
- M. Kamigaito, J. Nakashima, K. Satoh and M. Sawamoto, Macromolecules, 2003, 36, 3540–3544 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, Macromolecules, 2000, 33, 5405–5410 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, Macromolecules, 2000, 33, 5830–5835 CrossRef CAS.
- S. V. Kostjuk and F. Ganachaud, Macromolecules, 2006, 39, 3110–3113 CrossRef CAS.
- S. V. Kostjuk, A. V. Radchenko and F. Ganachaud, Macromolecules, 2007, 40, 482–490 CrossRef CAS.
- A. V. Radchenko, S. V. Kostjuk, I. V. Vasilenko, F. Ganachaud, F. N. Kaputsky and Y. Guillaneuf, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6928–6939 CrossRef CAS.
- A. V. Radchenko, S. V. Kostjuk and F. Ganachaud, Polym. Chem., 2013, 4, 1883–1892 RSC.
- S. V. Kostjuk, S. Ouardad, F. d. r. Peruch, A. Deffieux, C. Absalon, J. E. Puskas and F. o. Ganachaud, Macromolecules, 2011, 44, 1372–1384 CrossRef CAS.
- S. V. Kostjuk, A. V. Radchenko and F. Ganachaud, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4734–4747 CrossRef CAS.
- S. P. Lewis, J. Chai, S. Collins, T. J. Sciarone, L. D. Henderson, C. Fan, M. Parvez and W. E. Piers, Organometallics, 2009, 28, 249–263 CrossRef CAS.
- S. P. Lewis, N. J. Taylor, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2003, 125, 14686–14687 CrossRef CAS PubMed.
- R. T. Mathers and S. P. Lewis, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1325–1332 CrossRef CAS.
- Q. Huang, Y. Wu and J. Dan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 546–556 CrossRef CAS.
- S. P. Lewis, J. A. Richards and K. Damodaran, J. Polym. Sci., 2020, 58, 885–902 CrossRef CAS.
- S. P. Lewis, L. D. Henderson, B. D. Chandler, M. Parvez, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2005, 127, 46–47 CrossRef CAS PubMed.
- S. V. Kostjuk, F. Ganachaud, A. V. Radchenko and I. V. Vasilenko, Macromol. Symp., 2011, 308, 1–7 CrossRef CAS.
- J. Zhang, Y. Wu, K. Chen, M. Zhang, L. Gong, D. Yang, S. Li and W. Guo, Polymers, 2019, 11, 500 CrossRef PubMed.
- A. N. Frolov, S. V. Kostjuk, I. V. Vasilenko and F. N. Kaputsky, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3736–3743 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2007, 129, 2420–2421 CrossRef CAS PubMed.
- P. Dimitrov, J. Emert, J. Hua, S. Keki and R. Faust, Macromolecules, 2011, 44, 1831–1840 CrossRef CAS.
- A. Kanazawa, S. Shibutani, N. Yoshinari, T. Konno, S. Kanaoka and S. Aoshima, Macromolecules, 2012, 45, 7749–7757 CrossRef CAS.
- L. Sipos, P. De and R. Faust, Macromolecules, 2003, 36, 8282–8290 CrossRef CAS.
- K. Verebélyi and B. Iván, Poly, 2012, 53, 3426–3431 CrossRef.
- Y.-b. Wu, J. Mao, W.-l. Guo, L.-f. Gong, S.-x. Li, P. Ren and F. Xiao, Acta Polymerica Sinica, 2013, 909–914 CAS.
- R. Faust and J. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1847–1869 CrossRef CAS.
- J. Puskas and M. Lanzendörfer, Macromolecules, 1998, 31, 8684–8690 CrossRef CAS.
- J. p. Kennedy and R. R. Phillips, J. Macromol. Sci., Chem., 1970, 4, 1759–1784 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04334j |
| This journal is © The Royal Society of Chemistry 2021 |