
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIndolylbenzothiadiazoles as highly tunable fluorophores for imaging lipid droplet accumulation in astrocytes and glioblastoma cells†
Kilian Colas a,
Karl O. Holmbergb,
Linus Chiangc,
Susanne Doloczkia,
Fredrik J. Swartlingb and
Christine Dyrager*a
a,
Karl O. Holmbergb,
Linus Chiangc,
Susanne Doloczkia,
Fredrik J. Swartlingb and
Christine Dyrager*a
aDepartment of Chemistry – BMC, Uppsala University, Box 576, 75123 Uppsala, Sweden. E-mail: christine.dyrager@kemi.uu.se
bDepartment of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, 75185 Uppsala, Sweden
cDepartment of Chemistry, University of the Fraser Valley, V2S7M8, Abbotsford, BC, Canada
First published on 6th July 2021
Abstract
We present an extensive photophysical study of a series of fluorescent indolylbenzothiadiazole derivatives and their ability to specifically image lipid droplets in astrocytes and glioblastoma cells. All compounds in the series displayed positive solvatochromism together with large Stokes shifts, and π-extended derivatives exhibited elevated brightness. It was shown that the fluorescence properties were highly tunable by varying the electronic character or size of the N-substituent on the indole motif. Three compounds proved capable as probes for detecting small quantities of lipid deposits in healthy and cancerous brain cells. In addition, all twelve compounds in the series were predicted to cross the blood–brain barrier, which raises the prospect for future in vivo studies for exploring the role of lipid droplets in the central nervous system.
Introduction
Lipid droplets (LDs) are ubiquitous cellular organelles that serve as reservoirs for natural lipids, such as triglycerides and cholesterol esters.1 They tend to be excessively abundant in cancer cells, which can use them to reduce excess lipid toxicity, or mobilize them as energy sources to survive tumor starvation.2–7 Excessive LD accumulation has been recognized as a cancer hallmark,3,5 and specific imaging of LDs using fluorescent probes has become highly attractive for studying altered lipogenesis in cancer cell biology.8 Dysregulated LD accumulation (excessive or insufficient) has also been linked to various neurodegenerative disorders,9,10 including Alzheimer's,11 Huntingon's12 and Parkinson's13 diseases, as well as amyotrophic lateral sclerosis (ALS).14 While their presence has been observed or suggested in most brain cells,9 the mechanisms behind their formation and mobilization remain unclear and the available data on LDs in healthy vs. cancerous brain tissue is still somewhat contradictory.2,4,6,7,15 Hence, there is a growing demand for bright and LD-specific fluorophores that are useful for staining LDs in glial cells and brain cancer, such as probes that can detect and monitor small quantities of LDs for studying lipid dynamics in the central nervous system (CNS).8,16–19Herein we report the synthesis, photophysical characterization and cell imaging properties of a series of indolylbenzothiadiazole (InBTD) derivatives. Several of these compounds stained LDs with excellent signal-to-background ratios in both cancerous and healthy brain cells (glioblastoma and astrocytes). The photophysical characteristics of the compounds were extensively studied and correlated to structural diversity, electronical effects and to torsion angle variations between the BTD unit and the indole motif. All compounds in the series were predicted to cross the blood–brain barrier (BBB), which raises the prospect of studies in higher organisms.
Results and discussion
The 2,1,3-benzothiadiazole (BTD) motif has gained popularity as an electron-deficient acceptor unit in donor–acceptor (D–A) fluorophores – compounds that can be used in materials science and/or bioimaging. In general, BTD-based fluorophores exhibit several advantageous features, such as non-toxicity, pronounced solvatochromism, redshifted emission profiles and large Stokes shifts.20 The latter two are especially relevant for bioimaging since they reduce the risk of background noise that originate from autofluorescence or backscattering from the excitation source.21In this study, we anticipated that an electron-rich indole unit could be an attractive donor counterpart to the BTD core.20,22 Especially for CNS related bioimaging as it is found in numerous bioactive compounds such as the amino acid tryptophan, the neurotransmitter serotonin, and in various psychotropic drugs – all of which have BBB permeability.23,24 InBTDs have so far mainly been reported by the Ito group for their interesting mechanochromic properties (Fig. 1, top).25,26 Thus, in this work we wanted to explore their utility as imaging agents for fluorescent cell microscopy and also study how different N-substituents on the indole motif affect the photophysical properties (Fig. 1, bottom). We hypothesized that the size of the N-substituent would regulate the fluorescence efficiency through the BTD–indole torsion angle (θ), while the electronic nature, electron-donating groups (EDGs) or electron-withdrawing groups (EWGs), would impact the D–A character of the fluorophores. Accordingly, we synthesized twelve InBTDs (1–12, Scheme 1A) with such features, of which four included an additional aromatic motif to extend the π-conjugation and thereby obtain increased molar absorptivity.
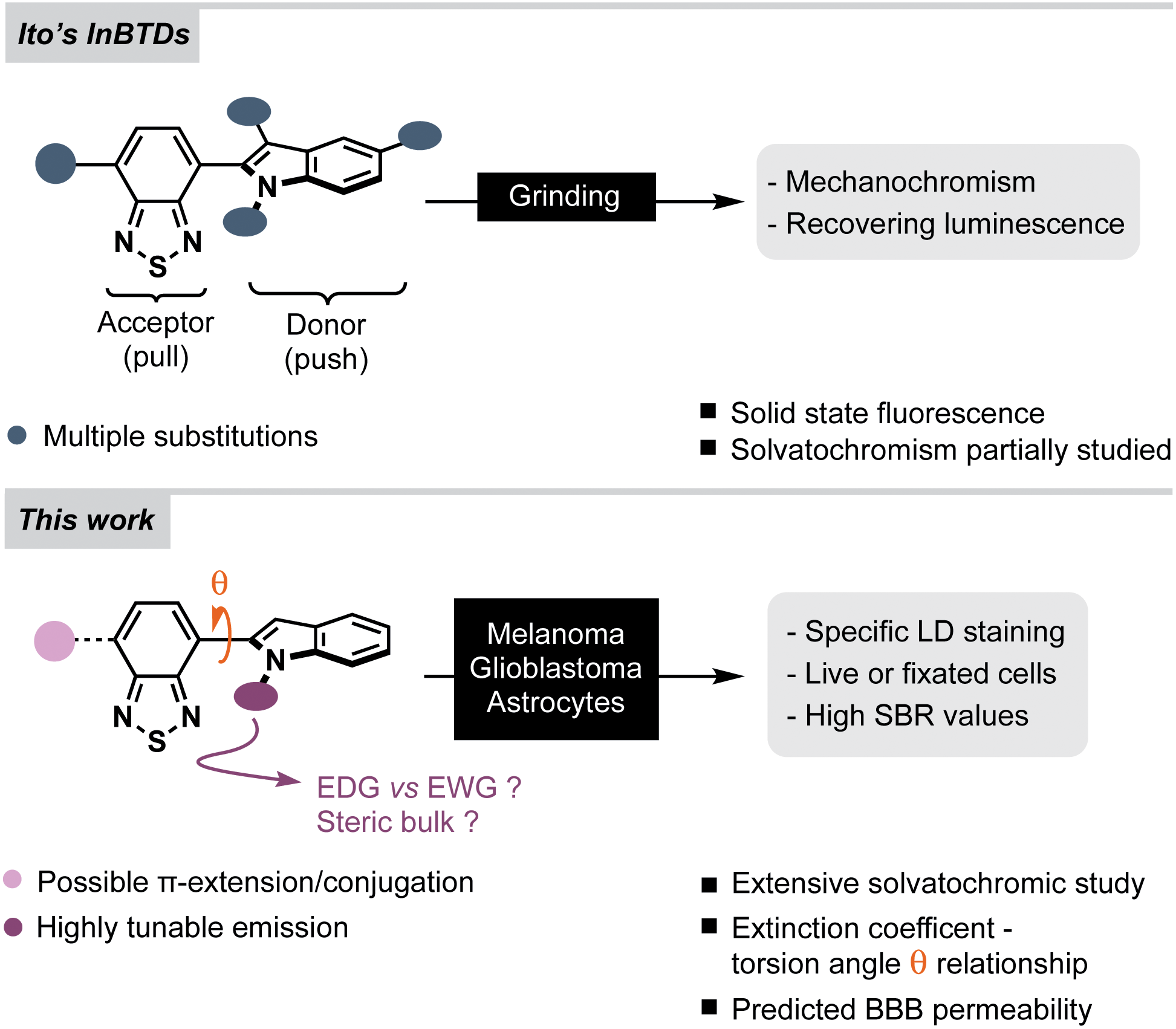 | ||
| Fig. 1 (Top) Previous studies of the mechanochromic properties of indolylbenzothiadiazoles, InBTDs.25,26 (Bottom) This work: extensive photophysical characterization in multiple solvents and intracellular lipid droplet (LD) staining in melanoma, glioblastoma and astrocytes. SBR = signal-to-background ratio. | ||
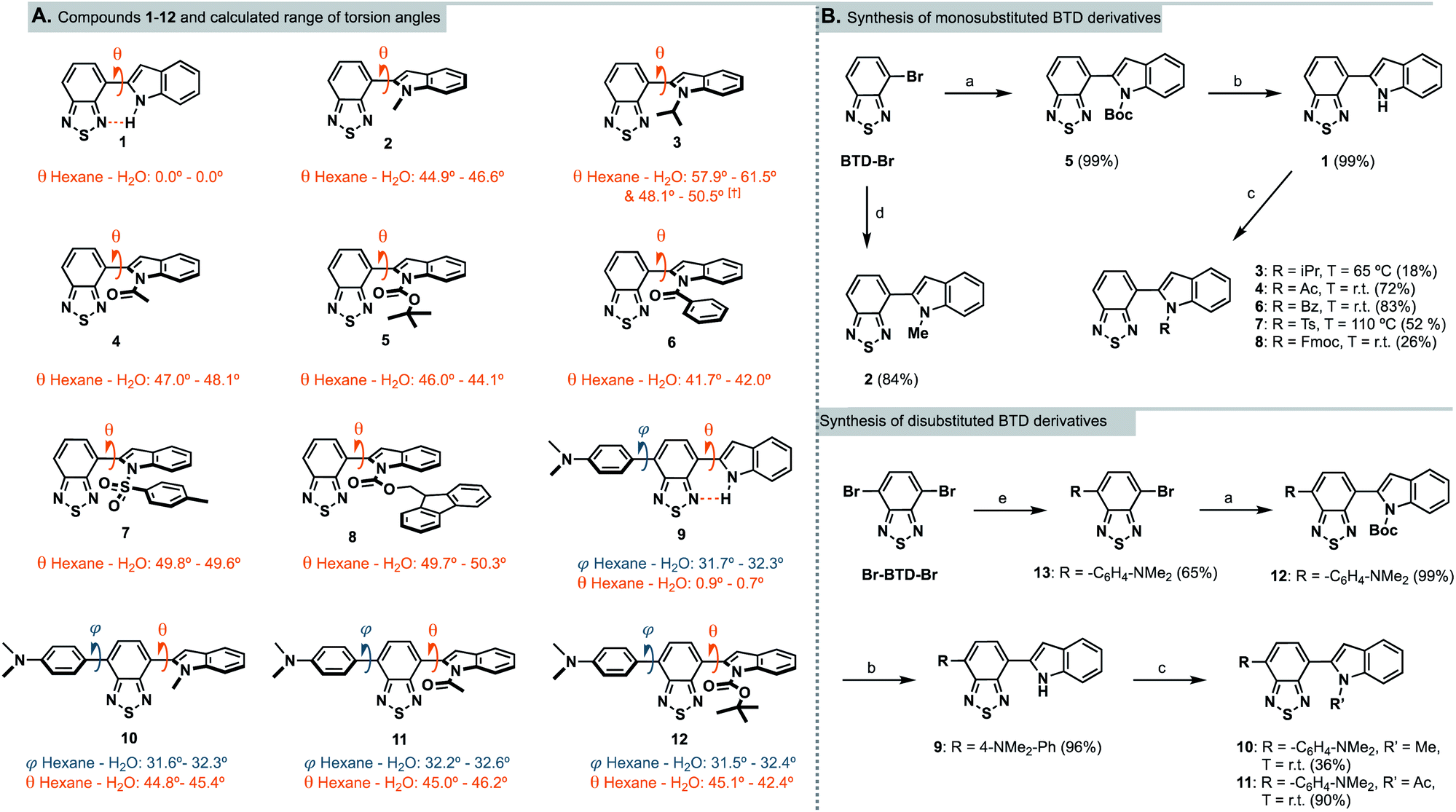 | ||
Scheme 1 (A) Chemical structures of InBTDs 1–12 and their calculated torsion angles in hexane and water (least to most polar solvent investigated).† Two stable conformers found. (B) Synthetic routes for the monosubstituted (top) and the disubstituted BTD derivatives (bottom). Reagents and conditions: (a) N-Boc-indole-2-boronic acid, PEPPSI-Ipr, K2CO3, toluene/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 80 °C, 2 h; (b) trifluoroacetic acid, DCM, 0 °C to r.t., 16 h; (c) R–Cl or R–Br, NaH, 15-crown-5, DMF, 0 °C to indicated T, 16 h; (d) 1-methyl-2-indoleboronic acid pinacol ester, PEPPSI-Ipr, K2CO3, toluene/MeOH (1:1), 80 °C, 2 h; (e) 4-(N,N-dimethylamino)phenylboronic acid pinacol ester, Pd(PPh3)4, Na2CO3 (2 M aq.), toluene/EtOH (1:10), 85 °C, 16 h. :1), 80 °C, 2 h; (b) trifluoroacetic acid, DCM, 0 °C to r.t., 16 h; (c) R–Cl or R–Br, NaH, 15-crown-5, DMF, 0 °C to indicated T, 16 h; (d) 1-methyl-2-indoleboronic acid pinacol ester, PEPPSI-Ipr, K2CO3, toluene/MeOH (1:1), 80 °C, 2 h; (e) 4-(N,N-dimethylamino)phenylboronic acid pinacol ester, Pd(PPh3)4, Na2CO3 (2 M aq.), toluene/EtOH (1:10), 85 °C, 16 h. | ||
DFT calculations
Geometry optimization of InBTDs 1–12 (Scheme 1A) were performed without symmetry constraints using the hybrid M06 functional and 6-31g* basis set combination,27–29 and the corresponding polarized continuum model (PCM)30 for the corresponding solvents (hexane, toluene, THF, DMSO, acetonitrile, isopropanol, MeOH and H2O). For full computational details, see ESI pp. 27–34.† This combination of functional and basis set has previously been used to investigate the electronic structures of similar BTD-containing compounds.31 All compounds in the series (1–12) exhibited a calculated BTD-to-indole bond distance of ca. 1.46 Å. Compounds 1 and 9 exhibited no or negligible twisting (θ = 0.0 and <1.0°, respectively) between the aromatic BTD and the unprotected indole ring due to intramolecular hydrogen bonding between the H-N-indole and the BTD fragment (further discussed below). All other compounds (2–8, 10–12) displayed torsion angles (θ) greater than 40°, which can be attributed to the absence of hydrogen bonding and increased steric bulk of the N-substituent. Compounds 9–12 also showed twisting between the 4-(N,N-dimethylamino)phenyl and the BTD unit (φ) of ca. 30°. Vertical excitation energies were calculated using time-dependent density functional theory (TD-DFT) at the same level of theory with the corresponding PCM. In all cases, vertical excitations originated from HOMO → LUMO transitions, where the HOMO was delocalized over the entire molecule, and the LUMO was predominantly BTD-based (ESI Fig. S18†). The disubstituted compounds (9–12) were predicted to exhibit red-shifted transitions in comparison to their monosubstituted counterparts (1, 2, 4 and 5). Furthermore, compounds 1–8 were predicted to exhibit blue-shifted absorptions in more polar solvents, while a far less pronounced trend was predicted for the compounds containing the 4-(N,N-dimethylamino)phenyl substituent (9–12). These predicted trends were proven to be in good agreement with experimental UV-Vis spectra collected in the corresponding solvents (vide infra).Synthesis
Following literature precedents,25,32 compounds 2 and 5 were prepared in excellent yields (84 and 99%, respectively) using a Suzuki–Miyaura cross-coupling protocol. Accordingly, 4-bromo-2,1,3-benzothiadiazole (BTD-Br) and the appropriate indolyl-boronic acid were coupled using PEPPSI-Ipr as the catalyst (Scheme 1B, top). To introduce other N-substituents on the indole – when the boronic acids were not commercially available – we found it more straightforward to proceed via N-functionalization of the unsubstituted indole motif. Hence, the Boc group in compound 5 was removed with trifluoroacetic acid to quantitatively provide the N-H derivative 1. Subsequently, 1 was deprotonated with sodium hydride and treated with various alkyl, acyl and sulfonyl chlorides, which initially resulted in very low yields. Noting the intense purple color of the reaction mixture upon deprotonation, we hypothesized that the conjugate base of 1 may strongly chelate to the sodium cation from the hydride base and hinder reactivity towards the electrophiles. Consequently, one equivalent of 15-crown-5 was added to abstract the sodium counterion, resulting in improved yields.The N-acetyl and N-benzoyl derivatives 4 and 6 were obtained in high yields (72 and 83%, respectively) at room temperature, while tosylation required heating at 110 °C to provide 7 in 52% yield (observed in NMR as a mixture of rotamers). InBTDs with bulkier substituents remained challenging to synthesize. Nonetheless, the isopropyl derivative 3 could be obtained in 18% yield upon heating to 65 °C (the boiling point of the electrophile). On the other hand, heating proved detrimental to the preparation of the Fmoc-protected 8, which was obtained in 26% yield at room temperature. Unlike previously reported InBTD derivatives,25,26 the compounds in this series were left unfunctionalized on the C-3 position of the indole. As a consequence, competing C-3 reactivity was observed when attempting other synthetic strategies. In particular, transition-metal catalyzed C–N coupling reactions led to dimerization of 1 at the C-3 position, presumably due to a strong directing group effect of the BTD core. Likewise, the use of anhydrides as electrophiles mostly resulted in C-3 acylation.
To improve the molar absorptivities of the compounds,33 we introduced an additional aromatic substituent at the 4-position of the BTD core starting from 4,7-dibromo-2,1,3-benzothiadiazole, Br-BTD-Br (Scheme 1B, bottom). First, Suzuki–Miyaura coupling conditions were used to prepare the 4-(N,N-dimethylamino)phenyl substituted bromo-BTD intermediate 13 in 65% yield. A second coupling afforded the Boc-protected InBTD derivative 12, followed by deprotection to yield 9 in quantitative yields. The addition of 15-crown-5 was again crucial to prepare both the methyl- and acetyl-functionalized compounds 10 and 11 in 36% and 90% yield, respectively.
Photophysical characterization
The photophysical properties of compounds 1–12 were investigated by UV-absorption and fluorescence spectroscopy in a broad variety of solvents of different polarity (Table 1 and ESI Fig. S1–S12†). Due to solubility issues, isopropanol was used instead of methanol, and measurements in water were excluded since the analysis could not be performed reliably. Several photophysical trends and behaviors could be noticed when changing the nature of the indole N-substituent. For the monosubstituted compounds 2–8, a clear correlation was observed between the molar extinction coefficients (ε) and the calculated torsion angles, which is best represented in hexane where the smallest solvent effects are expected (Fig. 2, top). As θ increases, the conjugation of the D–A system is weakened and ε decreases. Accordingly, the isopropyl derivative 3 (θ = 57.9°) showed the lowest ε value (2500 M−1 cm−1) and the benzoyl derivative 6 (θ = 41.7) the highest (8200 M−1 cm−1). The tosyl-derivative 7 displayed a clear exception to this trend (θ = 49.8°, ε = 7900 M−1 cm−1), likely due to conjugation of the sulfonyl group to the aromatic π-system. Also, the unprotected N-H compound 1, which calculations suggest should be flat (θ = 0°), showed similar ε values as for many of the other derivatives and no increased values caused by planarization. The disubstituted compounds 9–12, which are larger conjugated systems, displayed up to three times higher ε values than the monosubstituted analogues. Moreover, while the ε values differ between the derivatives in the series, they were rather uniform regardless of solvent for each compound.| Solvent | Comp. | λAmax [nm] | λEmax [nm] | Stokes shifta [nm] | ε [M−1 cm−1] | ΦF | Comp. | λAmax [nm] | λEmax [nm] | Stokes shift [nm] | ε [M−1 cm−1] | ΦF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Stokes shift in nm = λEmax − λAmax.b No emission detected.c Not determined due to poor solubility. | ||||||||||||
| Hexane | 1 | 452 | 509 | 57 | 6600 | 0.02 | 7 | 368 | 480 | 112 | 7900 | 0.17 |
| Toluene | 448 | 542 | 94 | 8400 | 0.06 | 368 | 495 | 127 | 4700 | 0.60 | ||
| THF | 434 | 565 | 131 | 8000 | 0.17 | 358 | 503 | 145 | 5000 | 0.50 | ||
| DMSO | 429 | 608 | 179 | 6700 | 0.02 | 354 | 524 | 170 | 8100 | 0.55 | ||
| MeCN | 424 | 601 | 177 | 6800 | 0.02 | 350 | 517 | 167 | 5700 | 0.55 | ||
| i-PrOH | 436 | 600 | 164 | 8000 | 0.07 | 358 | 529 | 171 | 5300 | 0.19 | ||
| Hexane | 2 | 415 | 517 | 102 | 5500 | 0.86 | 8 | 368 | 492 | 124 | 5100 | 0.69 |
| Toluene | 415 | 555 | 140 | 4300 | 0.36 | 370 | 516 | 146 | 3800 | 0.63 | ||
| THF | 408 | 575 | 167 | 4700 | 0.16 | 365 | 523 | 158 | 4700 | 0.57 | ||
| DMSO | 404 | 623 | 219 | 4700 | 0.01 | 367 | 554 | 187 | 3600 | 0.15 | ||
| MeCN | 395 | 609 | 214 | 4600 | 0.01 | 361 | 546 | 185 | 3800 | 0.41 | ||
| i-PrOH | 408 | n.e.b | — | 5100 | — | 364 | 552 | 188 | 4300 | 0.09 | ||
| Hexane | 3 | 414 | 518 | 104 | 2500 | 0.63 | 9 | 494 | 568 | 74 | n.d.c | 0.57 |
| Toluene | 411 | 555 | 144 | 300 | 0.20 | 491 | 604 | 113 | 2200 | 0.21 | ||
| THF | 401 | 478 | 177 | 1200 | 0.10 | 495 | 649 | 154 | 21400 |
0.11 | ||
| DMSO | 396 | n.e.b | — | 400 | — | 478 | 558 | 80 | 2700 | 0.02 | ||
| MeCN | 385 | n.e.b | — | 1200 | — | 487 | 697 | 210 | 17000 |
0.01 | ||
| i-PrOH | 402 | n.e.b | — | 400 | — | 490 | 661 | 171 | 8300 | <0.01 | ||
| Hexane | 4 | 382 | 499 | 117 | 5400 | 0.18 | 10 | 460 | 564 | 104 | n.d.c | 0.75 |
| Toluene | 381 | 520 | 139 | 5200 | 0.22 | 464 | 604 | 140 | 1300 | 0.40 | ||
| THF | 376 | 537 | 161 | 6000 | 0.15 | 463 | 638 | 175 | 6200 | 0.20 | ||
| DMSO | 373 | 571 | 198 | 3900 | 0.07 | 464 | 668 | 204 | 1400 | <0.01 | ||
| MeCN | 367 | 563 | 196 | 3000 | 0.07 | 452 | 680 | 228 | 6600 | 0.02 | ||
| i-PrOH | 373 | 569 | 196 | 5500 | 0.01 | 461 | 631 | 170 | 1400 | <0.01 | ||
| Hexane | 5 | 371 | 488 | 117 | 4500 | 0.79 | 11 | 452 | 560 | 108 | n.d.c | 0.89 |
| Toluene | 373 | 510 | 137 | 6300 | 0.88 | 464 | 612 | 148 | 13700 |
0.43 | ||
| THF | 369 | 521 | 152 | 5000 | 0.68 | 462 | 648 | 186 | 13700 |
0.15 | ||
| DMSO | 367 | 553 | 186 | 5000 | 0.48 | 464 | 562 | 98 | 13100 |
0.02 | ||
| MeCN | 363 | 548 | 185 | 5100 | 0.46 | 452 | 694 | 242 | 9800 | 0.02 | ||
| i-PrOH | 368 | 557 | 189 | 5700 | 0.07 | 460 | 608 | 148 | n.d.c | 0.01 | ||
| Hexane | 6 | 384 | 495 | 111 | 8200 | 0.23 | 12 | 441 | 551 | 110 | n.d.c | 0.85 |
| Toluene | 385 | 518 | 133 | 5900 | 0.24 | 452 | 599 | 147 | 11100 |
0.56 | ||
| THF | 381 | 531 | 150 | 5200 | 0.16 | 452 | 635 | 183 | 13000 |
0.22 | ||
| DMSO | 377 | 563 | 186 | 5600 | 0.11 | 458 | 562 | 104 | 13200 |
0.02 | ||
| MeCN | 372 | 555 | 183 | 6400 | 0.10 | 445 | 693 | 248 | 12000 |
0.02 | ||
| i-PrOH | 378 | 562 | 184 | 2900 | 0.02 | 449 | 632 | 183 | n.d.c | 0.01 | ||
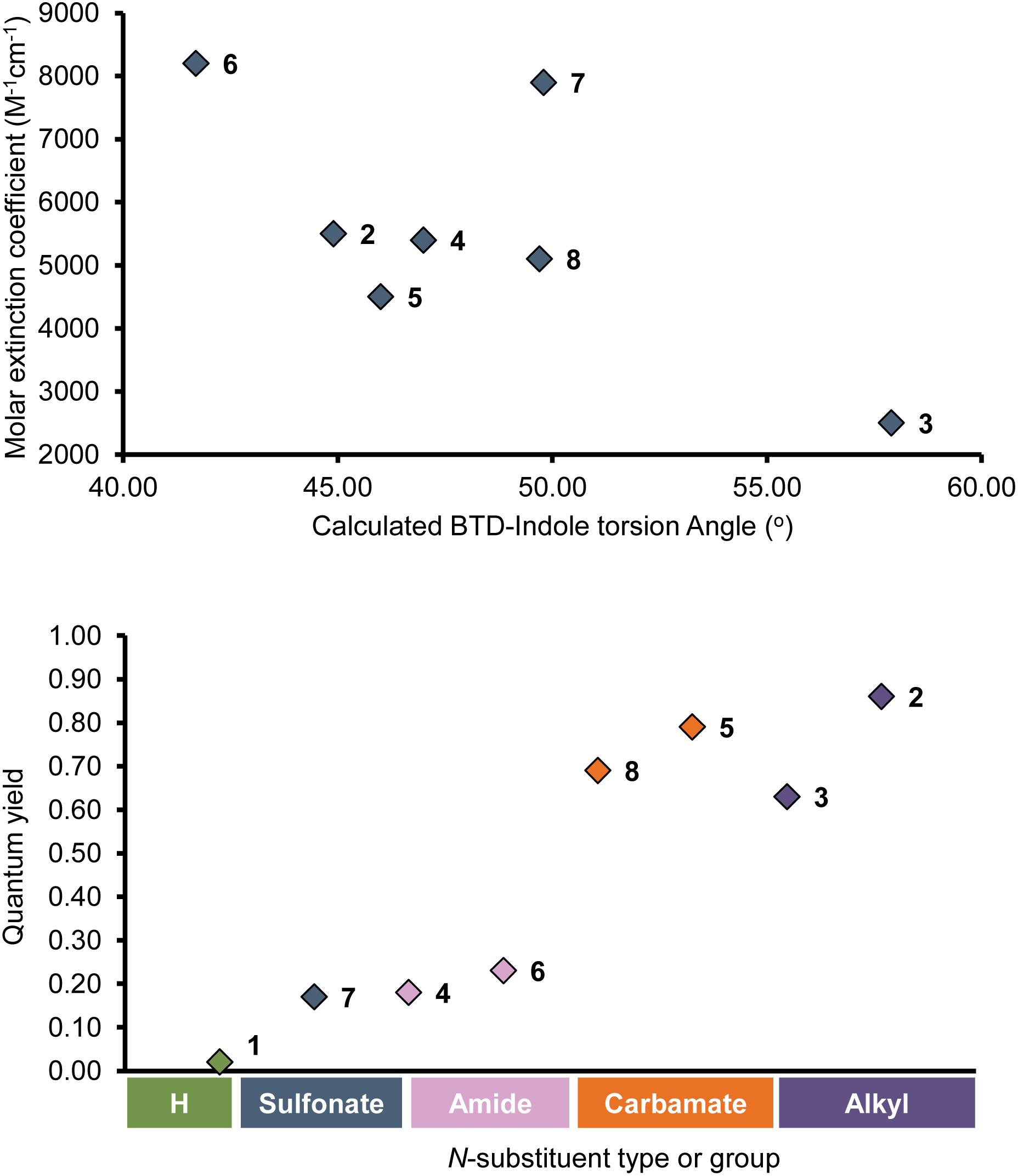 | ||
| Fig. 2 (top) Correlation between molar extinction coefficients (ε) and calculated torsion angles (θ) in hexane for InBTDs 2–8; (bottom) representation of the relationship between quantum yield Φ and the nature of the indole N-substituent in hexane for InBTDs 1–8. | ||
However, a few exceptions were observed for 3, 9 and 10 where ε varied dramatically in different solvents (e.g., 9, ε = 2200 M−1 cm−1 in toluene vs. 21400 in THF). Further comparing the monosubstituted derivatives 1–8 in hexane revealed that the fluorescence quantum yield ΦF was highly affected by the electronic character of the indole N-substituent (Fig. 2, bottom). Compounds 2 and 3 with EDGs (alkyls, methyl or isopropyl) showed excellent quantum yields (ΦF = 0.85 and 0.63) while 4, 6 and 7 with EWGs (amide or sulfonate) gave much lower values (ΦF = 0.18, 0.23 and 0.17). As an exception, the carbamate derivatives 5 and 8 showed very high fluorescence efficiency (ΦF = 0.79 and 0.69), despite the EWG character of the N-protecting group. The unprotected N-H derivative 1 displayed low fluorescent quantum yields in all solvents (ΦF = 0.02–0.07) except THF (ΦF = 0.17). As previously reported, this is likely due to strong intramolecular hydrogen-bonding.25,34–36 Furthermore, the photophysical behavior for the disubstituted analogue 9 differed from 1, even though the calculated torsion angles θ were close to zero in all solvents (θ = 0.7°–0.9°). Instead, it followed the trends of compounds bearing electron-donating N-substituents on the indole motif (i.e., 2, 3 and 10). The same trend also applied for the disubstituted derivatives with EWGs on the indole (11 and 12), showing that the quantum yields for these were highly affected by the additional donor motif, 4-(N,N-dimethylamino)phenyl. Compounds 2, 3 and 9–12 displayed good to excellent quantum yields in hexane, which gradually decreased with increasing solvent polarity becoming largely quenched in DMSO, acetonitrile and isopropanol. For the isopropyl derivative 3, the quenching was so pronounced that no emission could be detected in these solvents. As these compounds are the strongest D–A (2, 3) or D–A–D (9, 10) systems in the series, this behavior can be ascribed to a stronger stabilization of their excited states in polar environments.37–43 This is in line with the strong positive solvatochromism observed in all cases,43 where emission maxima gradually increased with solvent polarity, resulting in large Stokes shifts. On the other hand, the absorption maxima (λAmax) were generally unaffected by the solvent, although a slight negative solvatochromism was observed, followed by a small uptick in isopropanol. This suggests that solvent stabilization effects are also at play in the ground state.
Other trends were observed for the monosubstituted compounds 4–8 that contain protecting groups with electron-withdrawing character on the indole motif. Their emission and absorption profiles were more blue-shifted and their quantum yields generally decreased with solvent polarity. However, the latter was either less pronounced (e.g., in DMSO and acetonitrile for 5 and 8) or more irregular (4, 6 and 7). This indicates that the withdrawing nature of the N-substituent partially negates the donation character of the indole group in the push–pull system. Nevertheless, they still displayed strong positive solvatochromism and remarkably large Stokes shifts (e.g., 189 nm for 5 in isopropanol). We could also observe that the carbamate derivatives 5 and 8 had high or excellent quantum yields in most of the solvents. They even displayed high values in acetonitrile (ΦF = 0.46 and 0.41, respectively).
Lippert–Mataga (L–M) plots of all compounds (ESI Fig. S13 and S14†) further support solvatochromism as the main cause for the observed spectral shifts, as the correlation between Stokes shifts and solvent polarizability are linear.44,45 However, for the disubstituted compounds 9–12, some aberrations could be seen. First, a clear deviation from the linear trend of 9 in DMSO stems from hydrogen bonding between the solvent and the unprotected indole.35,36 The reason why this was observed for 9, and not for the monosubstituted analogue 1, presumably arise from the presence of a weaker intramolecular hydrogen bond (θ ≠ 0°, Scheme 1A) – caused by the presence of the additional donor substituent, 4-(N,N-dimethylamino)phenyl. Measurements of compounds 10–12 in isopropanol also deviated from the linear trend in the L–M plots. This was again likely due to hydrogen bonding, but this time between the protic solvent and the 4-(N,N-dimethylamino)phenyl motif. Furthermore, the emission profiles of 11 and 12 in DMSO displayed a very pronounced dual emission (ESI Fig. S11 and S12†). This a is a well-known phenomenon in D–A fluorophores that can be ascribed to a locally excited (LE) state and an intramolecular charge transfer (ICT) state – the latter being stabilized by polar solvents.37,46,47 The involvement of ICT states are typically associated with solvatochromism and decreased quantum yields with increasing solvent polarity,37,39 which is consistent with our observations. Moreover, the absorption spectra of 1–3 displayed distinct shoulders in hexane, and to a lesser extent in toluene and THF. This suggests the presence of two ground-state species that can be attributed to two different stable conformations. In the case of 1, with or without intramolecular hydrogen bonding and in the case of 3, the presence of steric bulk (isopropyl group) that generates rotamers. The latter is supported by our calculations (ESI p. 31†), which show two stable conformational isomers, one of which display the largest torsion angles in the series (θ = 57.9–61.5°) between the indole motif and the BTD unit. While the calculations do not indicate the same for compound 2, a similar effect may occur that is not reflected by the DFT model. In all three cases (1–3), a single emission band was observed, suggesting the existence of a single excited state. Thus, rapid rotamer interconversion in the excited state is less likely, as it would violate the non-equilibration of excited rotamers (NEER) principle.48 Although, such behavior has been reported in recent literature for D–A fluorophores.49,50 In a more subtle case, 7 showed slight shoulders in both the absorption and emission spectra, suggesting that two ground-state rotamers (which were observed in NMR) are converted to two distinct excited-state rotamers that do follow the NEER principle.
Cell studies
Several BTD-based fluorophores have shown to specifically stain LDs in cancer cells, including our recently reported dyes.32,51 We therefore wanted to explore the utility of InBTDs 1–12 as imaging agents for fluorescent cell microscopy. Three different cell lines were chosen for the study: malignant melanoma cells (SK-MEL-28), which contain a significant number of LDs.52 Glioblastoma cells (U1242MG) and normal human astrocytes (NHA) were also investigated, given the surge of interest of LDs in brain cancer,2,4,6,7 LDs role in neurodegenerative diseases9,11–14 as well as the prevalence of the indole motif in BBB-crossing substances.23,24 Live cells were incubated with compounds 1–12 (10 μM) for 24 h and subsequently fixated and costained with DAPI prior to fluorescence microscope imaging. The results showed that the InBTD derivatives 5, 9, 11 and 12, which exhibit the most pronounced brightness in apolar solvents (ε × ΦF ≥ 4000 M−1 cm−1 in hexane and toluene), were supreme in the series for obtaining clear and favorable signals in cells (Fig. 3 and ESI Fig. S15, S16†). For instance, the monosubstituted compound 5 gave a characteristic LD pattern in melanoma cells (ESI Fig. S15†) with a good signal-to-background ratio (SBR = 1.9) in the green channel.53 However, the signal contrast was much less favorable for 5 in glioblastoma cells and astrocytes (SBR = 1.8 and 1.7, respectively). This observation was likely attributed to (i) the lower amount and smaller size of LDs in these cells lines compared to melanoma cells and (ii) the modest brightness of 5 (e.g., 5544 M−1 cm−1 in toluene), which evidently is not optimal for detecting low quantities of LDs. Nevertheless, the brighter disubstituted compounds, 11 (Fig. 3, top) and 12 (ESI Fig. S15†), gave excellent results in all three cell lines, providing a clear LD staining pattern in the yellow channel53 with excellent signal distinction (for 11, SBR = 14.0, 5.0, and 5.6 in melanoma, glioblastoma and astrocytes, respectively). They also showed no or very low signal in the blue, green and red channels53 (ESI Fig. S17†), making them highly suitable for multicolor imaging with different fluorophores. Furthermore, compound 9 gave an unexpected worm-shaped pattern in the yellow and red channels53 (ESI Fig. S16†) when using our standard staining protocol (i.e., post-fixation after live cell treatment). Notably, the same pattern was observed in live-cell imaging experiments. While this could be due to precipitation in the cell medium, the shape of the putative aggregates seems to rule out crystallization. Instead, we propose that the unprotected indole moiety (made more nucleophilic by the electron donating 4-(N,N-dimethylamino)phenyl substituent) reacts with components of the cell culture medium. Nonetheless, 9 proved to be an excellent stain for fixed cells, providing a bright signal in the yellow channel,54 after 30 min treatment, with an LD-staining pattern comparable to the other probes (ESI Fig. S16†). All our cell imaging experiments with 5, 9, 11 and 12 resulted in punctate patterns that are consistent with intracellular LD staining from the literature.8,17,32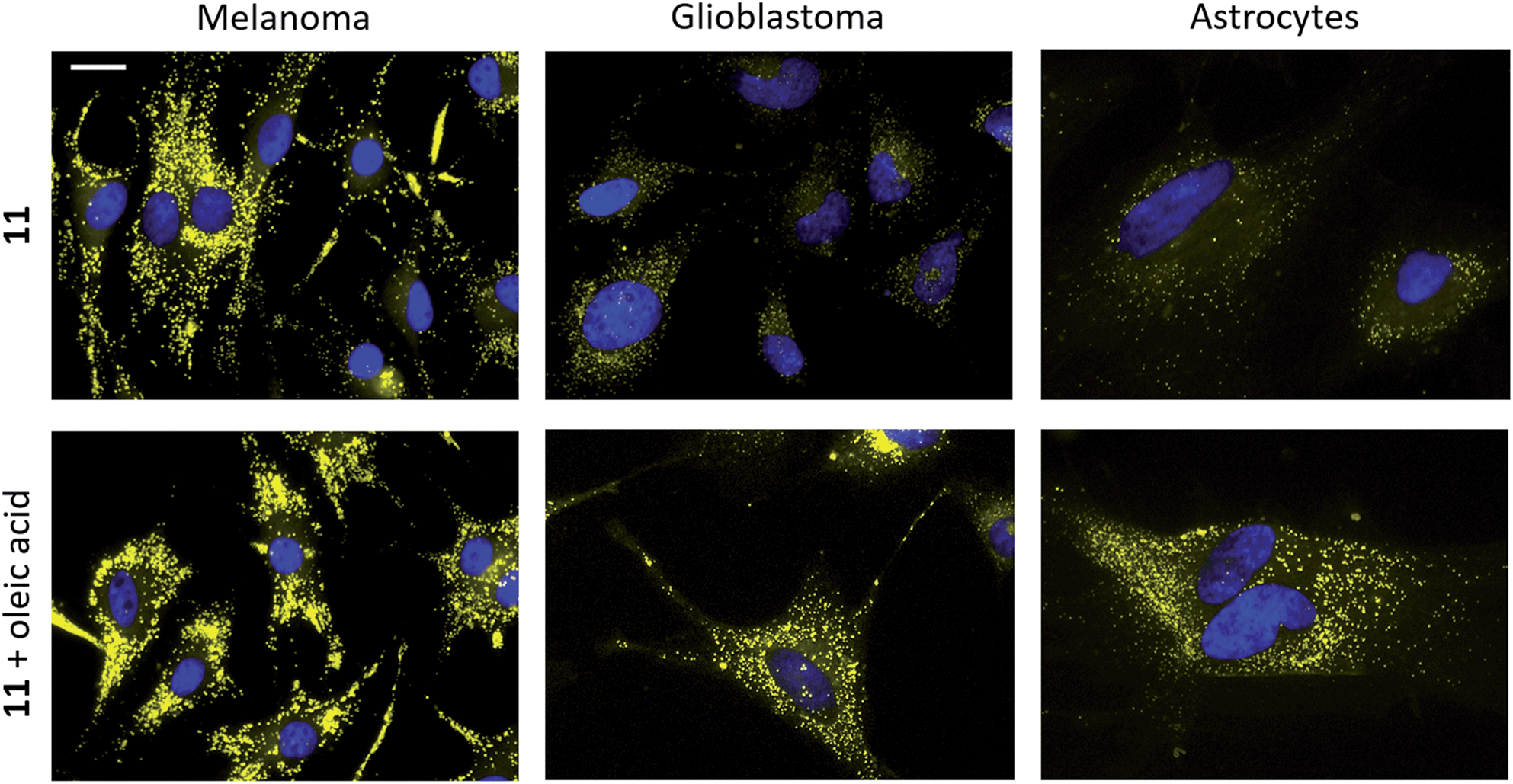 | ||
| Fig. 3 Staining of melanoma (SK-MEL-28, left), glioblastoma (U1242MG, middle) and normal human astrocytes (NHA, right) with 11 (10 μM, top and bottom) and oleic acid supplementation (100 μM, bottom). Staining was performed on live cells, which were fixated after 24 h incubation. Cell nuclei were stained with DAPI (seen in blue).54 Scale bar: 20 μm. | ||
To further verify LD staining, co-staining with standard fluorescent LD markers (e.g., Nile Red) could not be performed due to overlapping photophysical profiles. Instead, co-staining of compound 11 was performed using immunofluorescence with an antibody for adipophilin – a protein that is localized on the surface of LDs.17 The results clearly showed colocalization of 11 and adipophilin, which confirms LD staining (ESI Fig. S18†). Cells were also supplemented with oleic acid, a process known to strongly enhance LD formation.2,17 As depicted in Fig. 3, cell staining experiments were performed with 11 in the absence and presence of oleic acid (top and bottom panels, respectively). The latter showed a significant increase in both signal intensity and in the number of droplet-like items in all three cell lines, strongly supporting LD specific staining. The fluorescent cell images also revealed significant differences between the different cell lines. While LDs were more abundant and visibly larger in melanoma than in glioblastoma cells, they appeared to be distributed around the entire cell in a uniform fashion. In contrast, the healthy NHA exhibited much fewer LDs that seemed to be located in the periphery of the cells rather than close to the nucleus. The results clearly showed detectable LDs in NHAs, in accordance with the most recent literature.6,7,15,54 The observed differences between the cell lines were also consistent with mounting evidence that cancer cells rely on modified lipid metabolism and increased quantities of LDs to promote tumor survival by resisting cell starvation and lipid toxicity.3,5
The cell viability after treatment of 1–12 (10 μM) was investigated using the resazurin assay (Fig. 4, top). No significant signs of toxicity could be observed after 24 h incubation. However, the disubstituted compounds 9–12 showed a very modest decrease in viability compared to their monosubstituted counterparts. To explore the future potential of the compounds for in vivo CNS imaging, 1–12 were further evaluated using Xie's platform for BBB penetration (see ESI†).55 The model predicted that all compounds in the series could cross the BBB (Fig. 4, bottom), although with different grades of capacity. For example, the BBB+ scores for the brighter disubstituted compounds 9–12 were generally lower than for the majority of the monosubstituted derivatives 1–8 – an effect that suggestively originates from increased hydrogen bond ability in the presence of the 4-(N,N-dimethylamino)phenyl motif.
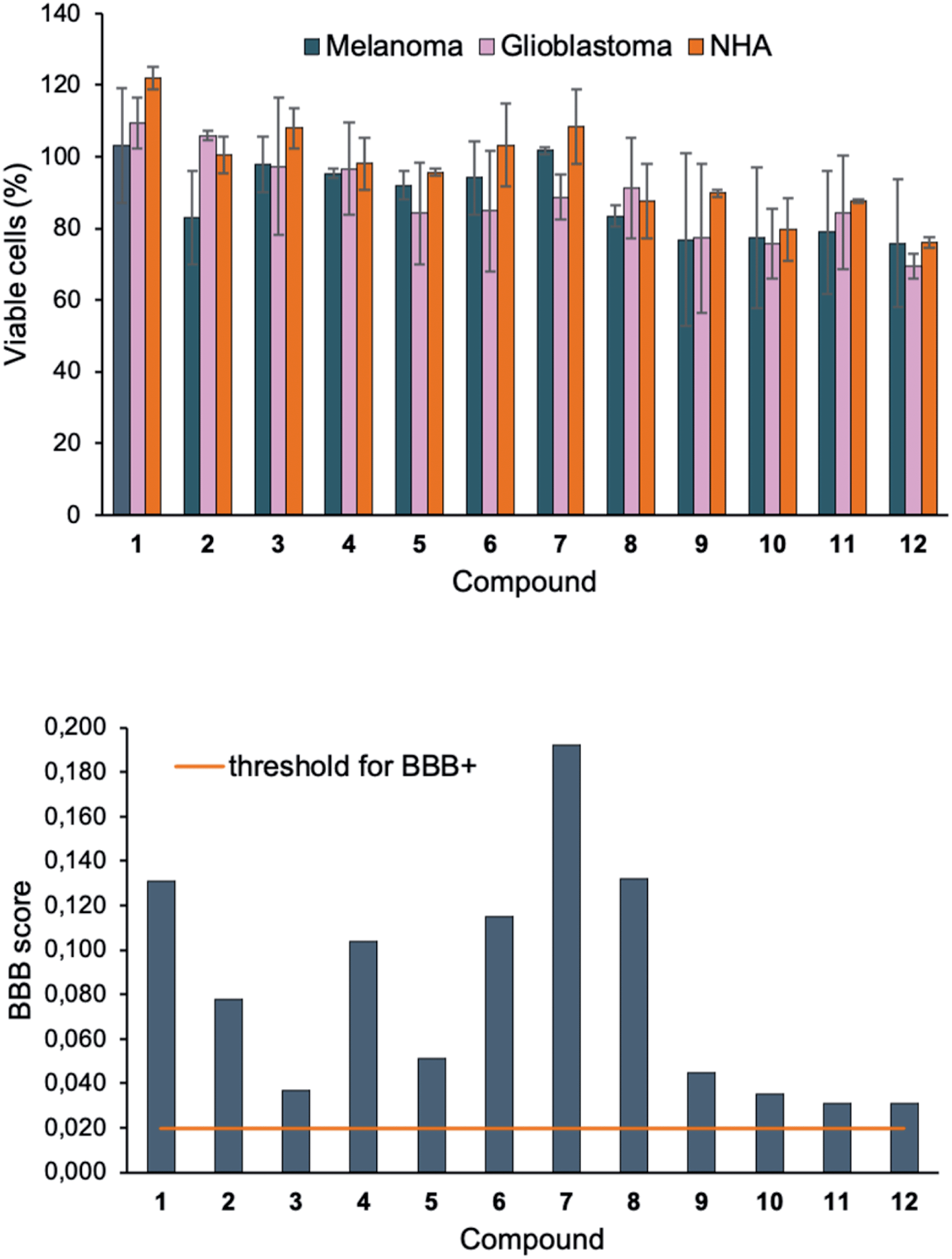 | ||
| Fig. 4 (Top) Cell viability of melanoma (SK-MEL-28, blue), glioblastoma (U1242MG, pink) and normal human astrocytes (NHA, orange) after treatment with 1–12 (10 μM) for 24 h, measured by resazurin staining assay. Results are represented as % of a DMSO control as a mean ± standard deviation of results obtained from triplicates and twice repeated independent experiments. (Bottom) Graphical representation of BBB+ scores for 1–12. The orange horizontal line represents the threshold for predicted positive BBB crossing. | ||
Conclusions
We have described a series of solvatochromic indolyl–BTD derivatives, whose fluorescence properties can be highly tuned via the N-substituent on the indole motif – either by varying its electronic nature (EDGs vs. EWGs) or steric bulk. The latter which affects the torsion angle between the aromatic units and thus the degree of conjugation within the molecules. Introduction of an electron-donating moiety, 4-(N,N-dimethylamino)phenyl, at the 4-position of the BTD core gave disubstituted derivatives with improved brightness and red-shifted emission profiles. Among those, compound 9, 11 and 12 proved to be excellent probes for specific imaging of lipid droplets in melanoma cells, and also for staining lower quantities of lipid accumulation in cancerous and healthy brain cells (glioblastoma and astrocytes).These compounds are easily synthesized, exhibit large Stokes shifts, allows for multicolor imaging and are predicted to cross the BBB. They also show superior emissive properties in hydrophobic environments and display suppressed emission in polar protic solvents – an ideal trait for staining lipid deposits with high contrast in cells. Notably, they do not suffer from unspecific staining or background artifacts (attributed to small Stokes shifts) – known limitations for commonly used and commercialized LD probes such as Nile Red and BODIPY-based derivatives.13 Thus, we believe that compound 9, 11 and 12 will be valuable additions to the available collection of LD-dyes,8 particularly for imaging LDs in brain tissue that require high specificity and excellent signal-to-background ratios.2,4,6,7,9,11–14 Also, their predicted BBB permeability raises the future prospects for in vivo studies to investigate altered lipogenesis in the CNS.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Swedish Research Council, Dnr: 2018-03524 (to CD), the Carl Trygger Foundation, Dnr: CTS 18:90 (to CD) and a NSERC Discovery Grant (to LC). ComputeCanada is acknowledged for access to computational resources. We also thank Prof. Katarina Edwards and her group for access to their spectrofluorometer and Dr Lukasz Pilarski for proof-reading the manuscript.Notes and references
- J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS.
- B. Taïb, A. M. Aboussalah, M. Moniruzzaman, S. Chen, N. J. Haughey, S. F. Kim and R. S. Ahima, Sci. Rep., 2019, 9, 19593 CrossRef.
- T. Petan, E. Jarc and M. Jusović, Molecules, 2018, 23, 1941 CrossRef PubMed.
- F. Geng and D. Guo, Int. Med. Rev., 2017, 3 DOI:10.18103/imr.v18103i18105.18443.
- A. L. S. Cruz, E. d. A. Barreto, N. P. B. Fazolini, J. P. B. Viola and P. T. Bozza, Cell Death Dis., 2020, 11, 105 CrossRef.
- C. N. Barber and D. M. Raben, Front. Cell. Neurosci., 2019, 13, 212 CrossRef CAS.
- B. C. Farmer, J. Kluemper and L. A. Johnson, Cells, 2019, 8, 182 CrossRef CAS.
- T. K. Fam, A. S. Klymchenko and M. Collot, Materials, 2018, 11, 1768 CrossRef PubMed.
- B. C. Farmer, A. E. Walsh, J. C. Kluemper and L. A. Johnson, Front. Neurosci., 2020, 14, 742 CrossRef.
- V. Teixeira, P. Maciel and V. Costa, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2021, 1866, 158820 CrossRef CAS PubMed.
- L. K. Hamilton, M. Dufresne, S. E. Joppé, S. Petryszyn, A. Aumont, F. Calon, F. Barnabé-Heider, A. Furtos, M. Parent, P. Chaurand and K. J. L. Fernandes, Cell Stem Cell, 2015, 17, 397–411 CrossRef CAS PubMed.
- M. Martinez-Vicente, Z. Talloczy, E. Wong, G. Tang, H. Koga, S. Kaushik, R. de Vries, E. Arias, S. Harris, D. Sulzer and A. M. Cuervo, Nat. Neurosci., 2010, 13, 567–576 CrossRef CAS PubMed.
- S. Fanning, D. Selkoe and U. Dettmer, npj Parkinson's Dis., 2020, 6, 3 CrossRef CAS.
- G. Pennetta and M. A. Welte, Dev. Cell, 2018, 45, 427–432 CrossRef CAS.
- M. S. Ioannou, J. Jackson, S.-H. Sheu, C.-L. Chang, A. V. Weigel, H. Liu, H. A. Pasolli, C. S. Xu, S. Pang, D. Matthies, H. F. Hess, J. Lippincott-Schwartz and Z. Liu, Cell, 2019, 177, 1522–1535 CrossRef CAS .e1514..
- X. Ren, F. Zhang, H. Luo, L. Liao, X. Song and W. Chen, Chem. Commun., 2020, 56, 2159–2162 RSC.
- L. L. Listenberger and D. A. Brown, Curr. Protoc. Cell Biol., 2007, 35, 24.22.21 Search PubMed.
- J. Rumin, H. Bonnefond, B. Saint-Jean, C. Rouxel, A. Sciandra, O. Bernard, J.-P. Cadoret and G. Bougaran, Biotechnol. Biofuels, 2015, 8, 42 CrossRef.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- D. Jiang, S. Chen, Z. Xue, Y. Li, H. Liu, W. Yang and Y. Li, Dyes Pigm., 2016, 125, 100–105 CrossRef CAS.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS.
- Y. Ban, Y. Murakami, Y. Iwasawa, M. Tsuchiya and N. Takano, Med. Res. Rev., 1988, 8, 231–308 CrossRef CAS PubMed.
- S. Ito, T. Yamada, T. Taguchi, Y. Yamaguchi and M. Asami, Chem.–Asian J., 2016, 11, 1963–1970 CrossRef CAS.
- S. Ito, T. Taguchi, T. Yamada, T. Ubukata, Y. Yamaguchi and M. Asami, RSC Adv., 2017, 7, 16953–16962 RSC.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- J. E. Barnsley, G. E. Shillito, C. B. Larsen, H. van der Salm, L. E. Wang, N. T. Lucas and K. C. Gordon, J. Phys. Chem. A, 2016, 120, 1853–1866 CrossRef CAS PubMed.
- H. Appelqvist, K. Stranius, K. Börjesson, K. P. R. Nilsson and C. Dyrager, Bioconjugate Chem., 2017, 28, 1363–1370 CrossRef CAS.
- Y. Lee, A. Jo and S. B. Park, Angew. Chem., Int. Ed., 2015, 54, 15689–15693 CrossRef CAS.
- C. Shang, G. Wang, K. Liu, Q. Jiang, F. Liu, P.-T. Chou and Y. Fang, Angew. Chem., Int. Ed., 2020, 59, 8579–8585 CrossRef CAS.
- G. Wiosna, I. Petkova, M. S. Mudadu, R. P. Thummel and J. Waluk, Chem. Phys. Lett., 2004, 400, 379–383 CrossRef CAS.
- J. Waluk, Acc. Chem. Res., 2003, 36, 832–838 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- K. Bhattacharyya and M. Chowdhury, Chem. Rev., 1993, 93, 507–535 CrossRef CAS.
- G. Haberhauer, Chem.–Eur. J., 2017, 23, 9288–9296 CrossRef CAS.
- Z. Zhang, G. Zhang, J. Wang, S. Sun and Z. Zhang, Comput. Theor. Chem., 2016, 1095, 44–53 CrossRef CAS.
- C. Zhong, Phys. Chem. Chem. Phys., 2015, 17, 9248–9257 RSC.
- P. Suppan, J. Photochem. Photobiol., A, 1990, 50, 293–330 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 CrossRef CAS.
- L. Ernst, Z. Phys. Chem., 1956, 6, 125–128 CrossRef.
- D. Liese and G. Haberhauer, Isr. J. Chem., 2018, 58, 813–826 CrossRef CAS.
- S. Sasaki, G. P. C. Drummen and G.-i. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- J. K. Whitesell, M. A. Minton and V. D. Tran, J. Am. Chem. Soc., 1989, 111, 1473–1476 CrossRef CAS.
- A. Cesaretti, B. Carlotti, F. Elisei, C. G. Fortuna and A. Spalletti, Phys. Chem. Chem. Phys., 2018, 20, 2851–2864 RSC.
- A. Cesaretti, B. Carlotti, F. Elisei, C. G. Fortuna, G. Consiglio and A. Spalletti, Phys. Chem. Chem. Phys., 2017, 19, 5262–5272 RSC.
- K. Colas, S. Doloczki, A. Kesidou, L. Sainero-Alcolado, A. Rodriguez-Garcia, M. Arsenian-Henriksson and C. Dyrager, ChemPhotoChem, 2021, 5 DOI:10.1002/cptc.202100040.
- C. Giampietri, S. Petrungaro, M. Cordella, C. Tabolacci, L. Tomaipitinca, A. Facchiano, A. Eramo, A. Filippini, F. Facchiano and E. Ziparo, Int. J. Mol. Sci., 2017, 18, 1271 CrossRef.
- The blue, green, yellow and red color channels of the microscope respectively correspond to the commonly used stains DAPI, FITC, Y3 and Y5 and are their excitation/emission windows are as follows: blue (Ex 325–375 nm, Em 435–485 nm); green (Ex 460–500 nm, Em 512–542 nm); yellow (Ex 532–558 nm, Em 570–640 nm) and red (Ex 590–650 nm, Em 662–738 nm).
- T. Smolič, P. Tavčar, A. Horvat, U. Černe, A. Halužan Vasle, L. Tratnjek, M. E. Kreft, N. Scholz, M. Matis, T. Petan, R. Zorec and N. Vardjan, Glia, 2021, 69, 1540–1562 CrossRef PubMed.
- H. Liu, L. Wang, M. Lv, R. Pei, P. Li, Z. Pei, Y. Wang, W. Su and X.-Q. Xie, J. Chem. Inf. Model., 2014, 54, 1050–1060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04419b |
| This journal is © The Royal Society of Chemistry 2021 |