
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper sulfide as the cation exchange template for synthesis of bimetallic catalysts for CO2 electroreduction†
Jinghan Li abc,
Junrui Libc,
Chaochao Dund,
Wenshu Chene,
Di Zhanga,
Jiajun Gua,
Jeffrey J. Urband and
Joel W. Ager*bc
abc,
Junrui Libc,
Chaochao Dund,
Wenshu Chene,
Di Zhanga,
Jiajun Gua,
Jeffrey J. Urband and
Joel W. Ager*bc
aState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
bJoint Center for Artificial Photosynthesis, Materials Sciences Division and Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: jwager@lbl.gov
cDepartment of Materials Science and Engineering, University of California, Berkeley, CA 94720, USA
dThe Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
eThe School of Environmental Science, Nanjing Key Laboratory of Advanced Functional Materials, Nanjing Xiaozhuang University, Nanjing, Jiangsu 211171, China
First published on 7th July 2021
Abstract
Among metals used for CO2 electroreduction in water, Cu appears to be unique in its ability to produce C2+ products like ethylene. Bimetallic combinations of Cu with other metals have been investigated with the goal of steering selectivity via creating a tandem pathway through the CO intermediate or by changing the surface electronic structure. Here, we demonstrate a facile cation exchange method to synthesize Ag/Cu electrocatalysts for CO2 reduction using Cu sulfides as a growth template. Beginning with Cu2−xS nanosheets (C-nano-0, 100 nm lateral dimension, 14 nm thick), varying the Ag+ concentration in the exchange solution produces a gradual change in crystal structure from Cu7S4 to Ag2S, as the Ag/Cu mass ratio varies from 0.3 to 25 (CA-nano-x, x indicating increasing Ag fraction). After cation exchange, the nanosheet morphology remains but with increased shape distortion as the Ag fraction is increased. Interestingly, the control (C-nano-0) and cation exchanged nanosheets have very high faradaic efficiency for producing formate at low overpotential (−0.2 V vs. RHE). The primary effect of Ag incorporation is increased production of C2+ products at −1.0 V vs. RHE compared with C-nano-0, which primarily produces formate. Cation exchange can also be used to modify the surface of Cu foils. A two-step electro-oxidation/sulfurization process was used to form Cu sulfides on Cu foil (C-foil-x) to a depth of a few 10 s of microns. With lower Ag+ concentrations, cation exchange produces uniformly dispersed Ag; however, at higher concentrations, Ag particles nucleate on the surface. During CO2 electroreduction testing, the product distribution for Ag/Cu sulfides on Cu foil (CA-foil-x-y) changes in time with an initial increase in ethylene and methane production followed by a decrease as more H2 is produced. The catalysts undergo a morphology evolution towards a nest-like structure which could be responsible for the change in selectivity. For cation-exchanged nanosheets (CA-nano-x), pre-reduction at negative potentials increases the CO2 reduction selectivity compared to tests of as-synthesized material, although this led to the aggregation of nanosheets into filaments. Both types of bimetallic catalysts are capable of selective reduction of CO2 to multi-carbon products, although the optimal configurations appear to be metastable.
Introduction
CO2 electroreduction (CO2R) has become one of the most promising strategies towards achieving a carbon-neutral environment. Provided that it is powered by a renewable energy source, it can sustainably convert the greenhouse gas CO2 into fuels like methanol and ethanol, and commodity chemicals such as ethylene.1,2 Cu has been of intense interest as an electrocatalyst for this reaction, as it is selective for CO2 reduction over water reduction and can produce C2+ products, due to its positive adsorption energy for H* and more optimal binding energy for CO2 and related intermediates, compared to other metals.3–6 Still, it has been difficult to control selectivity to a single CO2 reduction product. To this end, many research groups investigated alloys and bimetallic configurations of Cu with other metals to attempt to tune the overall catalyst performance.3There are two main conceptual strategies for Cu-based alloy and bimetallic electrocatalysts. One approach is to create a tandem catalyst mechanism via combining Cu with other CO-producing elements like Ag or Au. In this concept, the crucial intermediate CO made on the second metal surface can transfer to Cu to be further reduced.7–12 For example, polycrystalline copper foil with Au nanoparticles favors the generation of oxygenates over hydrocarbons at low overpotentials.13 Increased CO concentration achieved by Ag nanoparticles on oxide-derived Cu nanowires may also open another pathway, namely *CO + *CHx coupling towards increased ethanol generation.14 A second approach is to change the local electronic structure of Cu by alloying with the other elements in order to tune the binding strength towards intermediates.15–19 For example, Ag atoms in the bimetallic Cu–Ag catalyst create a diversity of binding configurations compared with pure Cu that facilitates the production of ethanol.20 The compressive surface strain induced by Ag reduces the H* adsorbates, leading to the selective suppression of HER and favors the production of multi-carbon oxygenates.21 Ag2Cu2O3, with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio between Ag and Cu, can be used to produce bimetallic catalysts with a known composition and uniform distribution on the atomic scale. When applied to CO reduction, catalysts of this type can achieve 92% faradaic efficiency towards C2+ products at 600 mA cm−2.22
:1 stoichiometric ratio between Ag and Cu, can be used to produce bimetallic catalysts with a known composition and uniform distribution on the atomic scale. When applied to CO reduction, catalysts of this type can achieve 92% faradaic efficiency towards C2+ products at 600 mA cm−2.22
In this context, a facile strategy to introduce another element of specific concentration mixed with Cu on a variety of catalyst morphologies would be beneficial. This motivated us to investigate the cation exchange method, whereby a guest metal is introduced in the ion-form to replace the host metal ion in the compounds partially or entirely. This chemical conversion method has been widely employed to metal sulfides and oxides to achieve metastable facets, heteroatom doping, and introducing defect and strain, and also can be used to make multi-metal catalysts.23 Specific to CO2 reduction, the choice of the parent compound used for cation exchange may influence catalyst performance. We note here the reported enhancement in C2+ product selectivity for oxide-derived copper as compared to metallic copper, some of which has been attributed to increased roughness and grain boundaries which form as a result of in situ reduction of the oxide starting material.24–27 Bearing this in mind, we hypothesized that Cu sulfide could serve as a convenient cation exchange template for the formation of bimetallic CO2R electrocatalysts. We further hypothesized that the depletion of sulfur which occurs at the negative potential used to drive CO2R could lead to morphology changes which could be beneficial for control of selectivity.11,28
To test these hypotheses, we developed a cation exchange method to accommodate two common catalyst designs for CO2 reduction: nanoparticles and foil electrodes. Ag was selected as the second metal as it is selective for the production of CO, which is believed to be the key intermediate for the formation of C2+ products. For creating nanoscale catalysts, we prepared Cu sulfide nanosheets (C-nano-0) by colloidal synthesis, while for modifying Cu surfaces, Cu sulfides were directly grown on Cu foil using electro-oxidation followed by sulfurization. In both cases, the Ag/Cu mass ratio of the catalysts could be controlled at the cation exchange step. For nanosheets, the Ag/Cu mass ratio can reach 25 with the original structure remaining nearly intact. Cation exchange on surface-modified Cu foils (C-foil-x) produces well-dispersed Ag at low concentrations but leads to Ag particle nucleation at higher concentrations. Compared with C-nano-0 controls, CO2 reduction on moderately cation exchanged Ag/Cu sulfide nanosheets (CA-nano-2) increases the selectivity to C2+ products at −1.0 V vs. RHE. The selectivity for CO2 reduction of cation exchanged foils increases and then decreases over a period of 16 hours. Both the nanosheets and copper foil catalysts undergo noticeable morphology changes during the CO2 reduction, which may explain why the product distributions change as the CO2 reduction proceeds.
Synthesis procedures
The synthesis methods are summarized here; full details are in ESI.†Cu sulfide nanosheets (C-nano-0)
C-nano-0 was synthesized with a modified colloidal synthesis recipe (Scheme 1a, see also Table 1 for sample nomenclature).29,30 Typically, 257 mg copper(I) thiocyanate (CuSCN) was dispersed in 25 mL oleylamine (OAM). The mixture was first degassed and heated in N2 to 240 °C for 30 min. The synthesized nanosheets were then washed with hexanes and ethanol to remove the surface ligands and dispersed in hexanes for storage.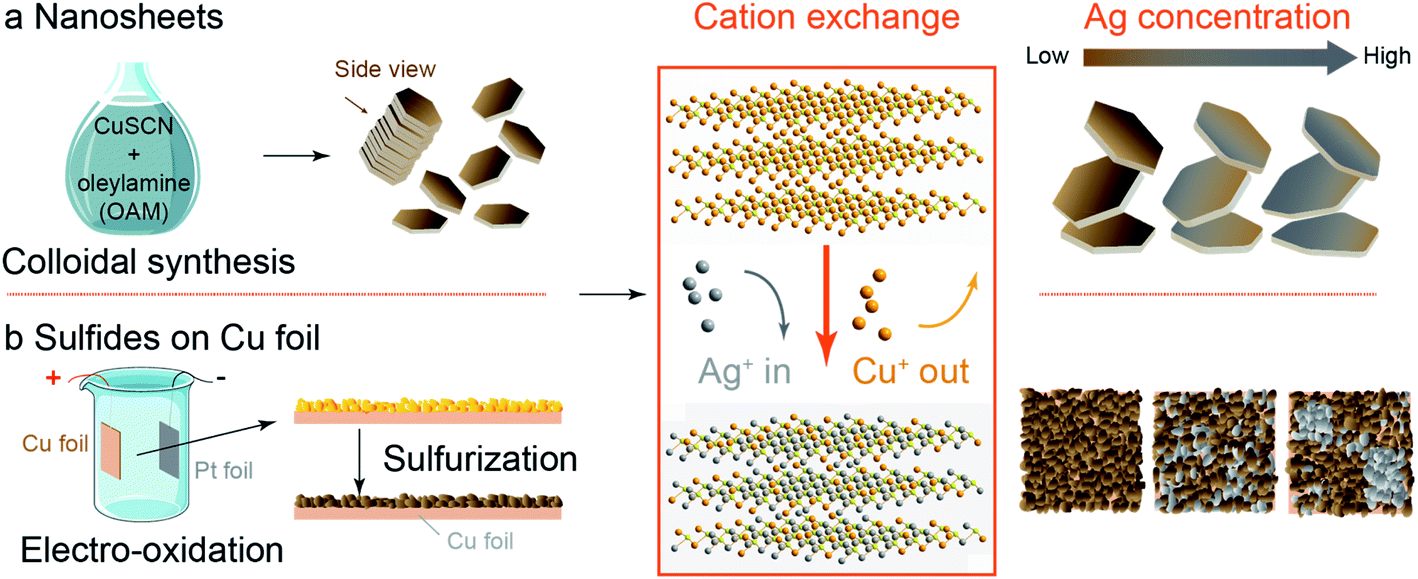 | ||
| Scheme 1 Synthetic strategies for Ag/Cu sulfide catalysts. (a) Cu sulfide nanosheets (C-nano-0, 100 nm lateral dimension, 14 nm thick) were obtained through colloidal synthesis with CuSCN in oleylamine (OAM). (b) Cu sulfides on Cu foil (C-foil-x) were obtained through electro-oxidation in 1 M NaOH to produce an oxide layer of a few 10 s of microns thick followed by sulfurization with 0.1 M Na2S. After cation exchange where Ag+ replaces the Cu+ in the Cu sulfides, Ag/Cu sulfide nanosheets (CA-nano-x) remain nanosheet structure with some distortion in shape as the Ag/Cu mass ratio ranges from 0.3 to 25; while for C-foil-x, Ag nucleates at higher Ag concentration, that impedes the uniform distribution of Ag and Cu. | ||
| Label | C-nano-0 (mg) | AgNO3 (mg) | Ag/Cu mass ratio |
|---|---|---|---|
| C-nano-0 | — | 0 | — |
| CA-nano-1 | 30 | 15 | 0.3 |
| CA-nano-2 | 30 | 30 | 0.8 |
| CA-nano-3 | 30 | 45 | 2.3 |
| CA-nano-4 | 30 | 90 | 25 |
Cu sulfides on Cu foil (C-foil-x)
Cu sulfides on Cu foil (C-foil-x, see Table 2 for sample nomenclature) were synthesized with a two-step electro-oxidation/sulfurization process (Scheme 1b). Cu foil was first cleaned and etched by 4 M HCl. After that, Cu(OH)2 was grown on the Cu foil by electro-oxidation in 1 M NaOH. The electrode was then immersed in 0.1 M Na2S at 90 °C for 12 h to obtain Cu sulfides.31 The current density set during the electro-oxidation process affects the grain size, as will be discussed later. Carbon substrates including carbon paper and carbon cloth with deposited Cu as the Cu source and chemical oxidation for the growth of Cu(OH)2 were also tried but were less successful; see ESI for details (Fig. S1–S6†).| Label | Current density (mA cm−2) | AgNO3 (mg) |
|---|---|---|
| C-foil-10 | 10 | — |
| C-foil-20 | 20 | — |
| C-foil-30 | 30 | — |
| C-foil-40 | 40 | — |
| CA-foil-20-10 | 20 | 10 |
| CA-foil-20-20 | 20 | 20 |
| CA-foil-20-30 | 20 | 30 |
| CA-foil-20-40 | 20 | 40 |
| CA-foil-40-40 | 40 | 40 |
Cation exchange method
For nanosheet samples, the hexanes dispersion containing C-nano-0 was added to an OAM solution (7 mL) containing the Ag precursor AgNO3.29 The solution was first degassed and heated to 50 °C in N2 and kept for another 30 min to complete the cation exchange reaction. The nanosheets were then washed with ethanol and hexanes and dispersed in hexanes for storage. The samples are denoted CA-nano-x as shown in Table 1.To perform cation exchange for the Cu sulfides on Cu foil (C-foil-x), AgNO3 was added to OAM with N2 bubbled to the solution in small Petri-dish. After the solution was heated to 50 °C and well mixed, C-foil-x was placed in the solution and kept for another 30 min. The electrode was then cleaned with ethanol and hexanes and dried with N2 flow. The Ag/Cu sulfides on Cu foil were named CA-foil-x-y with x denoting the current density and y the relative Ag fraction (Table 2). As discussed later, the concentration of Ag+ in the cation exchange solution affected the dispersion of Ag, with uniform distributions being formed at low concentrations and Ag particles nucleating on the surface at high concentrations.
Electrochemical characterization and product analysis
Electrode preparation and CO2 reduction
For nanosheet samples (C-nano-0 and CA-nano-x), the catalysts were first anchored on carbon black at a 1:1 mass ratio of catalyst to carbon. The catalyst was dispersed in ethanol and water, and Nafion was added as the binder. After sonication, the homogeneous catalyst ink was drop cast on glassy carbon substrates (GC) followed by drying overnight at room temperature. Typically, the catalysts were tested with the loading of 0.6 mg on an electrode area of 0.785 cm2. Sulfides on Cu foil (C-foil-x and CA-foil-x-y) were tested directly without further modification. The CO2 reduction was conducted in a three-electrode system with 0.05 M K2CO3 as the electrolyte and Pt and Ag/AgCl (saturated KCl) as the reference and counter electrode, respectively. The cathodic and anodic chambers were separated by an anion exchange membrane. CO2 was purged at 5 sccm to the cathodic chamber, and the test started after 15 min CO2 purging to ensure complete saturation, after which the electrolyte becomes 0.1 M bicarbonate. We note that the freshly prepared electrodes are not active for CO2R and initially favour H2 production. The time evolution of the catalysts and the induced changes in the product profiles under CO2 electroreduction conditions are thus discussed in detail below.
Product detection
The gas products, including H2, CO, methane, ethylene, and ethane, were detected by online gas chromatography (GC) using methods reported previously.32,33 Typically, GC sampling was started 5 min after the test began, and the results were given by the average of the second to the last sample. The electrolyte was collected after each test and analysed by nuclear magnetic resonance (NMR) for liquid products, including formate, methanol, ethanol, n-propanol, and other low-concentration C2+ products such as acetate, glycolaldehyde, allyl alcohol, acetaldehyde, acetone, and propionaldehyde.6Results and discussion
Cu sulfide nanosheets (C-nano-0)
TEM images of C-nano-0 made by colloidal synthesis are presented in Fig. 1. The nanosheets have a hexagonal shape with a lateral size of 100 nm and a thickness of 14 nm, as shown in Fig. 1a–c. As shown in the HRTEM image (Fig. 1d), the planes with the spacing of 1.95 Å and a 60° angle in between can be assigned to the (0 16 0) and (0 8 6) planes of monoclinic Cu7S4.34 In addition, the 3.28 Å lattice spacing in the side view HRTEM image matches the (16 0 0) plane (Fig. 1e). Some areas exhibit less contrast with no clear lattice; these areas may have a high defect concentration or be amorphous. The nanosheets have a large specific surface area which could be beneficial for catalytic activity.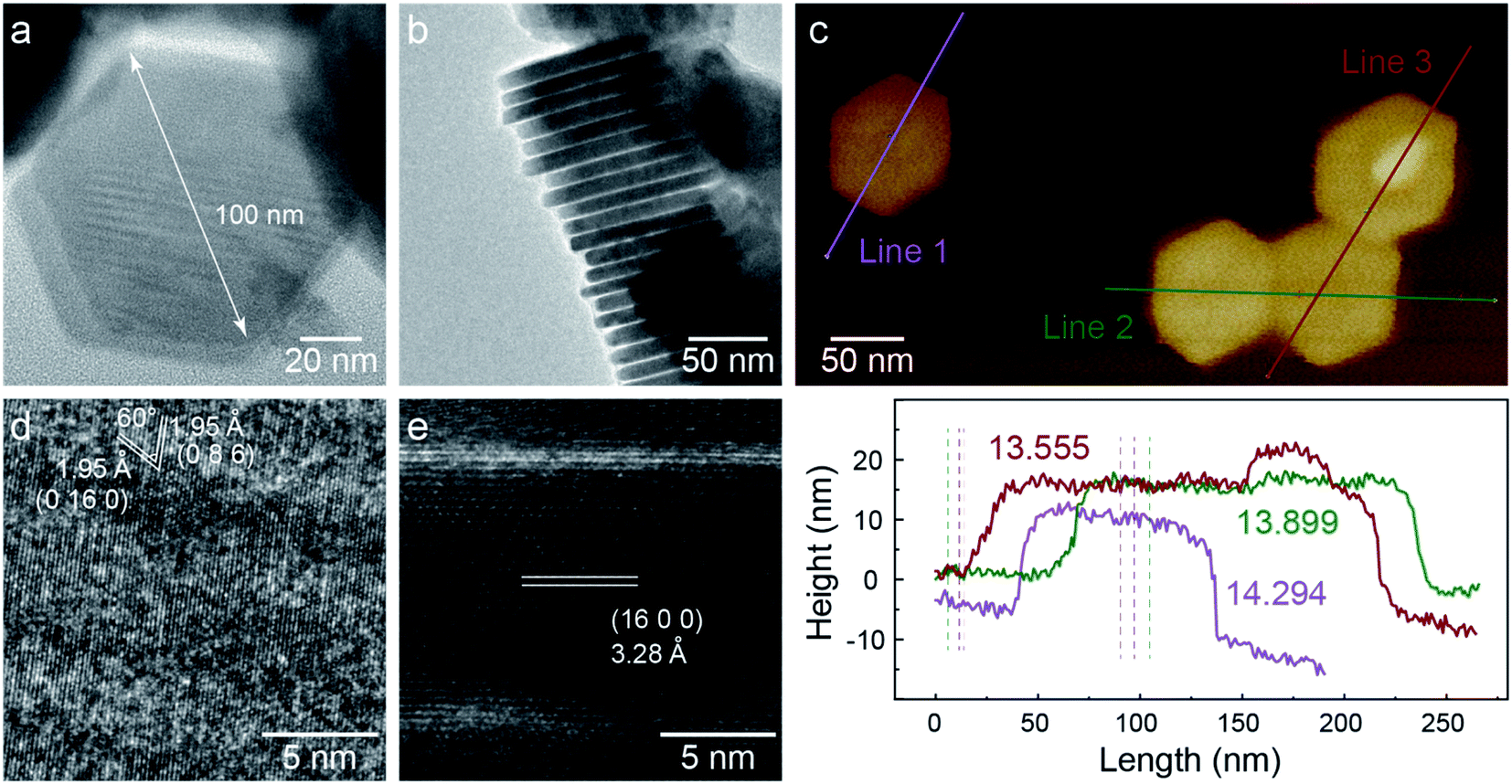 | ||
| Fig. 1 TEM characterization of C-nano-0. (a) C-nano-0 has a hexagonal shape with a lateral size of 100 nm. (b) Side-view of a stack of nanosheets. (c) AFM image and height information, showing the thickness of 14 nm. (d and e) High-resolution TEM (HRTEM) image shows the lattice structure of monoclinic Cu7S4. | ||
XPS spectra of C-nano-0 provide information about the surface condition of the nanosheets (Fig. S7 and S8†). The S/Cu atomic ratio was 0.67, slightly higher than the stoichiometric ratio of Cu7S4, supporting the Cu7S4 lattice structure with a sulfur-rich surface of C-nano-0. Most Cu in the nanosheets has the valence state of 1+, the deviation of the spectrum may result from the defects in the nanosheets and Cu2+.35,36 The existence of N, the peaks at 163 eV in the S2p spectrum, and the peak at 286 eV in the C1s spectrum indicate the presence of a small amount of residual ligand –SCN from the precursor CuSCN.37 It is also possible that OAM is present despite the washing steps designs to remove it; however, prior studies have shown that it does not block active sites for CO2 electroreduction.38
Bimetallic sulfide nanosheets (CA-nano-x)
The mass ratio of Ag/Cu concentration was well controlled from 0.3 (CA-nano-1) to 25 (CA-nano-4) (Table S1†). The ratio of Ag and Cu in samples were quantified by inductively coupled plasma mass spectroscopy (7900 ICP-MS, Agilent, ICP) using the He mode. The internal standard was Ge or Rh selected based on its first ionization potential and M/Z as compared to Cu or Ag, respectively. As shown in the XRD spectra (Fig. 2a), the crystal structure of the nanosheets undergoes a noticeable change as the Ag concentration increases. Prior to cation exchange, the nanosheets have the crystal structure of Cu7S4 (PDF #23-0958),39 in agreement with TEM. For a small amount of cation exchange (CA-nano-1), the crystal structure remains the same as the C-nano-0 with the most prominent peak in the XRD pattern at ∼48° being assigned to Cu7S4 (0 16 0). At higher Ag concentrations, a shift of the peak near ∼32° is observed from the yellow marked position for Cu7S4 to the purple marked Ag2S position. The appearance (CA-nano-2) and shift of the peak pointed by the arrow as Ag concentration increases support the gradual structural change. With the ratio of Ag/Cu reaches 25 (CA-nano-4), the structure completely changes to Ag2S, demonstrated by the peak at ∼34°, which can be assigned to (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 2 1) planes. The morphology also undergoes obvious change with large shape distortion and the nanosheet structure remains (Fig. 2b), while with Ag/Cu ratio less than 1, the hexagonal shape remains with only minor changes in shape or thickness (Fig. S9–S12†).
2 1) planes. The morphology also undergoes obvious change with large shape distortion and the nanosheet structure remains (Fig. 2b), while with Ag/Cu ratio less than 1, the hexagonal shape remains with only minor changes in shape or thickness (Fig. S9–S12†).
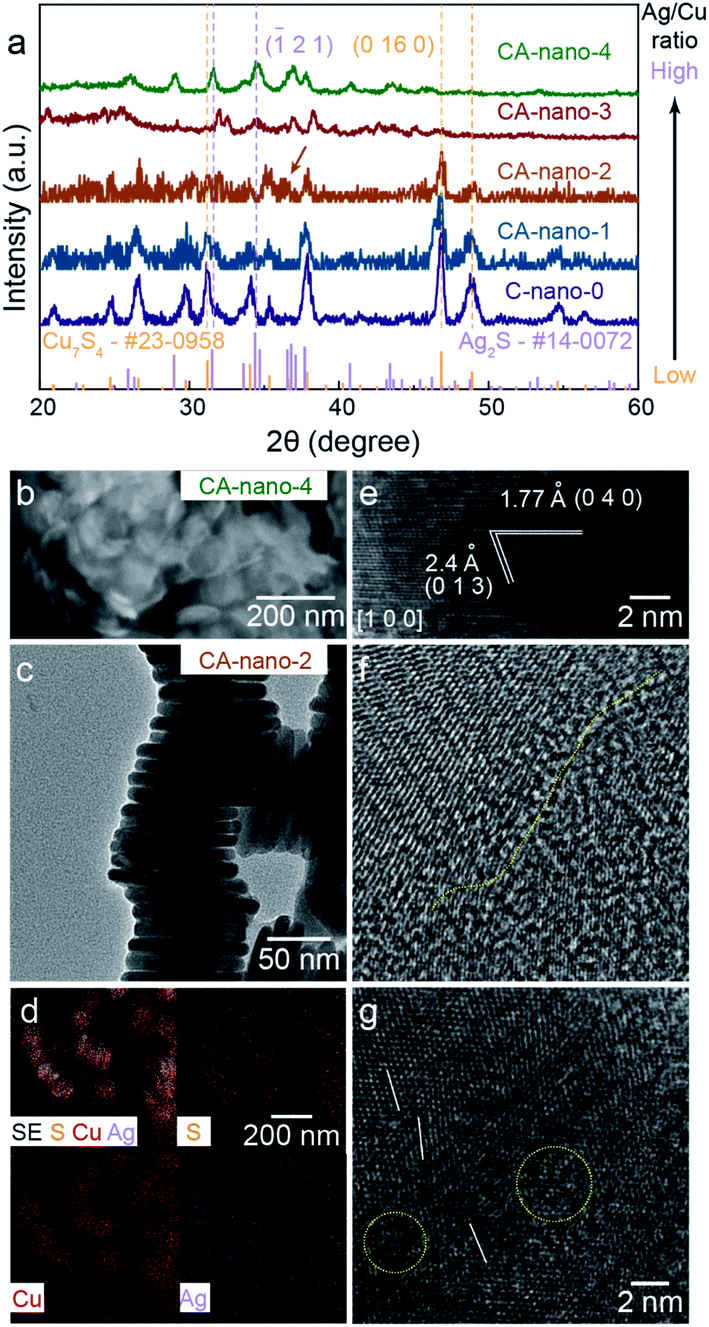 | ||
| Fig. 2 Lattice structure, morphology, and composition of sulfide nanosheets. (a) XRD spectra for C-nano-0 and CA-nano-x. Lattice structure evolves from Cu7S4 to Ag2S with increased Ag concentration. (b) SEM image for CA-nano-4, nanosheet structure remains with shape distortion. (c) Side-view TEM of stacked CA-nano-2, morphology is also shown in Fig. S13a.† (d) SEM and EDS mapping shows uniform distribution of Cu, Ag, and S within the nanosheets. (e) HRTEM image of CA-nano-2 shows the lattice structure of Ag2S viewed from direction [1 0 0]. (f and g) HRTEM images for the basal surface. Yellow and white marks denote the defect and boundary-rich surface. | ||
Fig. 2c–g show the morphology, elemental distribution, and typical lattice structure of CA-nano-2 (Ag/Cu = 0.8) as determined by SEM and TEM. The morphology uniformity of the nanosheets decreases compared with that before cation exchange but the enlarged SEM image with elemental mapping still provides evidence of the uniform distribution of Ag, Cu, and S, without any spatial separation (Fig. 2d).
HRTEM images obtained from the side-view demonstrate the existence of crystalline Ag2S. In Fig. 2e, observed from direction [1 0 0], the plane with the spacing of 1.77 Å and 2.4 Å can be assigned to the planes (0 4 0) and (0 1 3), respectively. Besides, it shows a composition of small crystals with different facets on the basal surface (Fig. 2f and g). The yellow line marks the boundary between two crystals that possess different structures, the white lines show the tilt of the lattice, and the yellow circles mark less-contrast areas indicating defect and amorphous regions. Combined with HRTEM images showing different lattices and the corresponding FFT patterns obtained from CA-nano-2 (Fig. S13†), one conclusion can be made is that crystal structure becomes more complicated due to the introduction of Ag and that the cation exchange does not have a simple outcome, a single crystal Ag2S or dominant exposed facets, for instance.
The complicated surface outcome from cation exchange might arise from a number of factors: the hexagonal shape of C-nano-0 triggers cation exchange from the corners and form separate grains connected by grain boundaries; the intrinsic poor crystallinity of the template leads to inconsistent reaction tendencies at different areas; the energy imposed by the low temperature (50 °C) for cation exchange is not enough for atoms to move towards the more crystalline structure.40 Therefore, as expected, CA-nano-2 has a complicated defect and boundary-rich structure.
Sulfide nanosheets for CO2 reduction
SEM analysis shows that the nanosheets were evenly dispersed on porous carbon before CO2R (Fig. S14†). Prior in situ work with copper oxide pre-catalysts has shown that under CO2R conditions, reduction of oxides to metallic Cu occurs prior to the formation of gas phase products.41 We thus expected that sulfide nanosheets could have a similar behaviour, with the initial current being due to non-faradaic processes as the catalyst is reduced. Therefore, pre-reduction at negative potentials may increase selectivity for CO2 reduction. Additionally, the pre-reduction may facilitate the removal of the surface ligands –SCN, which might block or change the activity of the catalytic sites. Previous research show such anionic ligands could be removed under negative potentials and may induce reconstruction of the nano-scale catalysts, which further influence the performance.42,43 Thus, for a consistent comparison, the electrodes were evaluated in the same potential sequence. All samples were tested for the same 1.5 h duration from the most positive potential (−0.2 V vs. RHE) to the most negative potential (−1.6 V vs. RHE), as shown in Fig. 3 where two different cation-exchanged samples CA-nano-2 and CA-nano-4 are evaluated and compared with C-nano-0 control. Cyclic voltammetry (CV) measurements were conducted for the electrodes to show both Ag and Cu in the nanosheets are electrochemical active (Fig. S15†).17 It is worth mentioning that the current densities between the three samples are of similar values and trends, such that all catalysts had similar mass transfer limits for CO2 availability at a given potential (Fig. S16†). The electrochemical impedance spectra (EIS) indicate a lower ion transport resistance for cation exchanged samples (CA-nano-2 and CA-nano-4) compared with Cu sulfides (C-nano-0) (Fig. S17†).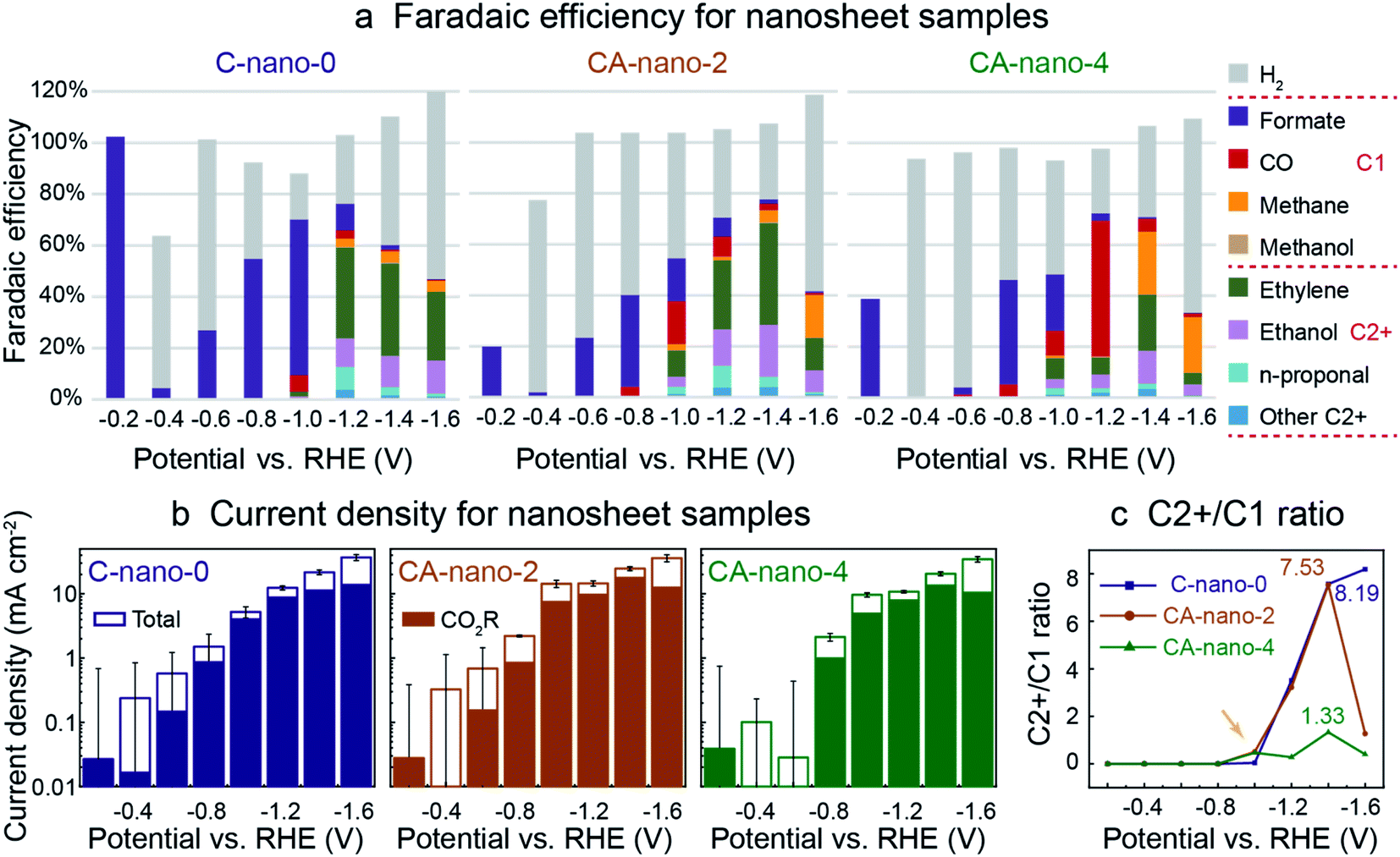 | ||
| Fig. 3 CO2 reduction performance for sulfide nanosheets: C-nano-0, CA-nano-2, and CA-nano-4. (a) Plot of faradaic efficiency (FE); note that all produce formate exclusively at low overpotentials (−0.2 V vs. RHE). (b) Total current density, solid portion of bar plot indicates partial current density for CO2 reduction products. Error bars of total current density at small negative potentials are relatively large as the current was fluctuating in time. The corresponding chronoamperometry (CA) plots are in Fig. S16.† (c) Plot for the ratio of C2+ to C1 products. Ag increases C2+ product generation at small negative potentials (−1.0 V vs. RHE). The numbers in the plot stand for the highest C2+/C1 ratio obtained for each catalyst. CO2 reduction was conducted in 0.05 M K2CO3 in an experimental sequence from more positive to more negative potentials. Catalyst loading was 0.6 mg in all cases. | ||
Interestingly, at −0.2 V vs. RHE, Fig. 3a, all nanosheet catalysts produced formate exclusively before the production of other potential 2e− products: CO and by-product H2 from HER. However, we note that the faradaic efficiency at −0.2 V vs. RHE cannot be measured precisely since the potentiostat current at this potential, ∼0.01 mA, is very small. Also, the larger current measured at the beginning of a run due to the non-faradic reduction of the catalyst surface sulfide or oxide layers, where not all the electrons were used for the formation of electrocatalytic CO2 reduction products, can lead to inaccuracy, especially for small negative potential regions, where large current fluctuations were observed as shown by the error bars (Fig. 3b). To assess whether formate was made just at the start or throughout the run, we tested CA-nano-2 again at −0.2 V vs. RHE after the electrode has been tested at more negative potentials (Fig. S18†). Formate was still the only product detected, although the initial current density was smaller compared with as-synthesized material. One explanation is the initial current was from the reduction of the surface oxidation layer formed in the environment after the previous test instead of the reduction of the catalyst as for fresh electrode.44 Formate was also the only product detected at −0.1 V and −0.3 V vs. RHE for nanosheets (Fig. S19†).
For all nanosheets, H2 appears as a product at −0.4 V vs. RHE. At larger negative potentials its FE decreases and FE for formate increases. For C-nano-0, CO appears at −1.0 V vs. RHE (FECO = 6%). C2+ products, including ethylene, ethanol, and C3 products like n-propanol, appear at −1.0 V vs. RHE in trace amounts and dominate at −1.2 V vs. RHE with the ratio of C2+ to C1 products of 3.51. This ratio further increases when a more negative potential is applied, 7.57 and 8.19 for −1.4 V and −1.6 V vs. RHE, respectively (Fig. 3c). However, this increase comes from the decrease of formate and methane, rather than increased production in C2+ products, and an increase in H2 production is also clearly shown since −1.0 V vs. RHE (Fig. S20†).
CA-nano-2 starts to produce noticeable C2+ products, including ethylene (10%), ethanol (4%), and n-propanol (3%) at −1.0 V vs. RHE, more positive than for C-nano-0. Also, the overall CO2 reduction products dominate at −1.4 V vs. RHE (77.7%), which is shifted from −1.2 V for C-nano-0; this may be attributed to better HER suppression at more negative potentials as a result of the Ag content.18 For CA-nano-2 at −1.4 V vs. RHE, the faradaic efficiency for C2+ products is 68.6%, and the ratio of C2+ over C1 products is 7.53. The C2+/C1 ratio is similar to that of the control (Fig. 3c), but with a smaller FE for H2. The introduction of Ag leads to an increase in CO production compare to the control, with the maximum FE reached −1.0 V vs. RHE. When a more negative potential is applied, the FE for CO decreases, which could be a result of CO diffusion to Cu where it is further reduced to C2+ products.8 At −1.6 V vs. RHE, where Ag has less contribution to reducing CO2 to CO but instead increases HER, methane becomes the dominant product.45
For CA-nano-4, where Ag/Cu ratio is 25, CO production from Ag becomes the primary product, especially at −1.2 V vs. RHE (53%), much higher than that of CA-nano-2 (8%). Also, methane is a dominant product at negative potentials (25% at −1.4 V vs. RHE). One explanation for the large increase in C1 products might be that the increased concentration of Ag breaks up the continuous Cu surface, making the CO–CO binding difficult.46 Thus, instead of making C2 products like ethylene and ethanol, conversion of the CO intermediate forms methane. Indeed, for this catalyst, the highest C2+/C1 ratio was only 1.33 (Fig. 3c). Similar to the other nanosheet samples, with more negative potential applied, H2 became the dominant product, which also caused a decrease in the current density for overall CO2 reduction at −1.6 V vs. RHE where more surface has been occupied by the H absorbent for H2, instead of performing CO2 reduction.
Sulfides on Cu foil. (C-foil-x and CA-foil-x-y)
For bimetallic sulfides on Cu foil, the crystal size of the Cu sulfide template (C-foil-x, x denoting current density, Table 2) can be controlled by the current density during the electro-oxidation process. Larger and more defined grains are produced at higher current densities (Fig. 4a). For cation-exchanged samples (CA-foil-x-y), the Ag/Cu mass ratio increases as more Ag precursor is added during the cation exchange (Fig. S21†). With lower Ag concentrations (CA-foil-20-10), the Ag and S were uniformly distributed across the surface (Fig. 4b and c). SEM image from the side view indicates the thickness of 10 s of microns, consistent with the calculated modified Cu thickness (Fig. 4d, ESI†). Also, compared with C-foil-20, the surface of the cation-exchanged counterpart CA-foil-20-10 transformed into a rippled structure composed of finer grains, with empty spaces between layers, demonstrated by zoomed-in images, that might be beneficial for the transport of reactants and products (Fig. 4e and f). However, with further increased Ag precursor AgNO3, Ag nucleation on the surface through the reduction of Ag+ to metallic Ag instead of exchanging Cu+ can be observed (CA-foil-20-40). Ag concentration at the newly merged flower-like flakes is clearly shown in Fig. 4g and S22.† When the particle size of Cu sulfides was too large (C-foil-40), the cation-exchanged counterpart CA-foil-40-40 with enough Ag precursor (40 mg) grow into a triangle structure (Fig. S23†). | ||
| Fig. 4 Morphology characterization for sulfides on Cu foil. (a) SEM images for Cu sulfides (C-foil-x) obtained from the sulfurization of Cu(OH)2 achieved by electro-oxidation at the current density of 10, 20, 30, and 40 mA cm−2. Morphology (b) and elemental distribution (c) from top view and side view (d–f) of bimetallic sulfides on Cu foil (CA-foil-x-y) with moderate Ag concentration and extra Ag concentration (g). | ||
Evolution of sulfide catalysts during the CO2 reduction
Bimetallic sulfides on Cu foil (CA-foil-x-y) with a variety of Ag concentrations were tested in the potential range from −0.8 V to −1.4 V vs. RHE. If considering ethylene as the target product, CA-foil-20-40 with high surface Ag concentration at −1.2 V vs. RHE gave the best performance (FEethylene = 34%, Fig. 5a and S24†). However, results of different runs show large variations in terms of both FE and current density, with selectivity to ethylene and liquid C2+ products initially improving and then declining (3 tests at −1.0 V vs. RHE and then 3 at −1.2 V vs. RHE, Fig. S25†). The morphology after reduction also shows obvious change from the fresh sample, which may come from the reaction between the active surface and the reactant and intermediates (Fig. 5b).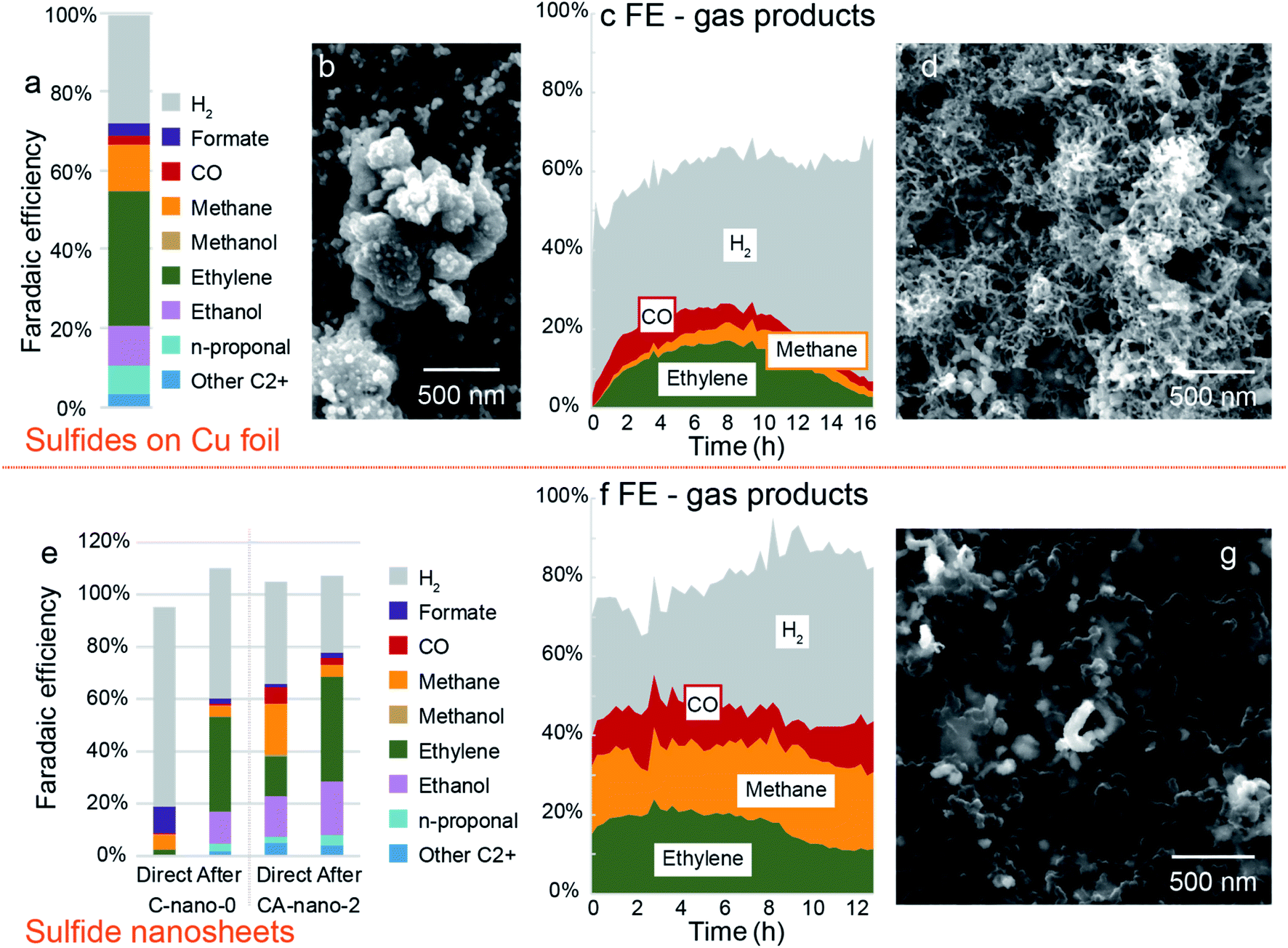 | ||
| Fig. 5 Evolution of catalysts and corresponding products change during CO2 reduction. (a) Faradaic efficiency plot for CA-foil-20-40 at −1.2 V vs. RHE. (b) Morphology of CA-foil-20-40 after CO2 reduction. Corresponding current plot is in Fig. S24.† (c) Faradaic efficiency for gas products. Sample: CA-foil-20-40. Test condition: −1.0 V vs. RHE for 16 h. Corresponding current plot is in Fig. S26.† (d) SEM image of CA-foil-20-40 after test. (e) Comparison of sulfide nanosheet C-nano-0 and CA-nano-2 between directly tested at −1.4 V vs. RHE (left bar plot) and tested after pre-reduction at other negative potentials (right bar plot). (f) Faradaic efficiency for gas products. Sample: CA-nano-4. Test condition: −1.4 V vs. RHE for 13 h. Corresponding current plot is in Fig. S29.† (g) SEM image of CA-nano-4 after test. | ||
To further investigate this performance change, as-synthesized CA-foil-20-40 was directly tested at −1.0 V vs. RHE for 16 h. The current was in the range of 8–12 mA cm−2 during the test, Fig. S26.† Faradaic efficiency for the gas products is plotted in Fig. 5c. CO was the first gas product of CO2 reduction to be observed and reaches a maximum in FE at 2 hours. Ethylene and methane increased with the consumption of CO, and FE for ethylene rose from 0% at first to 17% at 8 h. As the plot shows, the product profile undergoes continuous change during CO2 reduction, with the optimal working region for ethylene being between 6 to 10 h.
The evolution of the catalyst surface may explain the performance change.18,47 Fig. 5d is the SEM image of the catalyst after the 16 h reduction. The surface morphology changes from the particle shape into the nest structure composed of filaments with a diameter of ∼20 nm, which appears to have a higher surface area compared with the fresh sample. The increased roughness of the catalyst during the CO2 reduction might be one reason for the change of gas product distribution since the nest composed of the 1D filament structure possesses both high surface area and good electrical conductivity derived from the interconnected nature and structural stability compared with nanoparticle counterparts.48
C-foil-x also undergoes similar morphology change under CO2R conditions, with smaller grains (C-foil-10), the surface evolved into a nest structure composed of filaments (diameter: 40 nm). The time evolution of the catalyst is also influenced by the particle size of Cu sulfides on Cu foil, as the grain size increases as a higher current applied in the electro-oxidation step, the nest structure evolved from CO2R that possess high roughness and sufficient gas pathway, which might be beneficial, gradually disappears and is replaced by large particles (Fig. S27†). According to the research on oxide-derived copper. The surface copper oxide may be reduced to metallic copper under negative potential, while other groups have reported that the residual underlying oxygen is beneficial for CO2 reduction.25,49 The sulfides on Cu foil may also go through a similar process as Cu oxides, and the sulfur will eventually be depleted such that the catalyst will be metallic copper; thus the depletion of sulfur may result in the evolution of morphology and performance.50
Besides, as previously mentioned, for sulfides on the Cu foil electrode, there is a limit for the introduction of Ag because the cation exchange of Cu+ by Ag+ competes with the direct nucleation on the electrode surface. Thus, in order to create an Ag-rich surface by other means, we deposited 100 nm Ag by an E-beam evaporator onto the Cu sulfides layer. However, when tested at −1.1 V vs. RHE, the potential most beneficial for Ag foil to produce CO,51 the morphology change from the flat overlayer to more complex nest structures during the CO2 reduction still occurred and exposed the underlying Cu sites. As a result, the product distribution was very different from that expected from a silver-rich surface52 (Fig. S28†).
Activation of cation-exchanged nanosheets
For CA-nano-x nanosheets, when put directly under CO2 reduction conditions, the performance is far from ideal. Thus we tried to employ pre-reduction under other negative potentials as an activation method to improve CO2R selectivity. As shown in Fig. 5e, freshly prepared C-nano-0 and CA-nano-2 were directly placed at −1.4 V vs. RHE, and the results are compared with that after pre-reduction at other negative potentials. For direct, much more H2 was produced compared with the pre-reduced electrode results, the FE for H2 was 76% for C-nano-0 and 39% for CA-nano-2, while for pre-reduced samples, the number was only 50% and 30%, respectively. The product distribution of CO2 reduction also varies from the pre-reduced electrodes. For example, for fresh C-nano-0, the major CO2 reduction products are C1 species, including formate (10%) and methane (6%). In contrast, when the sample was tested at the same potential after pre-reduction, there were mainly C2+ products. The situation was similar for cation exchanged nanosheets. The total FE for C2+ products for fresh CA-nano-2 was 38.02%, only half the C2+ products observed for the pre-reduced electrode (69%).After CO2 reduction, as expected, the adjacent nanosheets were evolved into a filament structure with a diameter of ∼100 nm, similar to the diameter of the synthesized nanosheets, and the product profile changes accordingly (Fig. 5f and g). The change might be related to the surface ligands, which may affect electrochemical behavior and the stability of nanomaterials, yet more characterizations need to be done to confirm this.42,53,54 Additionally, for C-nano-0, the Cu was transformed into large clusters (∼500 nm, Fig. S30†), different from the filament structure of CA-nano-2. This might come from the difference in the CO/CO2 binding energy on Cu and Ag.55 With higher binding energy between the Cu and CO/CO2 facilitate the mobility of the anchored Cu, thus resulting in the aggregation. However, the Ag added tothe CA-nano-2 has smaller binding energy with CO/CO2 and confined the mobility, and the spatial separation of the nanosheets prevents further agglomeration.
Conclusions
We have demonstrated that Cu sulfides can be used as a template for cation exchange to achieve bimetallic Ag/Cu sulfide catalysts with a well-controlled Ag/Cu mass ratio by changing the concentration of Ag precursor AgNO3. For nanosheets, the Ag/Cu ratio can reach 25 with the nanosheet structure remaining, while it is difficult to produce an Ag-rich surface beginning with sulfides on Cu foil. Formate was the only product detected at low overpotentials (−0.2 V vs. RHE), and with the introduction of moderate Ag, nanosheet catalysts showed increased C2+ product generation for CO2 reduction. The product profiles appear to be influenced by CO availability controlled by Ag concentration, suggesting a possible tandem catalytic mechanism. The reconstruction of the catalyst during CO2 reduction increased the production of multi-carbon products.The cation exchange method can be further applied to other bimetallic or trimetallic chalcogenides like phase segregated Cu–Au sulfides,40 Cu–Ni selenides,56 Cu–Co sulfides nanoboxes,57 and CuInS2-doped ZnS,58 and could potentially be used for multifunctional photo/electrocatalysis. With modifications of ligands or additives during the cation exchange method, may realize the control of even vs. uneven distribution of two elements with the same overall concentration that can be employed as a great test field for mechanism investigation.
Author contributions
Jinghan Li and J. W. A. conceptualized the study, and JWA acquired funding for it. Jinghan Li and Junrui Li developed synthesis methodology and analytical chemistry methods. Jinghan Li performed all synthesis and conducted the electrochemical experiments, XRD, XPS, ICP, AFM, and SEM characterization and data interpretation. Chaochao Dun performed TEM characterization using funding acquired by Jeffrey J. Urban. Wenshu Chen performed synthesis. Di Zhang and Jiajun Gu provided advice on data analysis and mentorship. Jinghan Li and JWA wrote the original draft of the manuscript and all authors contributed to its final editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on the work supported by the Joint Center for Artificial Photosynthesis (JCAP), a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. Transmission electron microscopy was performed at the Molecular Foundry, which was supported by the Office of Science, Office of Basic Energy Sciences of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. J. L. acknowledges financial support from the China Scholarship Council and experimental help and support from group members during this challenging time.References
- O. S. S. Bushuyev, P. De Luna, C. T. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. O. Kelley and E. H. H. Sargent, What Should We Make with CO2 and How Can We Make It?, Joule, 2018, 2, 825–832 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Stability and Degradation Mechanisms of Copper-Based Catalysts for Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef PubMed.
- Y. Hori Electrochemical CO2 Reduction on Metal Electrodes, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, New York, NY, 2008, vol. 29, pp. 89–189 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces, Energy Environ. Sci., 2012, 5, 7050 RSC.
- G. Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, Sequential Cascade Electrocatalytic Conversion of Carbon Dioxide to C–C Coupled Products, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef.
- Y. Lum and J. W. Ager, Sequential Catalysis Controls Selectivity in Electrochemical CO2 Reduction on Cu, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- D. Ren, B. S. H. Ang and B. S. Yeo, Tuning the Selectivity of Carbon Dioxide Electroreduction toward Ethanol on Oxide-Derived CuxZn Catalysts, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Cu-Ag Tandem Catalysts for High-Rate CO2 Electrolysis toward Multicarbons, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, Structural Sensitivities in Bimetallic Catalysts for Electrochemical CO2 Reduction Revealed by Ag-Cu Nanodimers, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS.
- S. Lee, G. Park and J. Lee, Importance of Ag-Cu Biphasic Boundaries for Selective Electrochemical Reduction of CO2 to Ethanol, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Improved CO2 Reduction Activity towards C2+ Alcohols on a Tandem Gold on Copper Electrocatalyst, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- L. R. L. Ting, O. Piqué, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, Enhancing CO2 Electroreduction to Ethanol on Copper-Silver Composites by Opening an Alternative Catalytic Pathway, ACS Catal., 2020, 10, 4059–4069 CrossRef CAS.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Atomic Layer Deposition of ZnO on CuO Enables Selective and Efficient Electroreduction of Carbon Dioxide to Liquid Fuels, Angew. Chem., 2019, 131, 15178–15182 CrossRef.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, Electrochemical Activation of CO2 through Atomic Ordering Transformations of AuCu Nanoparticles, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS.
- X. Lv, L. Shang, S. Zhou, S. Li, Y. Wang, Z. Wang, T. Sham, C. Peng and G. Zheng, Electron-Deficient Cu Sites on Cu3Ag1 Catalyst Promoting CO2 Electroreduction to Alcohols, Adv. Energy Mater., 2020, 10, 2001987 CrossRef CAS.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Selective Reduction of CO to Acetaldehyde with CuAg Electrocatalysts, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Synergistic Geometric and Electronic Effects for Electrochemical Reduction of Carbon Dioxide Using Gold-Copper Bimetallic Nanoparticles, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton, E. H. Sargent, F. P. Garcia de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, Binding Site Diversity Promotes CO2 Electroreduction to Ethanol, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, Electrochemical CO2 Reduction over Compressively Strained CuAg Surface Alloys with Enhanced Multi-Carbon Oxygenate Selectivity, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- N. Martić, C. Reller, C. Macauley, M. Löffler, A. M. Reichert, T. Reichbauer, K.-M. Vetter, B. Schmid, D. McLaughlin, P. Leidinger, D. Reinisch, C. Vogl, K. J. J. Mayrhofer, I. Katsounaros and G. Schmid, Ag2Cu2O3 – a Catalyst Template Material for Selective Electroreduction of CO to C2+ Products, Energy Environ. Sci., 2020, 13, 2993–3006 RSC.
- T. Ling, M. Jaroniec and S. Qiao, Recent Progress in Engineering the Atomic and Electronic Structure of Electrocatalysts via Cation Exchange Reactions, Adv. Mater., 2020, 32, 2001866 CrossRef CAS PubMed.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Electrochemically Converting Carbon Monoxide to Liquid Fuels by Directing Selectivity with Electrode Surface Area, Nat. Catal., 2019, 1–7 Search PubMed.
- Y. Lum and J. W. Ager, Stability of Residual Oxides in Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction Investigated with 18O Labeling, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Evidence for Product-Specific Active Sites on Oxide-Derived Cu Catalysts for Electrochemical CO2 Reduction, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- Y. Huang, Y. Chen, T. Cheng, L.-W. W. Wang and W. A. Goddard, Identification of the Selective Sites for Electrochemical Reduction of CO to C2+ Products on Copper Nanoparticles by Combining Reactive Force Fields, Density Functional Theory, and Machine Learning, ACS Energy Lett., 2018, 3, 2983–2988 CrossRef CAS.
- W. T. Osowiecki, J. J. Nussbaum, G. A. Kamat, G. Katsoukis, M. Ledendecker, H. Frei, A. T. Bell and A. P. Alivisatos, Factors and Dynamics of Cu Nanocrystal Reconstruction under CO2 Reduction, ACS Appl. Energy Mater., 2019, 2, 7744–7749 CrossRef CAS.
- T. Kim, J. Park, Y. Hong, A. Oh, H. Baik and K. Lee, Janus to Core-Shell to Janus: Facile Cation Movement in Cu2−xS/Ag2S Hexagonal Nanoplates Induced by Surface Strain Control, ACS Nano, 2019, 13, 11834–11842 CrossRef CAS.
- J. Park, J. Park, J. Lee, A. Oh, H. Baik and K. Lee, Janus Nanoparticle Structural Motif Control via Asymmetric Cation Exchange in Edge-Protected Cu1.81S@IrxSy Hexagonal Nanoplates, ACS Nano, 2018, 12, 7996–8005 CrossRef CAS.
- S. Deng, Y. Shen, D. Xie, Y. Lu, X. Yu, L. Yang, X. Wang, X. Xia and J. Tu, Directional Construction of Cu2S Branch Arrays for Advanced Oxygen Evolution Reaction, J. Energy Chem., 2019, 39, 61–67 CrossRef.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Effects of Temperature and Gas-Liquid Mass Transfer on the Operation of Small Electrochemical Cells for the Quantitative Evaluation of CO2 Reduction Electrocatalysts, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- Y. Lum, T. Cheng, W. A. Goddard and J. W. Ager, Electrochemical CO Reduction Builds Solvent Water into Oxygenate Products, J. Am. Chem. Soc., 2018, 140, 9337–9340 CrossRef CAS.
- C. Zhang, C. Yan, Z. Xue, W. Yu, Y. Xie and T. Wang, Shape-Controlled Synthesis of High-Quality Cu7S4 Nanocrystals for Efficient Light-Induced Water Evaporation, Small, 2016, 12, 5320–5328 CrossRef CAS.
- X. Li, Z. Yao, J. Wang, D. Li, K. Yu and Z. Jiang, A Novel Flake-like Cu7S4 Solar Absorber for High-Performance Large-Scale Water Evaporation, ACS Appl. Energy Mater., 2019, 2, 5154–5161 CrossRef CAS.
- M. Sun, Z. Li, Q. Fang, S. Han, C. Cai, H. Li, W. Shen, X. Liu and Y. Fu, Room-Temperature Synthesized Porous Cu(OH)2/Cu7S4 Hybrid Nanowires as a High-Performance Electrode Material for Asymmetric Supercapacitors, J. Mater. Chem. A, 2020, 8, 724–734 RSC.
- N. Wijeyasinghe, A. Regoutz, F. Eisner, T. Du, L. Tsetseris, Y.-H. Lin, H. Faber, P. Pattanasattayavong, J. Li, F. Yan, M. A. McLachlan, D. J. Payne, M. Heeney and T. D. Anthopoulos, Copper(I) Thiocyanate (CuSCN) Hole-Transport Layers Processed from Aqueous Precursor Solutions and Their Application in Thin-Film Transistors and Highly Efficient Organic and Organometal Halide Perovskite Solar Cells, Adv. Funct. Mater., 2017, 27, 1701818 CrossRef.
- Z. Yin, C. Yu, Z. Zhao, X. Guo, M. Shen, N. Li, M. Muzzio, J. Li, H. Liu, H. Lin, J. Yin, G. Lu, D. Su and S. Sun, Cu3N Nanocubes for Selective Electrochemical Reduction of CO2 to Ethylene, Nano Lett., 2019, 19, 8658–8663 CrossRef CAS PubMed.
- J. Xu, J. Cui, C. Guo, Z. Zhao, R. Jiang, S. Xu, Z. Zhuang, Y. Huang, L. Wang and Y. Li, Ultrasmall Cu7S4@MoS2 Hetero-Nanoframes with Abundant Active Edge Sites for Ultrahigh-Performance Hydrogen Evolution, Angew. Chem., Int. Ed., 2016, 55, 6502–6505 CrossRef CAS PubMed.
- Y. Zhao, H. Wang, W. Zhao, X. Zhao, J.-J. Xu and H.-Y. Chen, Dark-Field Imaging of Cation Exchange Synthesis of Cu2−xS@Au2S@Au Nanoplates toward the Plasmonic Enhanced Hydrogen Evolution Reaction, ACS Appl. Mater. Interfaces, 2021, 13, 6515–6521 CrossRef CAS PubMed.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, Investigating the Role of Copper Oxide in Electrochemical CO2 Reduction in Real Time, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS PubMed.
- D. Kim, S. Yu, F. Zheng, I. Roh, Y. Li, S. Louisia, Z. Qi, G. A. Somorjai, H. Frei, L.-W. Wang and P. Yang, Selective CO2 Electrocatalysis at the Pseudocapacitive Nanoparticle/Ordered-Ligand Interlayer, Nat. Energy, 2020, 5, 1032–1042 CrossRef CAS.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Copper Nanoparticle Ensembles for Selective Electroreduction of CO2 to C2–C3 Products, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS.
- Y. Li, D. Kim, S. Louisia, C. Xie, Q. Kong, S. Yu, T. Lin, S. Aloni, S. C. Fakra and P. Yang, Electrochemically Scrambled Nanocrystals Are Catalytically Active for CO2-to-Multicarbons, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9194–9201 CrossRef CAS PubMed.
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, Efficient Methane Electrosynthesis Enabled by Tuning Local CO2 Availability, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heim, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, Electroreduction of Carbon Dioxide to Hydrocarbons Using Bimetallic Cu-Pd Catalysts with Different Mixing Patterns, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, Optimizing C-C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- M. M. Flores Espinosa, T. Cheng, M. Xu, L. Abatemarco, C. Choi, X. Pan, W. A. Goddard, Z. Zhao and Y. Huang, Compressed Intermetallic PdCu for Enhanced Electrocatalysis, ACS Energy Lett., 2020, 3672–3680 CrossRef CAS.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard and E. J. Crumlin, Subsurface Oxide Plays a Critical Role in CO2 Activation by Cu(111) Surfaces to Form Chemisorbed CO2, the First Step in Reduction of CO2, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CAS.
- M. B. McNeil and B. J. Little, Corrosion Mechanisms for Copper and Silver Objects in Near-Surface Environments, J. Am. Inst. Conserv., 1992, 31, 355–366 CrossRef.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- A. S. Varela, C. Schlaup, Z. P. Jovanov, P. Malacrida, S. Horch, I. E. L. Stephens and I. Chorkendorff, CO2 Electroreduction on Well-Defined Bimetallic Surfaces: Cu Overlayers on Pt(111) and Pt(211), J. Phys. Chem. C, 2013, 117, 20500–20508 CrossRef CAS.
- G. Collins, F. Davitt, C. O'Dwyer and J. D. Holmes, Comparing Thermal and Chemical Removal of Nanoparticle Stabilizing Ligands: Effect on Catalytic Activity and Stability, ACS Appl. Nano Mater., 2018, 1, 7129–7138 CrossRef CAS.
- M. Gao, Y. Zhu, Y. Liu, K. Wu, H. Lu, S. Tang, C. Liu, H. Yue, B. Liang and J. Yan, The Role of Adsorbed Oleylamine on Gold Catalysts during Synthesis for Highly Selective Electrocatalytic Reduction of CO2 to CO, Chem. Commun., 2020, 56, 7021–7024 RSC.
- Y. Ye, J. Qian, H. Yang, H. Su, K. J. Lee, A. Etxebarria, T. Cheng, H. Xiao, J. Yano, W. A. Goddard and E. J. Crumlin, Synergy between a Silver-Copper Surface Alloy Composition and Carbon Dioxide Adsorption and Activation, ACS Appl. Mater. Interfaces, 2020, 12, 25374–25382 CrossRef CAS.
- S. Kim, H. Mizuno, M. Saruyama, M. Sakamoto, M. Haruta, H. Kurata, T. Yamada, K. Domen and T. Teranishi, Phase Segregated Cu2−xSe/Ni3Se4 Bimetallic Selenide Nanocrystals Formed through the Cation Exchange Reaction for Active Water Oxidation Precatalysts, Chem. Sci., 2020, 11, 1523–1530 RSC.
- Y. Fang, D. Luan, Y. Chen, S. Gao and X. W. (David) Lou, Synthesis of Copper-Substituted CoS2@CuxS Double-Shelled Nanoboxes by Sequential Ion Exchange for Efficient Sodium Storage, Angew. Chem., 2020, 132, 2666–2670 CrossRef.
- A. T. Nguyen, F. Gao, D. Baucom and C. D. Heyes, CuInS2-Doped ZnS Quantum Dots Obtained via Non-Injection Cation Exchange Show Reduced but Heterogeneous Blinking and Provide Insights into Their Structure-Optical Property Relationships, J. Phys. Chem. C, 2020, 124, 10744–10754 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic strategies; electrochemical measurement details; supplemental catalyst characterization and CO2 reduction performance. See DOI: 10.1039/d1ra03811g |
| This journal is © The Royal Society of Chemistry 2021 |