
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free thermal organocatalytic pinacol coupling of arylaldehydes using an isonicotinate catalyst with bis(pinacolato)diboron†
Masamichi Yasui,
Kengo Hanaya,
Takeshi Sugai and
Shuhei Higashibayashi*
and
Shuhei Higashibayashi*
Faculty of Pharmacy, Keio University, 1-5-30 Shibakoen, Minato-ku, Tokyo 105-8512, Japan. E-mail: higashibayashi-sh@pha.keio.ac.jp
First published on 14th July 2021
Abstract
The metal-free thermal organocatalytic pinacol coupling of arylaldehydes has been developed. The intermolecular coupling of arylaldehydes catalyzed by t-butyl isonicotinate with bis(pinacolato)diboron as the co-reducing agent afforded 1,2-diphenylethane-1,2-diols. This reaction was also applicable to the intramolecular coupling of 1,1′-biphenyl-2,2′-dicarbaldehydes to afford 9,10-dihydrophenanthrene-9,10-diols. Various functional groups were tolerated under this coupling condition.
Pinacol coupling, a reductive coupling reaction between carbonyl groups, is one of the most useful carbon–carbon bond-forming reactions. Many kinds of coupling methods using stoichiometric amounts of metals or metal catalysts with co-reducing metals have been developed to date and applied to the syntheses of various biologically active compounds.1 A recent advance in pinacol coupling is the development of photoredox catalysts in combination with organic co-reducing agents. Coupling reactions by photoredox Ir or Ru catalysts with tertiary amines as co-reducing agents under visible light were reported by Rueping et al. and Yang et al., respectively.2 While these examples required precious metal catalysts, metal-free pinacol coupling using organic photoredox catalysts has also been achieved.3 Yanagida et al. and Sudo et al. developed the coupling reaction using poly(p-phenylene) or perylene as an organic photoredox catalyst with amines as co-reducing agents under visible light. Yao et al. reported the reaction using Eosin Y Na2 as a catalyst with a 1,4-dihydropyridine derivative as a co-reducing agent under visible light. In these examples, photoexcitation of the catalysts is an indispensable process to generate the strong one-electron reducing agents, since the organic co-reducing agents are weak electron donors. Given this background, the development of a metal-free organocatalytic pinacol coupling with an organic co-reducing agent without photoexcitation is recognized to be a challenging task. In order to realize such metal- and light-free organocatalytic pinacol coupling, we focused on the generation of a pyridine-boryl radical from 4-cyanopyridine and bis(pinacolato)diboron, reported by Li et al., and its application as a reducing agent for azo compounds, sulfoxides, and quinones.4a Li et al. also reported the addition reaction of ketyl radicals generated from aldehydes by a 4-(4-cyanophenyl)pyridine catalyst and bis(pinacolato)diboron to afford 1,1-diarylethylenes.4b Inspired by their reports, we initiated the development of pinacol coupling using a pyridine catalyst and a diboron as a co-reducing agent and achieved the pinacol coupling of arylaldehydes. Recently, Choi and Chung et al. also reported organocatalytic pinacol coupling of diaryl ketones with a pyridine catalyst and a diboron as a co-reducing agent.5 Here, we report inter- and intra-molecular organocatalytic pinacol coupling of arylaldehydes by an isonicotinate catalyst with bis(pinacolato)diboron (Scheme 1).
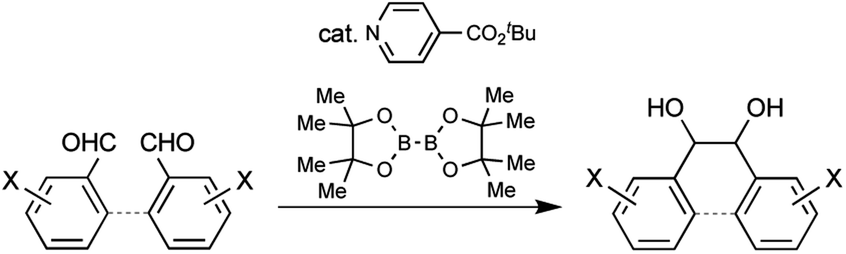 | ||
| Scheme 1 Inter- and intramolecular metal-free thermal organocatalytic pinacol coupling of arylaldehydes by a nicotinate catalyst and bis(pinacolato)diboron as the co-reducing agent. | ||
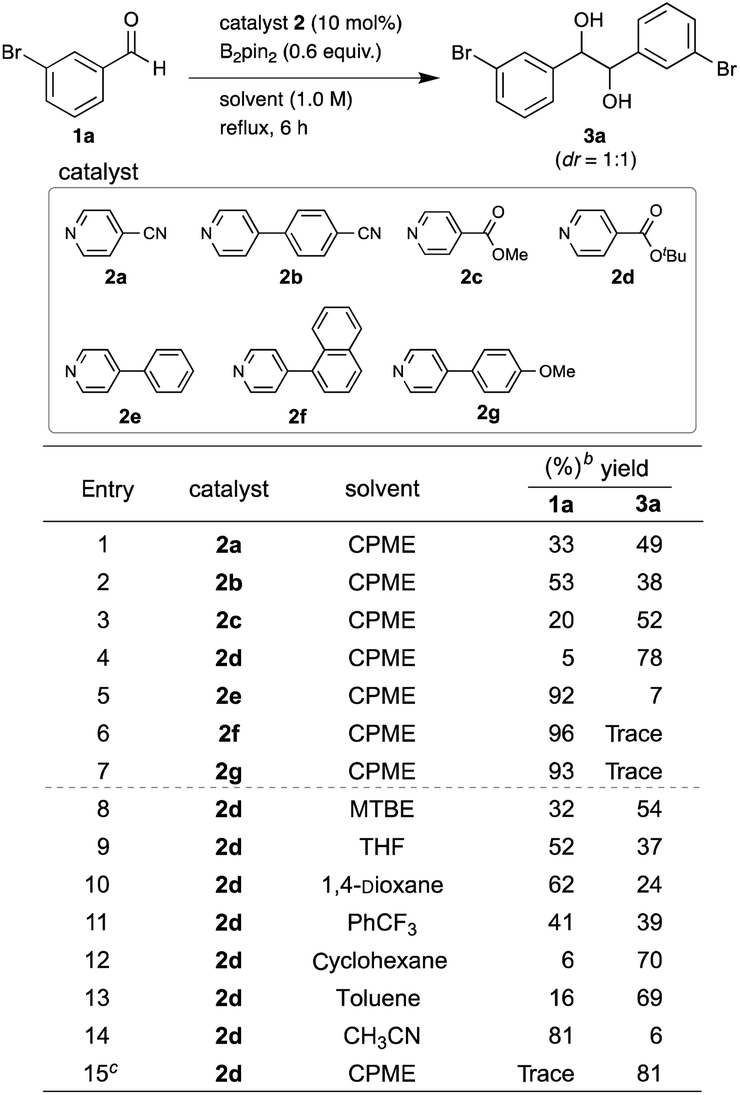
We initiated the screening of various pyridine catalysts 2 (10 mol%) with bis(pinacolato)diboron (B2pin2) (0.6 equiv.) in cyclopentyl methyl ether (CPME) for the pinacol coupling of arylaldehydes (Table 1). 3-Bromobenzaldehyde (1a) was chosen as the substrate, since we expected that the mild organic reducing agent would allow the desired coupling without dehalogenation in contrast to strong metal reducing agents. Using 4-cyanopyridine (2a)4a as a catalyst at reflux for 6 h, we succeeded in obtaining diol 3a in 49% yield with 33% recovery of the starting material 1a without dehalogenation (entry 1). Quenching with aq. KHF2 was effective for recovering the diol from the aqueous layer of the reaction mixture.5 The diastereometric ratio (dr) of the diol was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which was determined by 1H NMR spectrum. 4-(4-Cyanophenyl)pyridine (2b)4b was less efficient, giving 3a in 38% yield (entry 2). Using methyl isonicotinate (2c) reported by Choi and Chung et al.,5 the yield of 3a was increased up to 52% yield (entry 3). When t-butyl isonicotinate (2d)6a was used as a catalyst, the yield of 4a was further improved up to 78% yield (entry 4). In contrast, 4-phenylpyridine (2e),6b,c,d,e 4-(naphthalen-1-yl)pyridine (2f),6f and 4-(4-methoxyphenyl)pyridine (2g) showed lower reactivity, giving 3a in low yields (entries 5, 6 and 7). The catalyst screening results indicate that the electron-withdrawing group at the C4 position is crucial for the catalytic activity for this coupling. The higher catalytic activity of 2d than 2c (entries 4 vs. 3) could be due to the higher stability of catalyst 2d under the reaction conditions.6a Next, solvents were screened with catalyst 2d under reflux conditions (entries 4, 8–14). Among the eight screened solvents, CPME gave the best yield at 78% (entry 4). Cyclohexane and toluene also gave good yields (70% and 69% yields, respectively) (entries 12 and 13). Although 1,4-dioxane and PhCF3 were reported to be good solvents for the pinacol coupling of diaryl ketones by Choi and Chung et al.,5 they were not effective for this pinacol coupling of arylaldehydes, giving only 24 and 39% yields, respectively (entries 10 and 11). The yield of 3a was further improved up to 81% yield with 0.7 equiv. of B2pin2 (entry 15).
:1, which was determined by 1H NMR spectrum. 4-(4-Cyanophenyl)pyridine (2b)4b was less efficient, giving 3a in 38% yield (entry 2). Using methyl isonicotinate (2c) reported by Choi and Chung et al.,5 the yield of 3a was increased up to 52% yield (entry 3). When t-butyl isonicotinate (2d)6a was used as a catalyst, the yield of 4a was further improved up to 78% yield (entry 4). In contrast, 4-phenylpyridine (2e),6b,c,d,e 4-(naphthalen-1-yl)pyridine (2f),6f and 4-(4-methoxyphenyl)pyridine (2g) showed lower reactivity, giving 3a in low yields (entries 5, 6 and 7). The catalyst screening results indicate that the electron-withdrawing group at the C4 position is crucial for the catalytic activity for this coupling. The higher catalytic activity of 2d than 2c (entries 4 vs. 3) could be due to the higher stability of catalyst 2d under the reaction conditions.6a Next, solvents were screened with catalyst 2d under reflux conditions (entries 4, 8–14). Among the eight screened solvents, CPME gave the best yield at 78% (entry 4). Cyclohexane and toluene also gave good yields (70% and 69% yields, respectively) (entries 12 and 13). Although 1,4-dioxane and PhCF3 were reported to be good solvents for the pinacol coupling of diaryl ketones by Choi and Chung et al.,5 they were not effective for this pinacol coupling of arylaldehydes, giving only 24 and 39% yields, respectively (entries 10 and 11). The yield of 3a was further improved up to 81% yield with 0.7 equiv. of B2pin2 (entry 15).
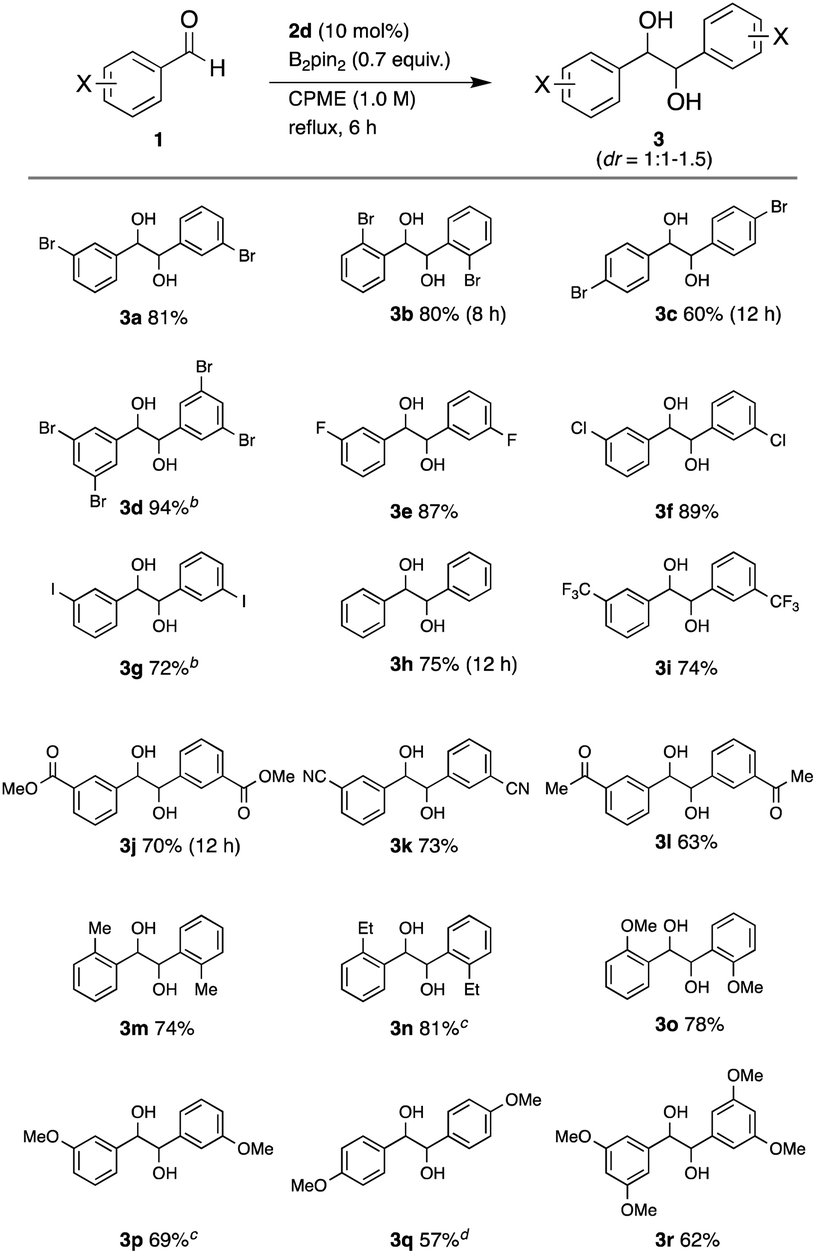
With the optimized reaction conditions in hand, we investigated the reaction scope on a series of substituted benzaldehydes (Table 2). The coupling of 2-bromobenzaldehyde (1b), 4-bromobenzaldehyde (1c), and 3,5-dibromobenzaldehyde (1d) gave the corresponding diols 3b, 3c, and 3d in 80%, 60%, and 94% yields, respectively. While the reaction of 1b required a slightly longer time (8 h) than that of 1a, the bromine atom at the ortho position did not inhibit the coupling, affording the high yield. Aldehyde 1c showed lower reactivity than 1a and 1b. The reaction was applicable to arylaldehydes with fluorine, chlorine, and iodine atoms at the 3-position, furnishing the coupling products 3e, 3f, and 3g in 87%, 89%, and 72% yields, respectively. The coupling product of simple benzaldehyde 1h was formed in 75% yield in 12 h. The reaction was applied to arylaldehydes with other electron-withdrawing groups. The reaction of 1i with a 3-trifluoromethyl group, 1j with a 3-methoxylcarbonyl group, 1k with a 3-cyano group, and 1l with an acetyl group afforded diols 3i, 3j, 3k, and 3l in 74%, 70%, 73%, and 63% yields, respectively. The ester, nitrile, and ketone functional groups remained intact under this condition, showing the high chemoselectivity of this coupling reaction. Applicability to electron-rich aldehydes was also investigated. The coupling of 2-methylbenzaldehyde (1m) gave the product 3min 74% yield. Encouraged by the tolerance of the reaction to steric hindrance, the reaction was applied to 2-ethylbenzaldehyde (1n). Although the reaction was slower (24 h), the desired product 3nwas obtained in 81% yield. Arylaldehydes 1o, 1p, 1q, and 1rwith strong electron-donating methoxy groups at the 2-, 3-, 4- and 3,5-positions also gave the coupling products 3o, 3p, 3q, and 3rin 78%, 69%, 57%, and 62% yields, respectively. The diastereomeric ratios were 1:1 to 1.5, which were determined by 1H NMR spectra. These results revealed that this coupling method has wide applicability regarding the functional groups on the aryl group. Particularly, it is noteworthy that the coupling of the ortho-substituted benzaldehydes also gave the desired products in high yields.
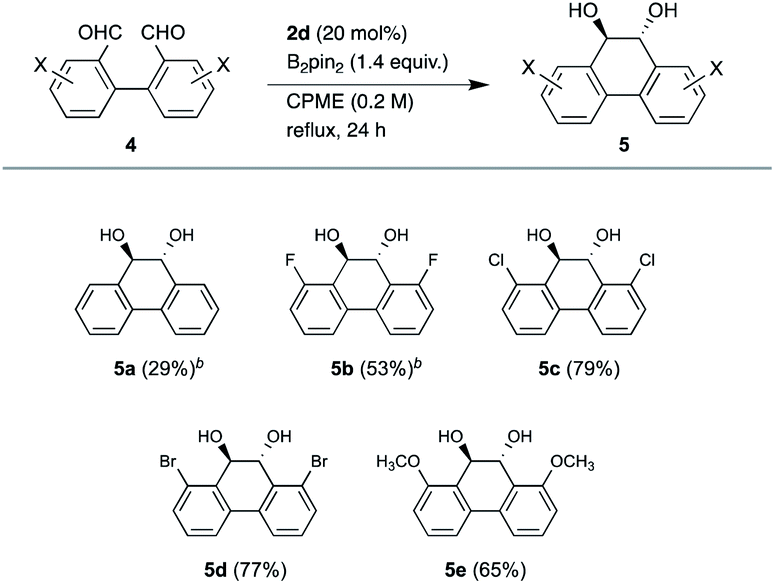
We further explored an intramolecular coupling of 1,1′-biphenyl-2,2′-dicarbaldehyde (4a), ortho-substituted 3,3′-dihalogenated derivatives 4b–4d and 3,3′-dimethoxy derivative 4e, which could be useful for syntheses of ortho-substituted phenanthrene derivatives (Table 3). The intramolecular reaction was conducted in 0.2 M for 24 h owing to the low solubility of 4 in CPME and the lower reactivity than that of the intermolecular reaction. While the coupling product 5a was formed in 29% yield, the coupling of halogenated derivatives gave better yields, 53% (5b), 79% (5c), and 77% (5d), respectively. Methoxy derivative 5e was also obtained in 65% yield. It seems that the intermolecular coupling caused the low yield of 5a owing to the less steric hindrance. Only trans isomers were formed for 5c-d and a small amount of cis isomers were generated for 5a and 5b (trans:cis = 10:1), which were determined by 1H NMR spectra. Again, this method was found to be suitable for the coupling of ortho-substituted derivatives.
According to the related previous reports,4,5 a pyridine-boryl radical formed from the pyridine catalyst 2d and B2pin2 could generate a ketyl radical of arylaldehydes 1 or 4. The radical reaction process was confirmed by the reaction of 1a with 2d and B2pin2 in the presence of TEMPO (1.0 equiv.), which recovered 1a in 97% yield. Two plausible reaction mechanisms are conceivable for the carbon–carbon bond formation after generation of the ketyl radical, which is the coupling of two ketyl radicals or the addition of keyl racial to an aldehyde followed by one-electron reduction with a pyridine-boryl radical.
The diols 3 and 5 can be derivatized to benzil and phenanthrene-9,10-diones. Thus, 1,8-dibromo-9,10-dihydrophenanthrene-9,10-diol 5d was converted to phenanthrene-9,10-dione 6d, which was further derivatized to 9,10-dimethoxyphenanthrene 7d (ref. 7) and phenanthrol[9,10-d]imidazole 8d (ref. 8) (Scheme 2). These 1,8-substituted phenanthrene derivatives are expected to serve as new building blocks for drug candidates having phenanthrene structures.9 Phenanthrene derivatives are also useful in many organic materials such as liquid crystals, fluorophores, redox materials, and nanostructures.10 Since materials with 1,8-substituted phenanthrene derivatives have not been developed yet, in contrast to widely utilized 2,7- or 3,6-substituted derivatives, these 1,8-substituted derivatives will be useful for the development of new organic materials.
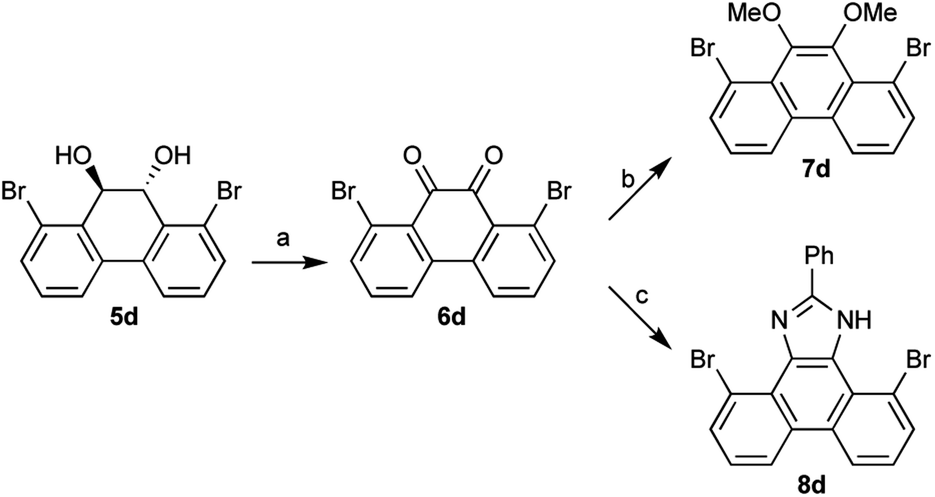 | ||
| Scheme 2 Conversion to 1,8-dibromophenanthrene derivatives. Reaction conditions: (a) PCC, SiO2, 1,2-dichloroethane, reflux, 12 h, 53%; (b) dimethyl sulfate, TBAB, sodium dithionite, THF/H2O, rt, 15 min, 60%; (c) benzaldehyde, NH4OAc, sulfamic acid, EtOH, 12 h, reflux, 77%. | ||
In summary, we established the metal-free thermal pinacol coupling of arylaldehydes by t-butyl isonicotinate as the catalyst and bis(pinacolato)diboron as the co-reducing agent. The reaction was effective for the coupling of arylaldehydes with various functional groups, particularly ortho-substituted arylaldehydes. The obtained 1,2-diarylethane-1,2-diols and dihydrophenanthrene-9,10-diols are expected to be useful building blocks to develop new drug candidates as well as organic materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP20K05499 (S. H.).Notes and references
- K. Suzuki and M. Tamiya, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, 2014, ch. 3.11, pp. 580–620 Search PubMed.
- (a) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 CrossRef CAS PubMed; (b) C.-M. Wang, P.-J. Xia, J.-A. Xiao, J. Li, H.-Y. Xiang, X.-Q. Chen and H. Yang, J. Org. Chem., 2017, 82, 3895 CrossRef CAS PubMed.
- (a) T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068 CrossRef CAS; (b) M. Zhang, W. D. Rouch and R. D. McCulla, Eur. J. Org. Chem., 2012, 6187 CrossRef CAS; (c) S. Okamoto, K. Kojiyama, H. Tsujioka and A. Sudo, Chem. Commun., 2016, 52, 11339 RSC; (d) J. Zhu, Y. Yuan, S. Wang and Z.-J. Yao, ACS Omega, 2017, 2, 4665 CrossRef CAS PubMed.
- (a) G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985 CrossRef CAS; (b) J. Cao, G. Wang, L. Gao, X. Cheng and S. Li, Chem. Sci., 2018, 9, 3664 RSC.
- J. Jo, S. Kim, J.-H. Choi and W.-J. Chung, Chem. Commun., 2021, 57, 1360 RSC.
- (a) W.-M. Cheng, R. Shang, B. Zhao, W.-L. Xing and Y. Fu, Org. Lett., 2017, 19, 4291 CrossRef CAS PubMed; (b) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed; (c) L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711 RSC; (d) L. Zhang, Z.-Q. Wu and L. Jiao, Angew. Chem., Int. Ed., 2020, 59, 2095 CrossRef CAS; (e) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124 CrossRef PubMed; (f) Y. Maekawa, Z. T. Ariki, M. Nambo and C. M. Crudden, Org. Biomol. Chem., 2019, 17, 7300 RSC.
- (a) M. Speck, D. Niethammer and M. O. Senge, J. Chem. Soc., Perkin Trans. 2, 2002, 455 RSC; (b) K. Paruch, L. Vyklicky and T. J. Katz, Org. Synth., 2003, 80, 227 CrossRef CAS; (c) H. Miyamura, F. Tobista, A. Suzuki and S. Kobayashi, Angew. Chem., Int. Ed., 2019, 58, 9220 CrossRef CAS.
- For selected examples, see: (a) N. Kishikawa, M. Wada, Y. Ohba, K. Nakashima and N. Kuroda, J. Chromatogr. A, 2004, 83, 1057 Search PubMed; (b) S. Damavandi, Heterocycl. Commun., 2011, 17, 79 CAS; (c) H.-N. Peng, X.-M. Peng, D.-G. Zheng, F. Yu and M. Rao, Heterocycl. Commun., 2011, 17, 223 CAS; (d) M. S. Jourshari, M. Mamaghani, F. Shirini, K. Tabatabaeian, M. Rassa and H. Langari, Chin. Chem. Lett., 2013, 24, 993 CrossRef; (e) H. Sharghi, M. Aberi and M. M. Doroodmand, Mol. Diversity, 2015, 19, 77 CrossRef CAS PubMed.
- (a) D. B. Hall, R. E. Homlin and J. K. Barton, Nature, 1996, 382, 731 CrossRef CAS; (b) M. Chu, R. Merzwa, I. Truumees, A. King, m. Patel, R. Berrie, A. Hart, N. Butkiewicz, B. Dasmahapatra, T.-M. Chan and M. S. Puar, Tetrahedron Lett., 1996, 37, 7229 CrossRef CAS; (c) M. Lepourcelet, Y.-N. P. Chen, D. S. France, H. Wang, P. Crews, F. Petersen, C. Bruseo, A. W. Wood and R. A. Shivdasani, Cancer Cell, 2004, 5, 91 CrossRef CAS; (d) S. Padhye, Z. Afrasiabi, E. Sinn, J. Fok, K. Mehta and N. Rath, Inorg. Chem., 2005, 44, 1154 CrossRef CAS PubMed; (e) B. Côté, L. Boulet, C. Brideau, D. Claveau, D. Ethier, R. Frenette, M. Gagnon, A. Giroux, J. Guay, S. Guiral, J. Mancini, E. Martins, F. Massé, N. Méthot, D. Riendeau, J. Rubin, D. Xu, H. Yu, Y. Ducharme and R. W. Friesen, Bioorg. Med. Chem. Lett., 2007, 17, 6816 CrossRef PubMed; (f) M. Iwatsuki, K. Otoguro, A. Ishiyama, M. Namatame, A. Nishihara-Tukashima, J. Hashida, T. Nakashima, R. Masuma, Y. Takahashi, H. Yamada and S. Ōmura, J. Antibiot., 2010, 63, 619 CrossRef CAS; (g) P. Cheng, K. Xu, Y. C. Chen, T. T. Wang, Y. Chen, C. L. Yang, S. Y. Ma, Y. Liang, H. M. Ge and R. H. Jiao, Tetrahedron Lett., 2019, 60, 1706 CrossRef CAS; (h) N. Bunbamrung, C. Intaraudom, A. Dramae, C. Thawai, S. Tadtong, P. Auncharoen and P. Pittayakhajonwut, Phytochemistry, 2020, 172, 112275 CrossRef CAS.
- (a) J.-P. Duan, P.-P. Sun and C.-H. Cheng, Adv. Mater., 2003, 15, 224 CrossRef CAS; (b) M.-S. Tsai, Y.-C. Hsu, J. T. Ling, H.-C. Chen and C.-P. Hsu, J. Phys. Chem. C, 2007, 111, 18785 CrossRef CAS; (c) H.-J. Kim, E. Lee, H. Park and M. Lee, J. Am. Chem. Soc., 2007, 129, 10994 CrossRef CAS PubMed; (d) Y.-T. Chang, S.-L. Hsu, M.-H. Su and K.-H. Wei, Adv. Mater., 2009, 21, 2093 CrossRef CAS; (e) D. G. Patel, I. M. Walton, J. M. Cox, C. J. Gleason, D. R. Butzer and J. B. Benedict, Chem. Commun., 2014, 50, 2653 RSC; (f) R. Francke and R. D. Little, J. Am. Chem. Soc., 2014, 136, 427 CrossRef CAS; (g) S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068 CrossRef CAS; (h) H. Yang, Y. Fu, S. Wan, G. D. Trahan, Y. Jin and W. Zhang, Chem. Sci., 2015, 6, 4049 RSC; (i) K. Urakawa, M. Sumimoto, M. Arisawa, M. Matsuda and H. Ishikawa, Angew. Chem., Int. Ed., 2016, 55, 7432 CrossRef CAS PubMed; (j) W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750 CrossRef CAS PubMed; (k) C. Lu, M. Zhang, D. Tang, X. Yan, Z. Y. Zhang, Z. Zhou, B. Song, H. Wang, X. Li, S. Yin, H. Sepehrpour and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7674 CrossRef CAS; (l) S. Kothavale, W. J. Chung and J. Y. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 18730 CrossRef CAS; (m) L. Chen, C. Chen, Y. Sun, S. Lu, H. Huo, T. Tan, A. Li, X. Li, G. Ungar, F. Liu and M. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10143 CrossRef CAS PubMed; (n) Z. Luo, Z. Yang, K. Cai, X. Fu, D. Zhang, Y. Ma and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 14854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04443e |
| This journal is © The Royal Society of Chemistry 2021 |