
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIndoleninyl-substituted pyrimido[1,2-b]indazoles via a facile condensation reaction†
Abdul Qaiyum Ramle*a,
Chee Chin Feib,
Edward R. T. Tiekink c and
Wan Jefrey Basirun*a
c and
Wan Jefrey Basirun*a
aDepartment of Chemistry, University of Malaya, Kuala Lumpur, 50603, Malaysia. E-mail: qaiyum@um.edu.my; jeff@um.edu.my
bNanotechnology and Catalysis Research Centre, University of Malaya, Kuala Lumpur, 50603, Malaysia
cResearch Centre for Crystalline Materials, School of Science and Technology, Sunway University, Bandar Sunway, Selangor Darul Ehsan 47500, Malaysia
First published on 14th July 2021
Abstract
A new series of pyrimido[1,2-b]indazoles bearing indolenine moieties was synthesized through a simple condensation reaction with up to 94% yield. The present method features the versatile formation of a pyrimidine ring with a broad range of substrates, great functional group compatibility and facile synthetic operation. The work offers opportunities in drug development as well as in materials science.
Introduction
Pyrimido[1,2-b]indazoles are a class of fused nitrogen-containing tricyclic skeletons that have received significant attention as pharmacologically important molecules, e.g. as anti-cancer agents,1 monoamine oxidase (MAO) inhibitors,2 PDE10A inhibitors,3 and for the treatment of hepatitis C virus (HCV) infection.4 Despite their high bioactive potential, the preparation of pyrimido[1,2-b]indazoles is rarely documented due to the limited synthetic options and strategies. Hence, the development of new drugs with diverse substituted pyrimido[1,2-b]indazoles with feasible methods for their synthesis remains challenging.A literature survey revealed that 3-amino-1H-indazole is an essential component for the preparation of pyrimido[1,2-b]indazole scaffolds. From a synthetic point of view, the condensation reaction of 3-amino-1H-indazole with various types of carbonyl compounds is the most frequent approach to access the structural motif.5 The addition of metal catalysts such as CuSO4·5H2O, Al(OTf)3, and Cu(OAc)2 also enhances the transformation of such products.6 However, this method is only feasible with specific functional groups, hence there is a limitation in the synthesis of structurally diverse derivatives.
Since several years ago, some efforts have resulted in the facile synthesis of pyrimido[1,2-b]indazoles. In 2017, Li et al. performed a one-pot, three-component reaction utilising mixtures of aromatic aldehydes, 3-amino-1H-indazoles and 3-oxopropanenitriles (Scheme 1a).7 Later, a cost-effective method by Balwe et al. resulted in the preparation of four compounds of (2-hydroxyphenyl)(pyrimido[1,2-b]indazol-3-yl)methanones at room temperature with excellent yields (Scheme 1b).4 Very recently, Jismy et al. investigated the treatment of 3-amino-1H-indazoles with 2-bromomalonaldehyde in ethanol in the presence of catalytic acetic acid which afforded the products in high yields (Scheme 1c).2
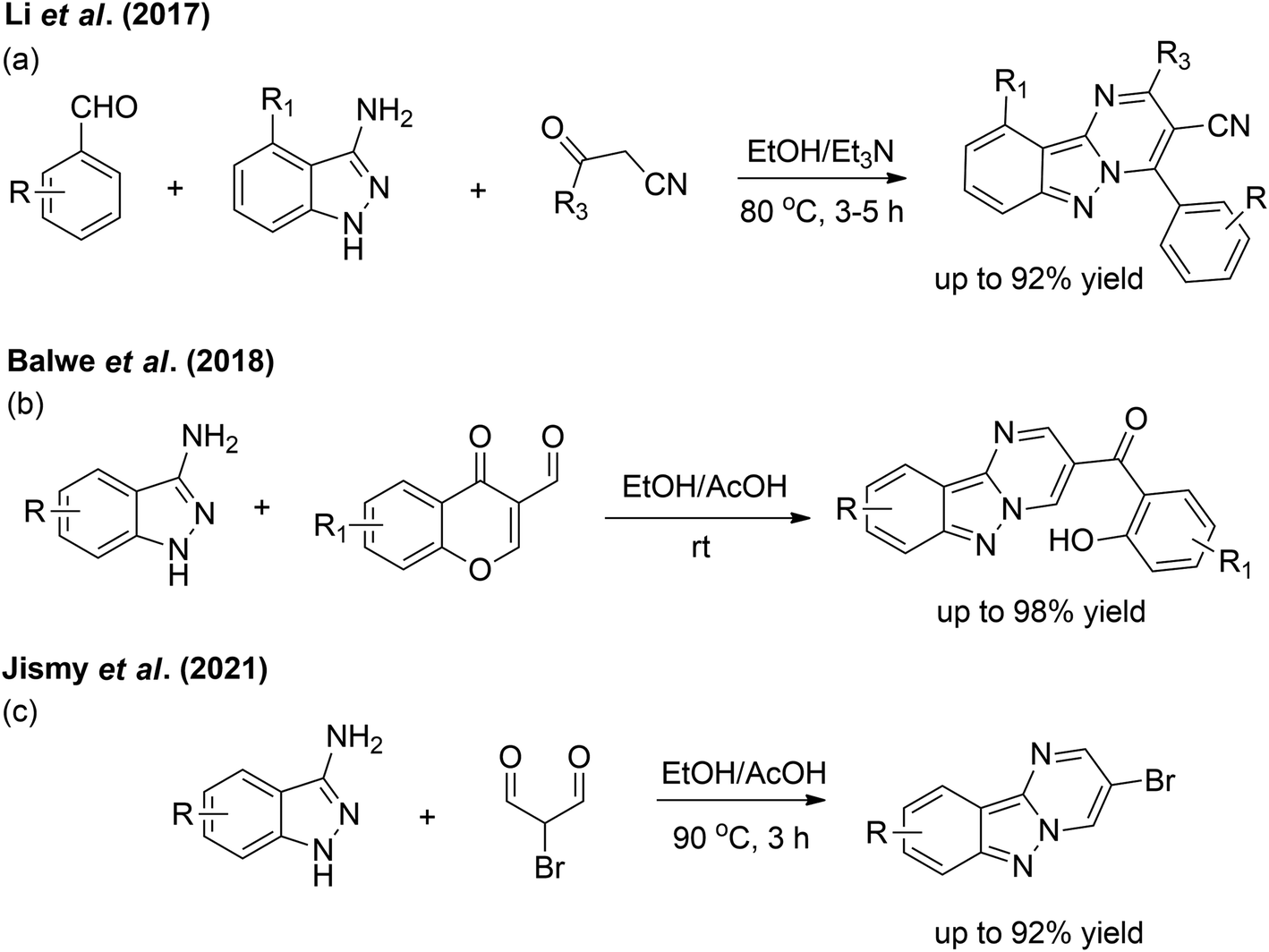 | ||
| Scheme 1 Efficient methods for the synthesis of substituted pyrimido[1,2-b]indazoles. | ||
With growing interest in this area, the present work reports for the first time a new series of pyrimido[1,2-b]indazoles featuring indolenine scaffolds. The compounds were prepared via a simple condensation reaction between the substituted diformyl indolenines and 3-amino-1H-indazoles catalysed by acetic acid. The results of this investigation are described herein.
Results and discussions
The starting materials, 1a–e were prepared according to previous methods.8 To begin the investigation, compounds 1a and 2a were selected as the model substrates. As tabulated in Table 1, preliminary results showed that the condensation reaction between 1a and 2a delivered the target product in 44% yield (entry 1). Treatment with a small amount of acetic acid and prolonged stirring at room temperature resulted in an 18% yield (entry 2). Further increase of the reaction temperature to 78 °C significantly improved the reaction efficiency to 61% yield (entry 3). On the other hand, the addition of a small amount of 37% hydrochloric acid catalyst to the reaction media decreased the product yield to only 12% (entry 4). However, the use of H2SO4 catalyst failed to yield the target product 3a (entry 5). Remarkably, increasing the amount of acetic acid catalyst afforded 3a in 68% yield (entry 6). It was observed that replacing ethanol with acetonitrile, dioxane or methanol solvents decreased the reaction efficiency and gave poor yields (entries 7–9).| Entry | Solvent/acid | Time (h) | Temperature (°C) | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: 1a (1 equiv.), 2a (1.1 equiv.) in 5.0 mL of solvent/acid. | ||||
| 1 | EtOH | 5 | 78 | 44 |
| 2 | EtOH/AcOH (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
72 | 25 | 18 |
| 3 | EtOH/AcOH (99:1) |
5 | 78 | 61 |
| 4 | EtOH/37% HCl (99:1) |
5 | 78 | 12 |
| 5 | EtOH/H2SO4 (99:1) |
5 | 78 | — |
| 6 | EtOH/AcOH (4:1) |
5 | 78 | 68 |
| 7 | MeCN/AcOH (4:1) |
5 | 82 | 5 |
| 8 | Dioxane/AcOH (4:1) |
5 | 101 | 16 |
| 9 | MeOH/AcOH (4:1) |
5 | 65 | 5 |
The substrate scope of 1a–e and 2a–d were investigated in optimised reaction conditions. As illustrated in Scheme 2, the reactions between 1a–e and 2a afforded the products 3a–e in reasonable yields (47–68%). Furthermore, the presence of a bromo group in substrate 2b was also compatible under this synthetic protocol, affording the desired products 3f–j in good yields (67–78%). It is interesting to note that the presence of a methoxy group in substrate 2c was also effective under typical conditions to yield the desired products 3k–o in moderate to high yields (61–94%). Substrate 2d, bearing a strong electron-withdrawing trifluoromethyl substituent, also reacted well with 1a–e to furnish the corresponding products 3p–t in good yields (50–79%). These results demonstrated that these facile syntheses were tolerable to various substituents regardless of their electronic nature, whether on the indolenine and indazole rings.
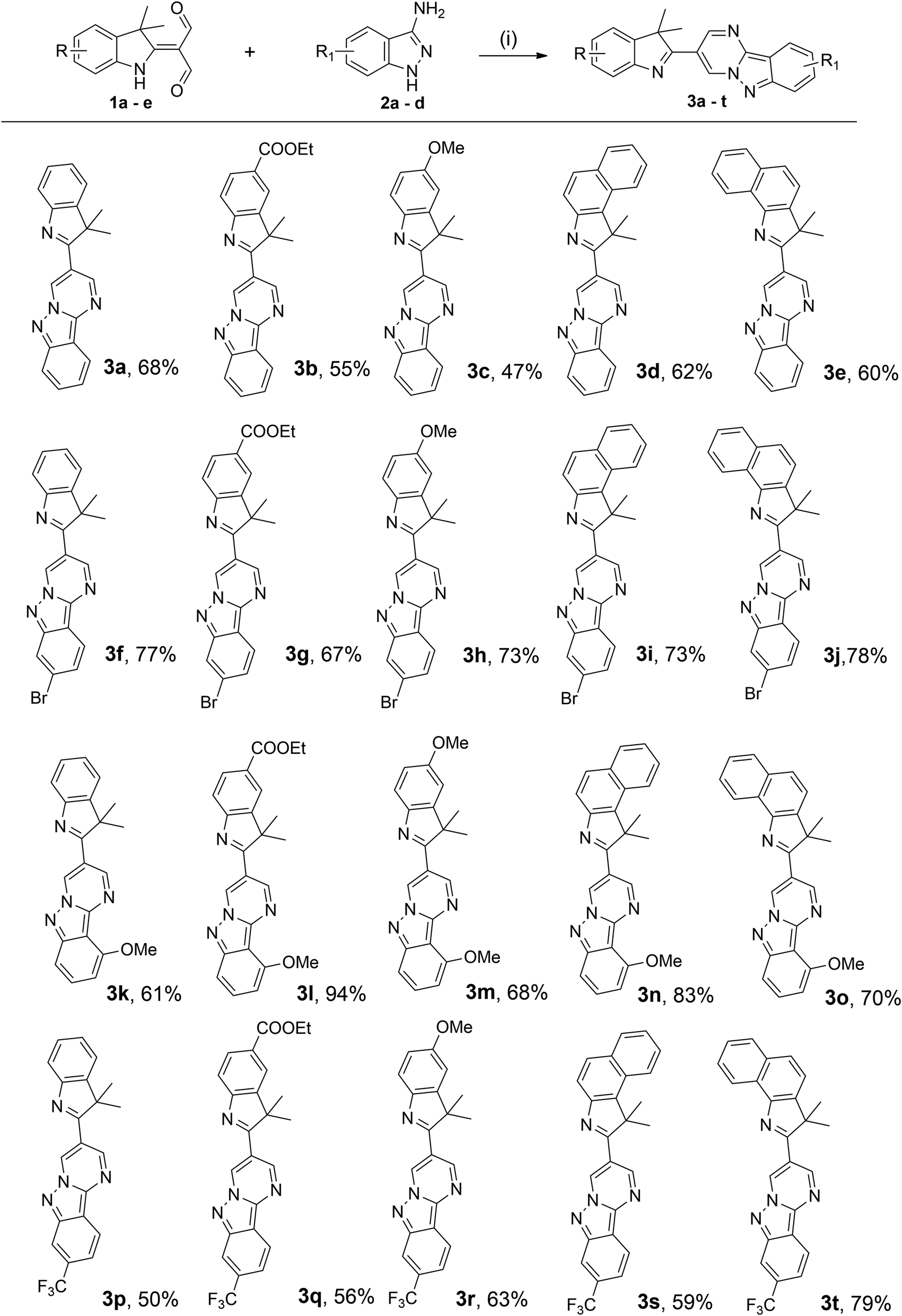 | ||
| Scheme 2 Substrate scope for the synthesis of 3. Reaction conditions: (i) 1a–e (1.0 equiv.) and 2a–d (1.1 equiv.) in ethanol/acetic acid (v/v = 4:1) at 78 °C for 5 h. Isolated yield. | ||
The 1H NMR spectra reveal two sets of doublets (J = 1.2–2.4 Hz) between δ 9.50–9.94 ppm which correspond to the two olefinic protons of the pyrimidine ring. The six methyl protons appear as singlets in the range of δ 1.64–1.96 ppm. The HRMS of the compounds show that the pseudo-molecular ions are in good agreement with the theoretical values.
The representative molecular structure, namely 3a, was elucidated by X-ray crystallography. The structure crystallises in the monoclinic space group P21/m with half a molecule comprising the crystallographic asymmetric unit. With the exception of the methyl substituents, the molecule lies on a crystallographic mirror plane, as illustrated in Fig. 1. The X-ray analysis indicates that the heteroatoms of the five-membered rings are syn. There is substantial delocalisation of the π-electron density in the pyrimido[1,2-b]indazole ring. Thus, there is evidence for the lengthening of the formally double bond lengths of C19–N4 [1.373(3) Å], C12–C14 [1.428(4) Å], C15–C16 [1.365(5) Å] and C17–C18 [1.356(4) Å]. Furthermore, the C11–N2 [1.319(4) Å] and C12–N2 [1.312(4) Å] bonds are experimentally equivalent to the pair of C13–N3 [1.364(4) Å] and C10–C13 [1.359(4) Å] bonds; the N3–N4 bond length is 1.341(3) Å. The details of the molecular packing are given in the ESI (Fig. S2 and S3†).
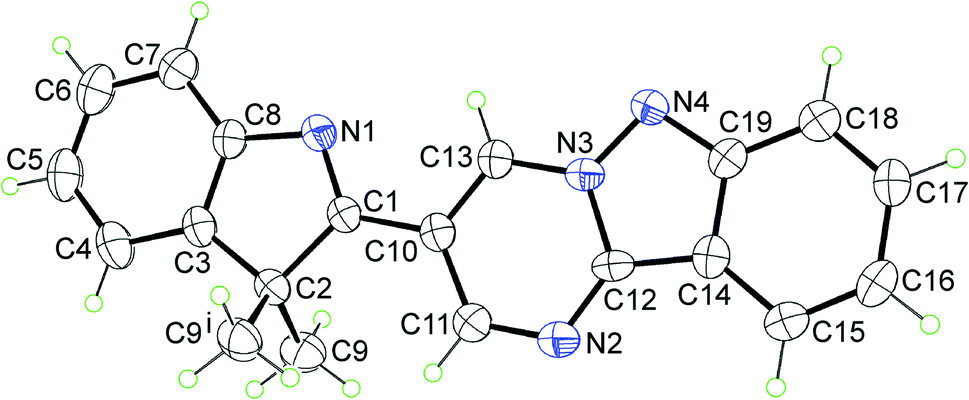 | ||
| Fig. 1 Molecular structure of 3a showing atom labelling scheme and displacement ellipsoids at the 35% probability level. The molecule, save the methyl groups, lies on a mirror plane; symmetry operation i: x, 1½ − y, z. | ||
A plausible mechanism for the reaction is proposed in Scheme 3. The first step involves the formation of intermediate I through the mono-condensation reaction between derivatives 1 and 2. The protonation at the aldehyde-oxygen atom produces intermediate II. Next, the donation of a lone-pair of electrons from the pyrazole–nitrogen atom to the carbocation centre generates a fused pyrimidine ring of intermediate III. The deprotonation of the pyrazole ring gives rise to intermediate IV. The protonation of the hydroxyl group produces intermediate V. The removal of a water molecule gives intermediate VI. Finally, the deprotonation of the indole generates the corresponding product 3.
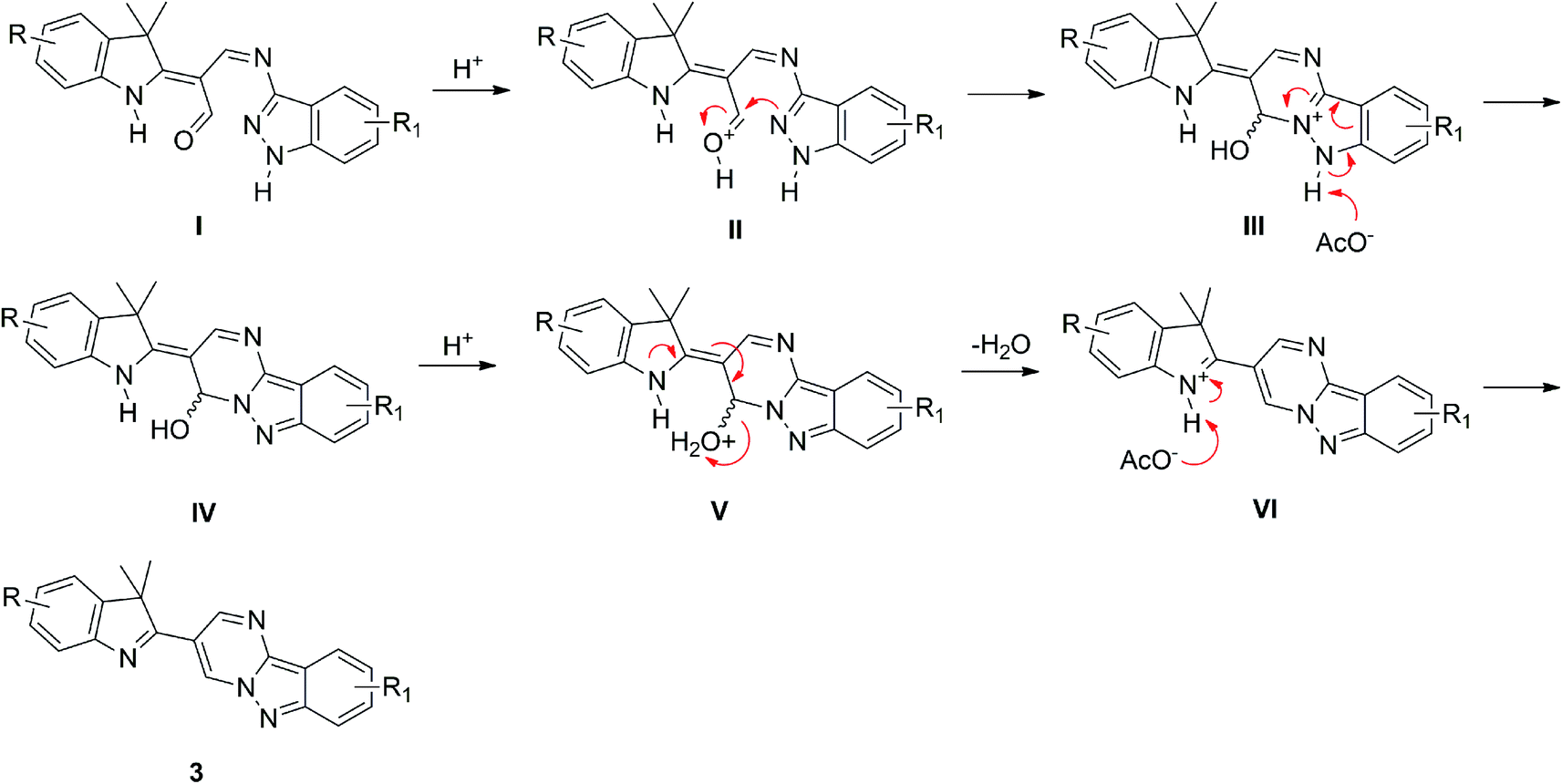 | ||
| Scheme 3 Plausible mechanism for the formation of 3. | ||
The UV-Vis absorption spectra of 3a in chloroform and 3d in other solvents are shown in Fig. 2. Compound 3a shows an intense absorption band at 328 nm, which is assigned to the π–π* transition. The optical band gap of the compound is 3.24 eV, which is determined from the absorption edge of the spectrum.9 In comparison, the spectrum 3d in chloroform displays a bathochromic shift to 350 nm due to the extension of the π-conjugation system. This results in the reduction of the optical band gap of 3d by 0.36 eV. Furthermore, the absorption spectra of 3d in other solvents such as acetic acid, dimethylformamide, methanol and tetrahydrofuran media are not significantly solvent-dependent, implying that the effect of solvent polarity is indistinguishable in the ground state. Details of the spectroscopic parameters for both compounds are summarized in ESI (Table S2†).
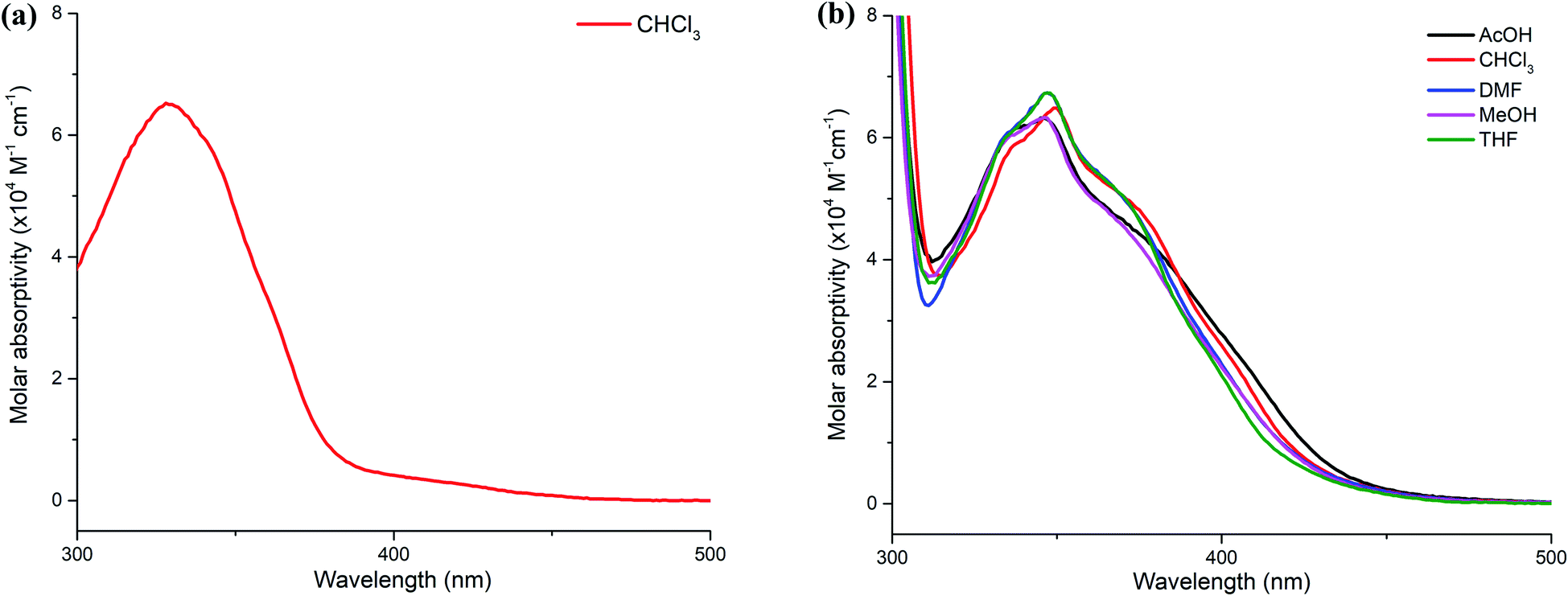 | ||
| Fig. 2 UV-Vis absorption spectra of (a) 3a in chloroform and (b) 3d in various solvents at a concentration of 14 μM. | ||
Conclusions
In summary, a new series of indoleninyl-substituted pyrimido[1,2-b]indazoles was successfully synthesised in good to high yields. The main advantages of this synthetic method are the simple operation, free from chromatographic purification and the ability to diversify the substituents on both the indolenine and indazole rings for the formation of the pyrimido[1,2-b]indazole scaffolds. The nitrogen-rich products are potent candidates for drug screening and biological studies. Further efforts to expand the synthetic scope of fused nitrogen tricyclic heterocycles are currently under investigation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
All authors wish to thank the University of Malaya (ST027-2019) and Sunway University Sdn. Bhd. (GRTIN-IRG-01-2021) for their financial support in this work.References
- T. Yakaiah, B. P. V. Lingaiah, B. Narsaiah, B. Shireesha, B. Ashok Kumar, S. Gururaj, T. Parthasarathy and B. Sridhar, Bioorg. Med. Chem. Lett., 2007, 17, 3445 CrossRef CAS PubMed.
- B. Jismy, A. El Qami, A. Pišlar, R. Frlan, J. Kos, S. Gobec, D. Knez and M. Abarbri, Eur. J. Med. Chem., 2021, 209, 112911 CrossRef CAS PubMed.
- A. Chino, R. Seo, Y. Amano, I. Namatame, W. Hamaguchi, K. Honbou, T. Mihara, M. Yamazaki, M. Tomishima and N. Masuda, Chem. Pharm. Bull., 2018, 66, 286 CrossRef CAS PubMed.
- S. G. Balwe and Y. T. Jeong, Org. Biomol. Chem., 2018, 16, 1287 RSC.
- (a) S. Annareddygari, V. R. Kasireddy and J. Reddy, J. Heterocycl. Chem., 2019, 56, 3267 CrossRef CAS; (b) W. Kong, Y. Zhou and Q. Song, Adv. Synth. Catal., 2018, 360, 1943 CrossRef CAS; (c) J. Palaniraja, S. Mohana Roopan, G. Mokesh Rayalu, N. Abdullah Al-Dhabi and M. Valan Arasu, Molecules, 2016, 21, 1571 CrossRef PubMed; (d) Y. M. Volovenko and V. A. Chuiguk, Chem. Heterocycl. Compd., 1974, 10, 859 CrossRef; (e) Q. Gao, X. Han, P. Tong, Z. Zhang, H. Shen, Y. Guo and S. Bai, Org. Lett., 2019, 21, 6074 CrossRef CAS; (f) A. M. Jadhav, S. G. Balwe, K. T. Lim and Y. T. Jeong, Tetrahedron, 2017, 73, 2806 CrossRef CAS; (g) S. G. Balwe, V. V. Shinde, A. A. Rokade, S. S. Park and Y. T. Jeong, Catal. Commun., 2017, 99, 121 CrossRef CAS.
- (a) J. Palaniraja, S. M. Roopan and G. M. Rayalu, RSC Adv., 2016, 6, 24610 RSC; (b) Y. Gu, F. Wu and J. Yang, Adv. Synth. Catal., 2018, 360, 2727 CrossRef CAS; (c) Y. Zhou, Y. Lou, Y. Wang and Q. Song, Org. Chem. Front., 2019, 6, 3355 RSC.
- L. Li, H. Xu, L. Dai, J. Xi, L. Gao and L. Rong, Tetrahedron, 2017, 73, 5358 CrossRef CAS.
- (a) A. Rashidi, A. Afghan, M. M. Baradarani and J. A. Joule, J. Heterocycl. Chem., 2009, 46, 428 CrossRef CAS; (b) A. Q. Ramle, H. Khaledi, A. H. Hashim, M. A. Mingsukang, A. K. Mohd Arof, H. M. Ali and W. J. Basirun, Dyes Pigm., 2019, 164, 112 CrossRef CAS; (c) H. A. Rothan, E. Amini, F. L. Faraj, M. Golpich, T. C. Teoh, K. Gholami and R. Yusof, Sci. Rep., 2017, 7, 45540 CrossRef CAS PubMed.
- A. Q. Ramle, E. R. T. Tiekink, C. C. Fei, N. M. Julkapli and W. J. Basirun, New J. Chem., 2021, 45, 1221 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082645. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04372b |
| This journal is © The Royal Society of Chemistry 2021 |