
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding enhancements of antibody functionalized natural and synthetic fibers†
Iqra Azeemab,
Marwa El yaagoubib,
Ana M. L. Sousa b,
Tai-De Lic,
Basit Yameen*a and
King Hang Aaron Lau*b
b,
Tai-De Lic,
Basit Yameen*a and
King Hang Aaron Lau*b
aDepartment of Chemistry and Chemical Engineering, Syed Babar Ali School of Science and Engineering (SBASSE), Lahore University of Management Sciences (LUMS), Lahore 54792, Pakistan. E-mail: basit.yameen@lums.edu.pk; aaron.lau@strath.ac.uk
bDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK
cAdvanced Science Research Center (ASRC) of Graduate Center, Department of Physics in City University of New York, CUNY, New York, NY 10031, USA
First published on 10th September 2021
Abstract
Development of low cost biosensing using convenient and environmentally benign materials is important for wide adoption and ultimately improved healthcare and sustainable development. Immobilized antibodies are often incorporated as an essential biorecognition element in point-of-care biosensors but these proteins are costly. We present a strategy of combining convenient and low-cost surface functionalization approaches for increasing the overall binding activity of antibody functionalized natural and synthetic fibers. We demonstrate a simple one-step in situ silica NP growth protocol for increasing the surface area available for functionalization on cotton and polyester fabrics as well as on nanoporous cellulose substrates. Comparing this effect with the widely adopted and low cost plant-based polyphenol coating to enhance antibody immobilization, we find that both approaches can similarly increase overall surface activity, and we illustrate conditions under which the two approaches can produce an additive effect. Furthermore, we introduce co-immobilization of antibodies with a sacrificial “steric helper” protein for further enhancing surface activities. In combination, several hundred percent higher activities compared to physical adsorption can be achieved while maintaining a low amount of antibodies used, thus paving a practical path towards low cost biosensing.
Introduction
Surface modification or functionalization is the process of adding chemical and physical functionalities on material surfaces to enable a wide variety of applications, including (bio)sensing,1–3 nanomedicine,4,5 biocatalysis,6 oil/water separation,7 and energy storage.8 Surface modification should ideally be facile, versatile, and chemically mild. To enable biomolecular applications like biosensing, surfaces are often functionalized by immobilization of proteins that can further bind specifically with other biomolecules.9 In particular, antibodies are commonly used to enable detection of diseases through their binding (i.e., biorecognition) of biomarkers.10–12Fabrics and other fibrous meshes represent an attractive direction in advanced materials not only because they are low-cost and light-weight, but also because their porous structures give them a combination of mechanical flexibility, toughness, and high internal surface area for surface modification.13 Cotton, an abundant fabric composed of natural cellulose fibers, additionally offers stability in both water and organic solvents, as well as degradability.14 Its potential to support biomolecular applications is illustrated by the wide report of the use of another cellulosic porous substrate—paper—for low-cost biosensing.15,16 Such devices, ranging from traditional dipsticks to lateral flow assays and microfluidic paper devices,17–19 may have special relevance to the ASSURED criteria (Affordable, Sensitive, Specific, User-friendly, Rapid and Robust, Equipment-free, Delivered) proposed by the World Health Organization (WHO) for public health-focused point-of-care medical diagnostics.20 Polyester, as a synthetic fabric, is another widely available and low-cost material. Functionalization of both cotton and polyester also holds significant appeal to wearable devices.21,22
In the context of nanotechnology, a range of nanoparticles (NPs) have been immobilized onto fabrics, including gold,23,24 silver,25 zinc oxide,26 titania,27 and silica.28,29 These modifications are aimed at imparting either some intrinsic property (e.g., UV protection,30 and catalysis and reactive species generation)31 or a physical effect. For example, NPs protruding out on a surface may be used to generate a superhydrophobic lotus leaf effect32,33 or simply to enhance the surface area available for further (bio)chemical functionalization.34–36 These NP properties have variously enabled remarkable biosensor detection limits, e.g., in the range of 100–104 CFU mL−1 for bacteria and 10−6 to 10−10 g mL−1 for antibodies. Notwithstanding, many recent studies have been aimed at combining reasonable sensitivity with other desirable goals such as the ASSURED criteria.37–42
Protein immobilization on chemically stable supports like cellulose often represents a number of trade-offs. Physical adsorption may be facile, but the amount immobilized may be low, and desorption can be an issue.6 Chemical activation may increase surface coupling but traditionally requires aggressive reagents43 (thus, other fabrics, e.g., polyesters, are also being considered).44,45 Enhanced protein immobilization through coatings of polymer brushes or hydrogel layers involves additional chemical steps.35,46 At the same time, strategies should be developed to use low amounts of proteins for immobilization while retaining high surface binding activity, especially for high-cost proteins (e.g., antibodies for detecting viruses like HPV, hepatitis C, and SARS-COV-2, etc.).
We hereby report the strategy of increasing the overall binding activity of antibody functionalized fiber surfaces by a combination of convenient surface modification techniques. Silica NPs (SNPs) were chosen for surface area enhancement since silica is a common material and SNPs can be conveniently synthesized. In fact, we have compared modification using pre-synthesized SNPs with in situ surface-initiated SNP growth. Polyphenol coatings were used to confer surface reactive groups since they have been demonstrated for diverse applications, including protein immobilization.47–49 They do not discolor the substrate and are much lower in cost compared with polydopamine.6,50 However, our recent work also showed that their chemical properties and the activity of proteins immobilized on them could vary depending on the substrate material.6 In this report, we have therefore studied how the combination of poly(tannic acid) (PTA) and SNPs may modify the final immobilized antibody activity. Co-immobilization with a sacrificial protein was further included to increase surface crowding and potentially reduce surface induced denaturation.51 Overall, results on cellulosic substrates—cotton and nanoporous regenerated cellulose (NRC)—were compared with those on polyester (PE). Given a suitable combination of (bio)chemical functionalization and NP surface area enhancement, we demonstrate that binding activity of the functionalized fibers can be significantly increased without increasing the amount of antibody used.
Results and discussion
Sample design
Tannic acid (TA), an abundant plant polyphenol, was used to form the polyphenol coating—TA crosslinks by oxidative polymerization and interacts with a surface to form PTA.50,52 The antibody used was a mouse immunoglobulin G (IgG). Serum albumin (SA) was chosen for co-immobilization as a sacrificial “steric helper” protein (see later discussion) because of its low cost and its common use as a blocking agent in immunoassays to reduce non-specific IgG binding.53 The different combinations of polyphenol coating and SNP surface modification studied are schematically shown in Fig. 1a, and the resulting sample types (labelled I–IV), functionalized with IgG alone or with SA co-immobilization, are shown in Fig. 1b.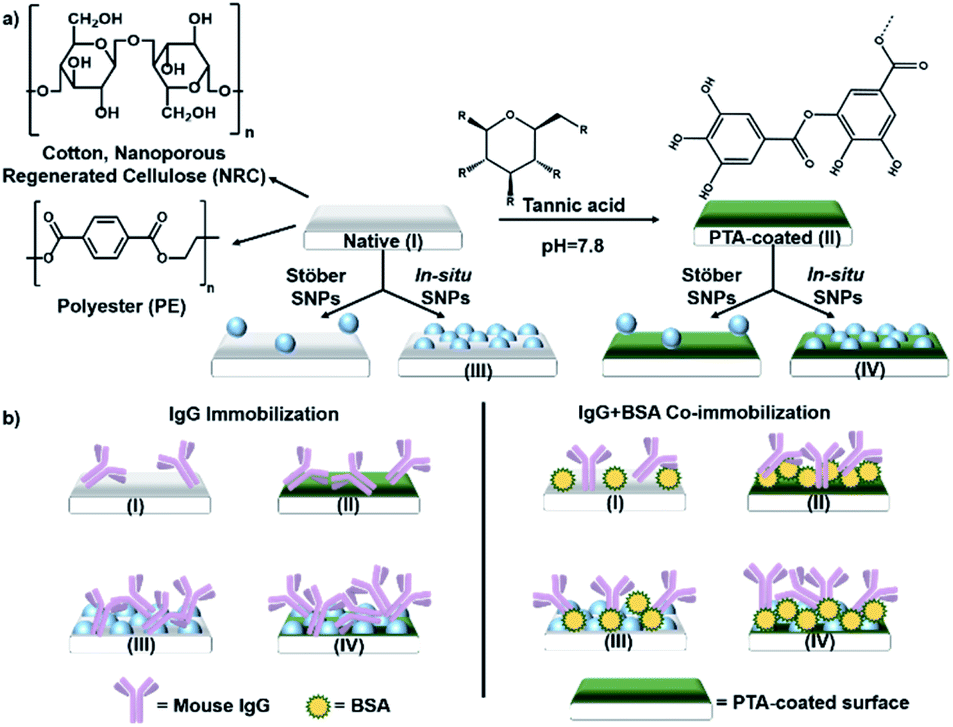 | ||
| Fig. 1 (a) Schematic of functionalization steps on cotton, polyester (PE), and nanoporous regenerated cellulose (NRC) substrates. Native surfaces are labelled as type I, and types II–IV are different modifications with and without poly(tannic acid) (PTA) coating and/or silica NP (SNP). (b) List of sample types with IgG alone or with SA co-immobilized. | ||
PTA and nanoparticle surface modification
PTA coatings were prepared by immersing 1 × 1 cm2 samples in a pH 7.8 buffered solution of tannic acid following our published protocol (see ESI† for details).6,50 A low concentration was chosen (0.03 mg mL−1) to prevent coating over adjacent fibers and obscuring the porous structure, especially in the nanoporous NRC. The presence of the polyphenol coating was confirmed by the established technique of silver staining (Fig. S1†) and corroborated by ATR-FT-IR (Fig. S2†) and XPS (Fig. S3 and S4†). The PTA thickness estimated from XPS data was 1.4 nm on cellulose and 5 nm on PE, within the range of our previous study.54 Moreover, environmental scanning electron microscopy (E-SEM) showed that the porous fiber morphologies for all samples were preserved (Fig. 2A–C show cotton, NRC, and PE, respectively; panels 1 and 2, in turn, show the morphologies before and after PTA coating). | ||
| Fig. 2 E-SEM images of native and functionalized fabrics. Panels (A1) & (A2) are native and PTA-coated cotton, whereas, (A3–A6) are PTA-coated cotton incubated with 0.03, 1, 10, and 20 mg mL−1 suspensions of StöberSNPs in that order. (A7) & (A8) are inSNPs modified native and PTA-coated cotton, respectively. (B1) & (B2) are native and PTA-coated NRC, while (B3) & (B4) are inSNPs modified native and PTA-NRC, respectively. (C1) & (C2) are native and PTA-coated PE, while (C3) & (C4) are inSNPs modified native and PTA-coated PE, respectively. Additional E-SEM images characterizing each sample are provided in Fig. S7 to S17.† | ||
Two approaches to SNP surface modification were compared using cotton samples. In the first, more conventional approach, SNPs were separately synthesized from a simple silica nanosol consisting of tetraethyloxysilane (TEOS), ammonia, ethanol, and water, following a method reported by Stöber.55 These “StöberSNPs” were collected, dried, and resuspended in water for surface attachment (see ESI† for complete protocols). E-SEM characterization showed that uniformly sized StöberSNPs ∼120 nm in diameter were synthesized (e.g., Fig. 2A3). Attachment on native cotton fibers (i.e., type I samples, Fig. 1a) was actually not expected since there was no obvious adhesion mechanism between the silica of StöberSNPs and bare cellulose surfaces, and indeed attachment was not observed. In contrast, Fig. 2A3–A6 show that attachment on PTA-coated cotton (i.e., type II “PTA” samples) was possible. In fact, increasing the suspended StöberSNP concentration from 0.03 to 20 mg mL−1 also led to increased attachment. Nonetheless, coverages were heterogeneous and relatively low.
This led to our second approach of growing in situ SNPs (inSNPs), which was conveniently achieved in one step by incubating samples directly in a modified TEOS nanosol solution (see ESI†). This apparently allowed silica nuclei in solution to attach on the fiber surfaces and continue to grow into inSNPs. E-SEM images showed that relatively uniform inSNP coverages could be achieved on both native and PTA-coated samples (Fig. 2A7 and A8). Image analysis (ESI Fig. S18–S20†) gave average diameters of 56 nm on native cotton and 39 nm on PTA-coated cotton (Table 1). These inSNPs were much smaller than the StöberSNPs and a good amount of NPs (27–30% surface coverage) was observed. Such a larger amount of smaller inSNPs should produce a higher surface area enhancement for further functionalization. Indeed, preliminary experiments showed that inSNPs led to higher binding activities compared to StöberSNP modified cotton (Fig. S21†). Therefore, the rest of the study proceeded only with inSNP modification.
| No PTA on fibers | With PTA on fibers | |||||
|---|---|---|---|---|---|---|
| Diameter | Particles per μm2 | Coverage/surf. area increase | Diameter | Particles per μm2 | Coverage/surf. area increase | |
| inSNP on cotton | 56 ± 11 nm | ∼140 | ∼27%/+54% | 39 ± 10 nm | ∼340 | ∼30%/+60% |
| inSNP on NRC | 54 ± 12 nm | ∼210 | ∼33%/+66% | 58 ± 14 nm | ∼110 | ∼20%/+40% |
| inSNP on PEs | ∼15 nm | ∼4000 | ∼70%/+140% | ∼15 nm | ∼4000 | ∼70%/+140% |
Using the same nanosol, NRC and PE were also successfully modified with inSNPs (Fig. 2B and C). On native NRC, the average particle diameter was ∼54 nm and the coverage was ∼33%, similar to growth on native cotton (Fig. 2B3 and Table 1). Growth on PTA-coated NRC gave a slightly lower surface coverage (∼20%; Fig. 2B4) but the NP diameters remained similar. On PE, the diameters were much smaller at ∼15 nm, and the coverage was much higher at ∼70% (Fig. 2C3 and Table 1). Growth on PTA-coated PE appeared very similar within the resolution limits of the E-SEM (Fig. 2C4).
The large differences in inSNP growth on the cellulose and PE substrates were likely due to differences in interfacial forces on these surfaces controlling the attachment of silica precursors. Moreover, we have previously observed that the properties of PTA can change depending on the nature of the substrate.6 Cotton actually incorporates several percent of other biomolecules (e.g., hemicellulose, waxes, etc.) that are absent on NRC.56,57 Together with a potential difference in PTA thickness, some differences in surface properties between PTA-coated cotton and NRC are to be expected, which would have contributed to the minor differences in inSNP growth observed on the two coated cellulosic substrates.
Overall, our inSNP methodology gave rise to relatively high and uniform NP surface coverages. The higher inSNP coverage relative to StöberSNP was also confirmed by energy dispersive X-ray (EDX) measurements on the imaged areas (Fig. S22†), which showed much more intense silicon peaks on inSNPs modified samples. A lower bound estimate of the surface area enhancement due to inSNPs can be obtained by assuming that the particles grow to hemispherical shapes that cover circular areas on the original substrate. This would imply surface area increases that are double the particle surface coverage (Fig. S23†). Accordingly, we expect at least 40–140% surface area enhancements depending on the substrate (Table 1), which should lead to corresponding increases in the amounts of proteins immobilized and higher overall binding activities.
IgG immobilization on inSNP
Samples were functionalized by immersing the 1 × 1 cm2 fabric pieces into pH 7.4 PBS containing 5 μg mL−1 of a polyclonal mouse IgG antibody at room temperature (see ESI†). This step is analogous to immobilizing a capture antibody for an ELISA sandwich assay, which commonly utilizes 1–10 μg mL−1 antibody solutions. However, such concentrations are much lower than the 10–1000 μg mL−1 commonly used in biosensor or nanomaterial surface functionalization,10,58,59 or the ∼2000 μg mL−1 required to saturate IgG surface coverage.51 The physiological pH used should promote reasonable biorecognition binding for most antibodies and can be broadly applied (pH below 6.5 and above 8.4 should be avoided).60–62To assay the binding capability of the immobilized IgG (i.e., its surface activity), we exposed the samples to a model protein binding partner—a 10 μg mL−1 solution of anti-mouse IgG conjugated to horseradish peroxidase (anti-IgG-HRP). This allowed us to use a sensitive HRP enzyme assay for characterization of immobilized IgG binding activity (Fig. 3a; see ESI† for details). Furthermore, we focus on characterizing surface activities rather than the amount of protein immobilized. Not only is binding activity the ultimate criterion for successful surface functionalization, our previous study also established that trends in the immobilization amount and surface activity are often at odds due to the complex factors governing activity retention (e.g., surface-induced denaturation).6
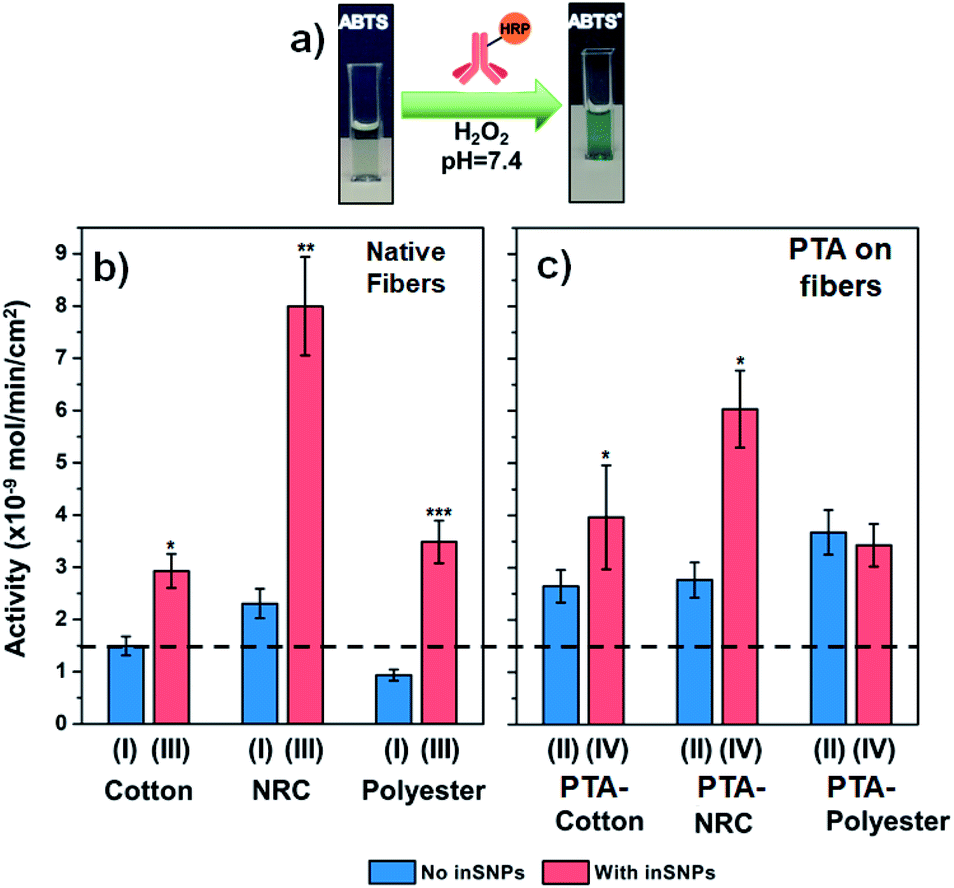 | ||
| Fig. 3 (a) Schematic of the HRP enzyme assay that indicates the level of anti-IgG-HRP binding to immobilized IgG. (b and c) Activities from bound anti-IgG-HRP measured on cotton, NRC, and PE, with and without inSNP. IgG immobilized from 5 μg mL−1 IgG. For ease of comparison, the dotted line drawn across the plots indicates the level of IgG binding enabled on native cotton (first bar on left). The Roman numerals refer to the substrate preparation procedure shown in Fig. 1. See Tables S7 to S10† for raw and normalized activity values. The Student's t-test was performed to calculate statistical significance for each pair of samples with and without inSNP: *, **, and ***, indicate P ≤ 0.1, ≤0.01, and ≤0.001, respectively (see Table S12† for values). | ||
We first analyzed the effect of inSNP modification on native surfaces without PTA (Fig. 3b). In these cases, IgG immobilization occurred by physical adsorption. On native cotton (type I samples), a surface activity of 1.5 nmol min−1 cm−2 was measured. We estimate that this level of activity corresponded to 170 ng cm−2 of active IgG adsorbed (Table S8†). In comparison, the activity increased 2-times to 2.9 nmol min−1 cm−2 on inSNP modified cotton (type III samples). On native NRC and PE, the increases with inSNP modification were even more dramatic, both at roughly 3.7-times (8.0 vs. 2.2 nmol min−1 cm−2 and 3.4 vs. 0.9 nmol min−1 cm−2, respectively). In comparison, StöberSNP modification on cotton only slightly increased the activity to 1.7 nmol min−1 cm−2 (Fig. S21†), and was not investigated further.
We were pleasantly surprised by the magnitude of the observed activity increases with inSNPs. For example, the +100% (i.e. 2-times) and +280% (i.e. 3.8-times) increases in activities observed on cotton and PE, respectively, were essentially double the corresponding +54% and +140% surface area enhancements (Table 1). Such differences are much larger than can be attributed to uncertainties in quantifying the inSNP surface area. One contribution to this “excess” enhancement might have been the high surface curvature of the inSNPs which, as shown in NP–protein interaction studies, can reduce steric hindrance between adjacent IgGs as well as enhance molecular transport, and hence increasing binding efficiency.63,64 A surface curvature effect would also be consistent with the higher enhancements observed for PE, which had much smaller ∼15 nm diameter inSNPs with even higher curvatures.
In the case of NRC samples, there could also be a potential contribution from changes in wettability. The native nanoporous NRC actually possess a ca. 5.6-times higher internal surface area compared to our cotton samples (Table S6:† 107 cm2 vs. 19 cm2 per sample), but the activity from adsorbed IgG was initially only 1.6-times higher (Fig. 3b: 2.2 vs. 1.5 nmol min−1 cm−2). After inSNP modification, however, the contrast in activity increased to 2.8-times (Fig. 3b: 8.0 on inSNP-NRC vs. 2.9 nmol min−1 cm−2 on inSNP–cotton). Although cellulose is considered “hydrophilic”, many of its hydroxyl groups participate in intra-molecular hydrogen bonding, and the advancing contact angle of regenerated cellulose is upwards of ∼40°.65,66 It may be that not all of the NRC nanoporous structure was initially wetted but wetting increased after inSNP modification because of silica's hydrophilicity, which enabled higher IgG immobilization. In comparison, wetting of the much larger cotton and PE microfibers is less challenging, and the initially lower activity on native PE compared to cotton is consistent with the ∼60% lower fiber surface areas of our PE vs. cotton samples (12 vs. 19 cm2 per sample; see Table S6†).
IgG immobilization with PTA
In further experiments, we observed that IgG surface activity can also be increased with PTA coating alone (i.e., type II samples in Fig. 3c vs. type I in Fig. 3b), to an extent similar to inSNP modification. The activity achieved on PTA–cotton was 1.8-times higher than on native cotton (2.6 vs. 1.5 nmol min−1 cm−2). On PTA–PE, the activity was 4-times higher than on native PE (3.6 vs. 0.9 nmol min−1 cm−2). The increase with PTA–NRC was smaller (2.7 vs. 2.2 nmol min−1 cm−2), which might have also been related to poor wetting inside NRC since coating with PTA is an aqueous process like IgG immobilization.Further inSNP modification on top of PTA coated cotton and NRC (type IV samples in Fig. 3c) led to some additive increases in binding. Growing inSNP on PTA–cotton resulted in a further 1.5-times increase in activity compared to PTA–cotton alone (4.0 vs. 2.6 nmol min−1 cm−2 for types IV vs. II in Fig. 3c), which represents a combined 2.6-times increase compared to native cotton (1.5 nmol min−1 cm−2 for type I, Fig. 3b). On NRC, although the final activity with inSNP on PTA–NRC was higher than on PTA–NRC alone, it was lower than for inSNP on native NRC. On PTA-coated PE, similar surface activities were observed with or without inSNP modification. These trends can also be seen in Fig. S24,† which replots the data according to the treatment step.
The smaller additive effect of inSNP on PTA–NRC could be due to the lower inSNP surface coverage and hence lower area enhancement on these samples (see Table 1). In the case of PE, inSNP coverage was very high (∼70%). Thus most of the PTA coating underlying the inSNPs would have been covered by the inSNPs. Furthermore, the nanoparticles were spaced very closely together (Fig. 2B4: ca. <15 nm) and it is likely that it became challenging for IgG, which has a comparable ∼10 nm hydrodynamic diameter,67 to immobilize on the PTA-coated spaces between inSNP particles. Thus including a PTA coating with inSNP modification would have had little effect. A second PTA coating on inSNPs was also attempted (Fig. S25†) but this also produced little additional effect, possibly due to a combination of the coating smoothing out high surface curvatures and differences between IgG immobilization on PTA and on silica. Higher activities could naturally also be achieved by using higher IgG concentrations during immobilization. However, this is an inefficient approach and would significantly increase costs. For example, increasing the IgG concentration used for immobilization on PTA-coated cotton by 3-fold to 15 μg mL−1 only increased the surface activity 1.3-times (Fig. S26†), barely matching the 1.5-times enhancement from adding inSNP on PTA-coated cotton (Fig. 3c).
IgG co-immobilization with SA
Activities of immobilized proteins is generally reduced by surface-induced denaturation—the spreading and unfolding of proteins driven by excess chemical coupling or by ubiquitous and non-specific van der Waals attraction with the surface.9,68 The effect is especially significant for large, flexible proteins such as the “Y-shaped” IgG. Therefore, in a second series of experiments, serum albumin (SA) was addded at increasing concentrations from 5 to 45 μg mL−1 to a constant concentration of 5 μg mL−1 IgG in the solution used for protein immobilization. We hypothesized that co-immobilization of the relatively small SA (66 kDa, vs. ca. 150 kDa for IgG),69 with no non-specific interaction with IgG,53 could act as a surface “steric helper”. Co-immobilization should limit IgG interactions to smaller patches of the surface between SA molecules, thus reducing spreading and denaturation while spacing IgG out to reduce steric hindrance. Any activity enhancement, however, would have to outweigh the reduction in the amount of IgG immobilized.Fig. 4 shows the surface activities with SA-IgG co-immobilization normalized to the original levels for each substrate type using only IgG. Three kinds of trends may be identified. The first is the desired increase in activity. For example, on inSNP modified cotton (Fig. 4a, type III), the activity increased by up to 1.8-times as the SA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IgG ratio progressively increased from 1:1 to 4:1 SA:IgG. Combined with the 2-times enhancement already present due to inSNP modification (Fig. 3b), there was in total a 3.6-times higher activity from combining inSNP modification with 4:1 SA:IgG co-immobilization. A smaller 1.25-times increase in activity from co-immobilization was also observed for 1:1 SA:IgG on inSNP modified PTA–cotton (Fig. 4a, type IV). This was however additional to the 1.5-times enhancement from inSNPs and the 1.7-times enhancement from PTA functionalization (Fig. 3c). Thus, the three enhancement mechanisms together gave a combined 3.2-times higher activity.
:IgG ratio progressively increased from 1:1 to 4:1 SA:IgG. Combined with the 2-times enhancement already present due to inSNP modification (Fig. 3b), there was in total a 3.6-times higher activity from combining inSNP modification with 4:1 SA:IgG co-immobilization. A smaller 1.25-times increase in activity from co-immobilization was also observed for 1:1 SA:IgG on inSNP modified PTA–cotton (Fig. 4a, type IV). This was however additional to the 1.5-times enhancement from inSNPs and the 1.7-times enhancement from PTA functionalization (Fig. 3c). Thus, the three enhancement mechanisms together gave a combined 3.2-times higher activity.
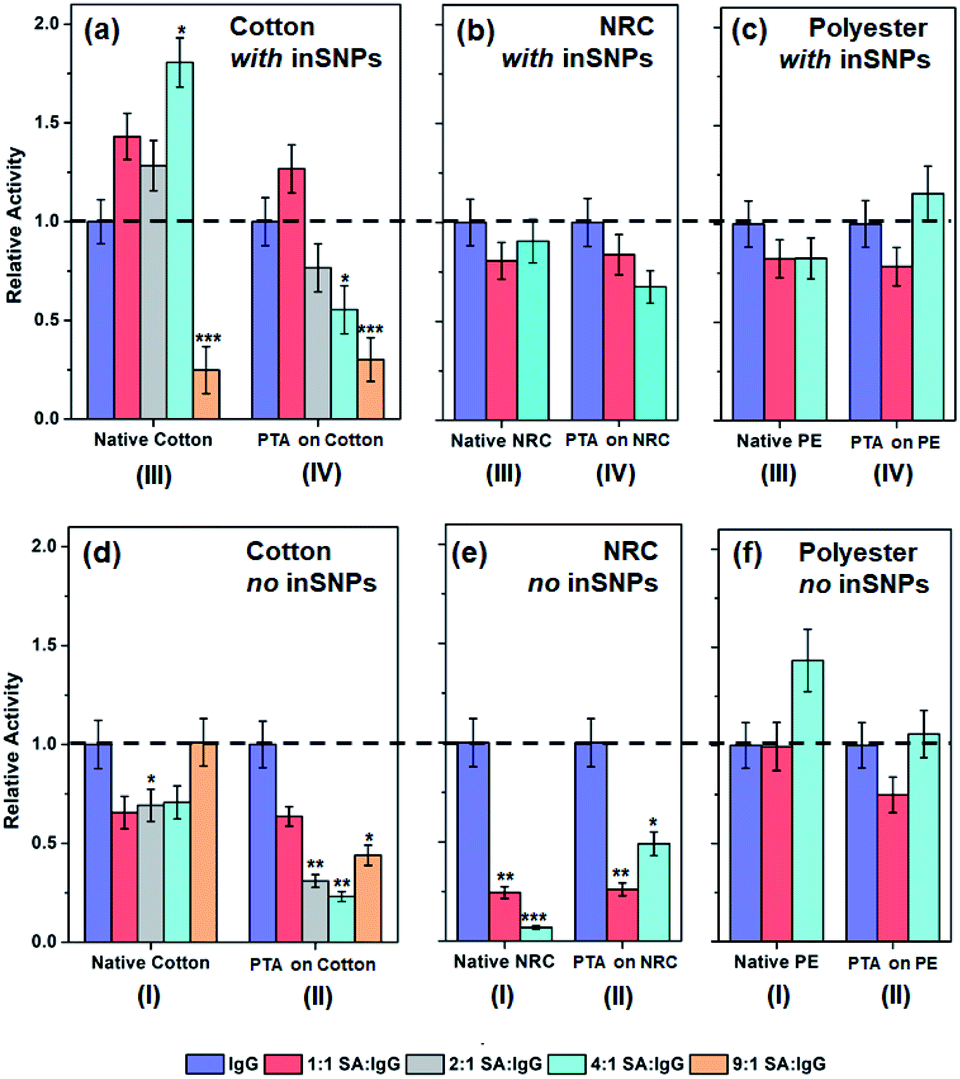 | ||
| Fig. 4 Relative binding activities of co-immobilization samples for (a and d) cotton-based surfaces with and without inSNPs prepared from 1:1 to 1:9 mixtures of IgG and SA (i.e. 5 μg mL−1 IgG mixed with 5 to 45 μg mL−1 SA). Whereas (b and e) NRC-based, and (c and f) PE-based surfaces with and without inSNPs prepared from 1:1 and 1:4 mixtures of IgG and SA. Values were normalized to the activities for pure IgG immobilized on native fibers (i.e. 1.5, 2.6, and 0.9 nmol min−1 cm−2 on cotton, NRC and PE, respectively; see Fig. 3b and c; see Tables S7 to S11† for raw activity values). The Student's t-test was performed w.r.t samples immobilized with 100% IgG within each category (*, **, and ***, indicate P ≤ 0.1, ≤0.01, and ≤0.001, respectively; see Table S13† for values). | ||
The second, opposite trend is decreasing activity with increasing SA:IgG. On the aforementioned inSNP modified cotton, a further increase to 9:1 SA:IgG ultimately reduced activity down to ∼20% of the pure IgG reference. A progressive decrease for inSNP modified PTA–cotton beyond 1:1 SA:IgG was also observed as the SA:IgG ratio further increased. Decreases were also observed on inSNP modified PTA–NRC (Fig. 4b, type IV) and on native NRC (Fig. 4e, type I).
The final kind of trend is a decrease followed by an increase in activity with increasing SA:IgG ratios, which was observed for the remaining surfaces. For example, on PTA-coated PE, the activity recovered as SA:IgG increased to 4:1 (Fig. 4c and f, types IV and II, respectively). Moreover, the activity finally increased by 1.4-times on native PE using this 4:1 SA:IgG co-immobilization (Fig. 4f, type I). This third trend was also prominent on cotton without inSNPs (Fig. 4d) and on both PTA-coated NRC and PE (Fig. 4e and f type II).
Overall, Fig. 4 shows that increases in activity with increasing SA:IgG ratios (i.e. higher SA co-immobilized) are possible for some combinations of surface preparations. Apart from modification of IgG binding efficiency due to the presence of co-immobilized SA, the competition between IgG and SA for immobilization on a given surface also contributed to whether an activity enhancement was observed. Physical adsorption of the larger IgG is actually preferred over SA since more non-specific van der Waals interactions with the surface can be contributed by a larger protein.51 Conversely, chemical coupling on a reactive PTA coating depends less on the size of the protein. Thus, activities on sample types I and III, which did not have PTA, were mostly higher with co-immobilization. The inSNP modified samples also exhibited relatively higher activities (Fig. 4a–c vs. d–f) since the inSNPs were not coated with PTA. Overall, the balance between inSNP modification and any changes in the substrate-dependent competitive immobilization of SA and IgG eventually determined the SA:IgG ratio at which activities may be enhanced.
Lastly, we conducted experiments to test for the activity retention of the immobilized proteins. After seven days' storage in pH 7.4 PBS at temperature 4 °C, Fig. S27† shows that higher activity retention was achieved for inSNP modified samples in combination with a high amount of BSA co-immobilization and no PTA coating, or with PTA but no BSA co-immobilization (e.g. ca. 84% activity retention on “Native Cotton” with inSNP and 4:1 BSA:IgG). However, more than 50% losses were observed for an intermediate level of BSA co-immobilization and samples without inSNP. Nonetheless, since our immobilization procedure is simple and can be carried out within 1 h at room temperature, field preparation of test substrates could prove effective. Overall, the storage test results also corroborate our overall findings that details of surface preparation may impact on IgG surface denaturation and that BSA co-immobilization may ameliorate conformational (and steric) effects.
Conclusion
Significant enhancements of surface binding activity of IgGs immobilized on cotton and polyester (PE) fabrics, as well as on nanoporous regenerated cellulose (NRC), were achieved by a combination of simple surface functionalization approaches. A convenient one-step methodology was demonstrated for growing silica NPs in situ (inSNPs) directly on fiber surfaces to enable much more homogeneous and higher density NP modification than achieved by the attachment of pre-synthesized SNPs. This inSNP modification was found to increase surface activities significantly more than expected from the increase in surface area alone, which we attributed mainly to NP curvature effects. Similar activity enhancements could alternatively be achieved with polyphenol (poly(tannic acid)) coatings, which provided a reactive surface for chemical coupling of proteins as opposed to immobilization by physical adsorption. Additive enhancements were further observed for certain combinations of inSNP modification, polyphenol coating, and serum albumin (SA) co-immobilization depending on the underlying substrate material. Overall, we have demonstrated the potential for enhancing immobilized antibody binding by combining convenient surface functionalization procedures without resorting to site-directed or oriented immobilization procedures. Since our strategy increases specific binding at all analyte concentrations, limits of detection would be expected to scale linearly with this increase if non-specific adsorption and other sources of detection noise remain relatively constant. The materials used are environmentally benign, and all methodologies consisted of simple and low-cost, one-step solution phase procedures. Together with the low antibody concentration used, our results are anticipated to expand the scope of low cost biosensing based on immobilized antibodies and the use of widely available fabrics and nanofiber materials.Author contributions
IA performed the investigation and drafted the manuscript. MEY, AMLS, T-DL and KHAL contributed measurements and data analysis. BY and KHAL acquired funding, supervised the researchers, as well as reviewed and edited the manuscript. KHAL conceptualized the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KHAL, BY, and IA acknowledge support from HFSP (RGY0074/2016). BY and IA acknowledge support from HEC for NRPU (Project No. 20-1740/R&D/10/3368, 20-1799/R&D/10-5302 and 5922), and LUMS for start-up fund and FIF grants. IA acknowledges the Commonwealth Scholarship Commission for award PKCN-2018-275. T.-D. L. was supported by the CBET funded by the National Science Foundation under Grant No 1604504.References
- J. B. Chen, H. Yousefi, C. R. Nemr, S. Gomis, R. Atwal, M. Labib, E. Sargent and S. O. Kelley, Adv. Funct. Mater., 2019, 1907701 Search PubMed.
- A. Loiseau, L. Zhang, D. Hu, M. Salmain, Y. Mazouzi, R. Flack, B. Liedberg and S. Boujday, ACS Appl. Mater. Interfaces, 2019, 11, 46462–46471 CrossRef CAS PubMed.
- G.-R. Han, H. Ki and M.-G. Kim, ACS Appl. Mater. Interfaces, 2019, 12, 1885–1894 CrossRef PubMed.
- Y. Ding, W. Li, F. Zhang, Z. Liu, N. Zanjanizadeh Ezazi, D. Liu and H. A. Santos, Adv. Funct. Mater., 2019, 29, 1802852 CrossRef.
- A. R. Armiento, L. P. Hatt, G. Sanchez Rosenberg, K. Thompson and M. J. Stoddart, Adv. Funct. Mater., 2020, 1909874 CrossRef CAS.
- A. M. L. Sousa, T. D. Li, S. Varghese, P. J. Halling and K. H. A. Lau, ACS Appl. Mater. Interfaces, 2018, 10, 39353–39362 CrossRef CAS PubMed.
- J. Zhang and S. Seeger, Adv. Funct. Mater., 2011, 21, 4699–4704 CrossRef CAS.
- S. M. Kang, M.-H. Ryou, J. W. Choi and H. Lee, Chem. Mater., 2012, 24, 3481–3485 CrossRef CAS.
- M. Hoarau, S. Badieyan and E. N. G. Marsh, Org. Biomol. Chem., 2017, 15, 9539–9551 RSC.
- S. Bagherbaigi, E. P. Córcoles and D. H. Wicaksono, Anal. Methods, 2014, 6, 7175–7180 RSC.
- J. Byun, S. Cho, J. Moon, H. Kim, H. Kang, J. Jung, E.-K. Lim, J. Jeong, H. G. Park and W. K. Cho, ACS Appl. Bio Mater., 2020, 3, 3631–3639 CrossRef CAS.
- J. Moon, J. Byun, H. Kim, J. Jeong, E. K. Lim, J. Jung, S. Cho, W. K. Cho and T. Kang, Macromol. Biosci., 2019, 19, 1800486 CrossRef PubMed.
- H. M. Wang, C. Wang, S. Y. Tao, J. S. Qiu, Y. X. Yu and M. F. Gu, ACS Sustainable Chem. Eng., 2016, 4, 992–998 CrossRef CAS.
- J. M. Krochta and C. L. De Mulder-Johnston, Biodegradable polymers from agricultural products, ACS Symposium Series, 1996, vol. 647, pp. 120–140 Search PubMed.
- C. Parolo and A. Merkoçi, Chem. Soc. Rev., 2013, 42, 450–457 RSC.
- T. Ozer, C. McMahon and C. S. Henry, Annu. Rev. Anal. Chem., 2020, 13, 85–109 CrossRef PubMed.
- G. Zhu, X. Yin, D. Jin, B. Zhang, Y. Gu and Y. An, TrAC, Trends Anal. Chem., 2019, 111, 100–117 CrossRef CAS.
- K. Mahato, B. Purohit, A. Kumar and P. Chandra, in Comprehensive Analytical Chemistry, Elsevier, 2020, vol. 89, pp. 163–188 Search PubMed.
- A. M. López-Marzo and A. Merkoçi, Lab Chip, 2016, 16, 3150–3176 RSC.
- H. Kettler, K. White and S. J. Hawkes, Mapping the landscape of diagnostics for sexually transmitted infections: key findings and recommendations, World Health Organization, 2004 Search PubMed.
- Q. Gao, Q. Zhu, Y. Guo and C. Q. Yang, Ind. Eng. Chem. Res., 2009, 48, 9797–9803 CrossRef CAS.
- J. Lin, X. Chen, C. Chen, J. Hu, C. Zhou, X. Cai, W. Wang, C. Zheng, P. Zhang and J. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 6124–6136 CrossRef CAS PubMed.
- C. Li, Y. Liu, X. Zhou and Y. Wang, J. Mater. Chem. B, 2020, 3582–3589 RSC.
- T.-T. Tsai, C.-Y. Huang, C.-A. Chen, S.-W. Shen, M.-C. Wang, C.-M. Cheng and C.-F. Chen, ACS Sens., 2017, 2, 1345–1354 CrossRef CAS PubMed.
- Z. Liu, L. Qi, X. An, C. Liu and Y. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 40987–40997 CrossRef CAS PubMed.
- M. Lee, G. Kwak and K. Yong, ACS Appl. Mater. Interfaces, 2011, 3, 3350–3356 CrossRef CAS PubMed.
- M. Zahid, E. L. Papadopoulou, G. Suarato, V. D. Binas, G. Kiriakidis, I. Gounaki, O. Moira, D. Venieri, I. S. Bayer and A. Athanassiou, ACS Appl. Bio Mater., 2018, 1, 1154–1164 CrossRef CAS.
- B. R. Knowles, P. Wagner, S. Maclaughlin, M. J. Higgins and P. J. Molino, ACS Appl. Mater. Interfaces, 2017, 9, 18584–18594 CrossRef CAS PubMed.
- L. Lao, L. Fu, G. Qi, E. P. Giannelis and J. Fan, ACS Appl. Mater. Interfaces, 2017, 9, 38109–38116 CrossRef CAS PubMed.
- T. I. Shaheen, M. E. El-Naggar, A. M. Abdelgawad and A. Hebeish, Int. J. Biol. Macromol., 2016, 83, 426–432 CrossRef CAS PubMed.
- M. Rehan, N. A. Abdel-Wahed, A. Farouk and M. M. El-Zawahry, ACS Sustainable Chem. Eng., 2018, 6, 5911–5928 CrossRef CAS.
- M. E. Yazdanshenas and M. Shateri-Khalilabad, Ind. Eng. Chem. Res., 2013, 52, 12846–12854 CrossRef CAS.
- X. Su, H. Li, X. Lai, L. Zhang, J. Wang, X. Liao and X. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 28089–28099 CrossRef CAS PubMed.
- J. Ma, S. Luan, L. Song, S. Yuan, S. Yan, J. Jin and J. Yin, Chem. Commun., 2015, 51, 6749–6752 RSC.
- W. Hu, Y. Liu, T. Chen, Y. Liu and C. M. Li, Adv. Mater., 2015, 27, 181–185 CrossRef CAS PubMed.
- R. Banerjee and A. Jaiswal, Analyst, 2018, 143, 1970–1996 RSC.
- E. Morales-Narváez, T. Naghdi, E. Zor and A. Merkoçi, Anal. Chem., 2015, 87, 8573–8577 CrossRef PubMed.
- Y. Zhao, D. Zeng, C. Yan, W. Chen, J. Ren, Y. Jiang, L. Jiang, F. Xue, D. Ji and F. Tang, Analyst, 2020, 145, 3106–3115 RSC.
- C. Parolo, A. de la Escosura-Muñiz and A. Merkoçi, Biosens. Bioelectron., 2013, 40, 412–416 CrossRef CAS PubMed.
- R. Chen, C. Ren, M. Liu, X. Ge, M. Qu, X. Zhou, M. Liang, Y. Liu and F. Li, ACS Nano, 2021, 15, 8996–9004 CrossRef CAS PubMed.
- X. Li and X. Liu, Adv. Healthcare Mater., 2016, 5, 1326–1335 CrossRef CAS PubMed.
- Y. Du, A. Pothukuchy, J. D. Gollihar, A. Nourani, B. Li and A. D. Ellington, Angew. Chem., Int. Ed., 2017, 56, 992–996 CrossRef CAS PubMed.
- K. N. Onwukamike, S. p. Grelier, E. Grau, H. Cramail and M. A. Meier, ACS Sustainable Chem. Eng., 2018, 7, 1826–1840 CrossRef.
- J. Zimmermann, F. A. Reifler, G. Fortunato, L. C. Gerhardt and S. Seeger, Adv. Funct. Mater., 2008, 18, 3662–3669 CrossRef CAS.
- J. Su, J. Noro, S. Silva, J. Fu, Q. Wang, A. Ribeiro, C. Silva and A. Cavaco-Paulo, React. Funct. Polym., 2019, 136, 25–33 CrossRef CAS.
- D. T. Marquez, J. Chawich, W. M. Hassen, K. Moumanis, M. C. DeRosa and J. J. Dubowski, ACS Omega, 2021, 6, 7286–7295 CrossRef CAS PubMed.
- W. Zhang, C. Ling, H. Liu, A. Zhang, L. Mao, J. Wang, J. Chao, L. J. Backman, Q. Yao and J. Chen, Chem. Eng. J., 2020, 125232 CrossRef CAS.
- S. A. Abouelmagd, F. F. Meng, B. K. Kim, H. Hyun and Y. Yeo, ACS Biomater. Sci. Eng., 2016, 2, 2294–2303 CrossRef CAS PubMed.
- C. Dhand, S. Harini, M. Venkatesh, N. Dwivedi, A. Ng, S. P. Liu, N. K. Verma, S. Ramakrishna, R. W. Beuerman, X. J. Loh and R. Lakshminarayanan, ACS Appl. Mater. Interfaces, 2016, 8, 1220–1232 CrossRef CAS PubMed.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef CAS PubMed.
- A. Kamyshny, S. Lagerge, S. Partyka, P. Relkin and S. Magdassi, Langmuir, 2001, 17, 8242–8248 CrossRef CAS.
- M. A. Rahim, S. L. Kristufek, S. Pan, J. J. Richardson and F. Caruso, Angew. Chem., Int. Ed., 2019, 58, 1904–1927 CrossRef CAS PubMed.
- R. Wang, X. Zhou, X. Zhu, C. Yang, L. Liu and H. Shi, ACS Sens., 2017, 2, 257–262 CrossRef CAS PubMed.
- A. M. Sousa, T.-D. Li, S. Varghese, P. J. Halling and K. H. Aaron Lau, ACS Appl. Mater. Interfaces, 2018, 10, 39353–39362 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- R. Nickerson, Ind. Eng. Chem., 1940, 32, 1454–1462 CrossRef CAS.
- M. Dochia, C. Sirghie, R. Kozłowski and Z. Roskwitalski, in Handbook of natural fibres, Elsevier, 2012, pp. 11–23 Search PubMed.
- C. Parolo, A. Sena-Torralba, J. F. Bergua, E. Calucho, C. Fuentes-Chust, L. Hu, L. Rivas, R. Álvarez-Diduk, E. P. Nguyen and S. Cinti, Nat. Protoc., 2020, 15, 3788–3816 CrossRef CAS PubMed.
- B. Della Ventura, M. Iannaccone, R. Funari, M. Pica Ciamarra, C. Altucci, R. Capparelli, S. Roperto and R. Velotta, PLoS One, 2017, 12, e0171754 CrossRef PubMed.
- D. Absolom and C. Van Oss, CRC Crit. Rev. Immunol., 1986, 6, 1–46 CAS.
- N. Hughes-Jones, B. Gardner and R. Telford, Immunology, 1964, 7, 72 CAS.
- A. E. Barnes, J. Immunol., 1966, 96, 854–864 CAS.
- K. Woeppel, X. Zheng and X. Cui, J. Mater. Chem. B, 2018, 6, 3058–3067 RSC.
- S. Kumar, I. Yadav, V. K. Aswal and J. Kohlbrecher, Langmuir, 2018, 34, 5679–5695 CrossRef CAS PubMed.
- F. Bartell and B. R. Ray, J. Am. Chem. Soc., 1952, 74, 778–783 CrossRef CAS.
- T. A. Dankovich and D. G. Gray, J. Adhes. Sci. Technol., 2011, 25, 699–708 CrossRef CAS.
- J. Mathes and W. Friess, Eur. J. Pharm. Biopharm., 2011, 78, 239–247 CrossRef CAS PubMed.
- W. Norde and T. Zoungrana, Biotechnol. Appl. Biochem., 1998, 28, 133–143 CAS.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2010, 31, 9527–9534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04645d |
| This journal is © The Royal Society of Chemistry 2021 |