
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
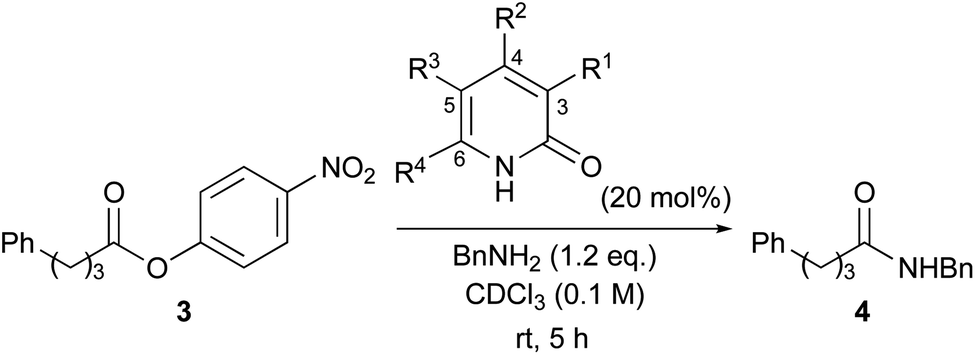
6-Halo-2-pyridone as an efficient organocatalyst for ester aminolysis†
Takeshi Yamada *,
Yusuke Watanabe and
Sentaro Okamoto*
*,
Yusuke Watanabe and
Sentaro Okamoto*
Department of Materials and Life Chemistry, Kanagawa University, 3-27-1 Rokkakubashi, Kanagawa-ku, Yokohama, 221-8686, Japan. E-mail: tyamada@kanagawa-u.ac.jp; okamos10@kanagawa-u.ac.jp
First published on 14th July 2021
Abstract
It was found that 6-halo-2-pyridones catalysed ester aminolysis in which not only reactive aryl esters but also relatively less reactive methyl and benzyl esters could be used as a substrate. The reaction could be performed without strictly dry and anaerobic conditions and the 6-chloro-2-pyridone catalyst could be recovered quantitatively after reaction. The method could be applied to dipeptide synthesis from methyl or benzyl esters of amino acids, where a high enantiomeric purity of the products was maintained. The mechanism involving dual activation of ester and amine substrates through hydrogen bonding between catalyst and substrates is proposed where 6-halo-2-pyridones act as a bifunctional Brønsted acid/base catalyst.
Introduction
An amide bond is one of the most frequently observed chemical bonds in biologically relevant and functional molecules. The development of chemically and economically efficient methods for creating amide bonds is an ongoing research topic among organic chemists. Generally, amide bonds can be formed from carboxylic acids and amines using a stoichiometric condensation reagent.1 However, this promising method has economic and atom-economic drawbacks. Therefore, the formation of amide bonds from amines and carboxylic acids under catalytic conditions has become a recent trend in organic chemistry.2 Another method for creating an amide bond is the acylation of an amine with acid derivatives, such as acid anhydrides, halides and others.3 In addition, several amine equivalents such as isocyanide4 and azide5 were applied to form amide bonds. However, these methods require tedious preparation of substrates unless they are ordinary simple reagents.The direct amidation of esters is also an effective method, particularly in the case of the elongation of a peptide from the C-terminus, and this is based on their availability from commercial sources and their ease of preparation. In the case of peptide elongation from the C-terminus, the ester was primarily hydrolysed prior to condensation with an amine in the presence of an appropriate stoichiometric amount of condensation reagent. Saponification of esters sometimes causes undesired reactions. Direct amidation of the ester avoids the need for a two-step process in this regard.
There are many reports on the direct amidation of esters with amines.6–14 Although many reactions are limited to relatively reactive phenyl esters or specific substrates, some reactions tolerate a broad range of substrates, such as the commonly used methyl or ethyl esters of amino acids, and provide the corresponding peptide without decreasing optical purity.6c–e,8e,9b However, these reactions require strict dryness and anaerobic conditions. In addition, many reactions require expensive or commercially unavailable catalysts that cannot be reused.
Ester aminolysis catalysed by neutral organomolecules such as N-heterocyclic carbenes,12 polyethers13 and 2-pyridones14 is an attractive reaction because of its mild reactivity and operational convenience. Ester aminolysis catalysed by 2-pyridone (1) was first reported by Rony and Openshaw's groups, independently.14 In this process, 2-pyridone (1) behaves as a bifunctional catalyst, the NH group activates an ester group to function as a Brønsted acid, and the CO group activates the amine as a Brønsted base.14,15 The proposed reaction mechanisms are described in Scheme 1, which shows three possible approaches. Path A involves four-membered ring transition state 1 (TS-1), and paths B and C include using eight-membered ring transition state 2 (TS-2) and 3 (TS-3). The NH group of 1 creates a hydrogen bond to aryloxygen to activate the ester in TS-2. The TS-3 approach includes the hydrogen bonding between NH group of 1 and ester carbonyl oxygen. A lactim tautomer, 2-hydroxy pyridine (2), can also be considered to function as a bifunctional catalyst.15 Although several mechanistic investigations have been conducted to date, only one study focused on the development of a highly reactive 2-pyridone type catalyst.15a,b We are interested in the ability of 2-pyridone as an acid–base conjugate catalyst, and we reported on the organocatalytic α-addition of isocyanide with aldehyde using 3,5,6-trifluoro-2-pyridone as a catalyst.16 In this manuscript, we report on organocatalytic ester aminolysis using 6-halo-2-pyridones as a reactive catalyst.
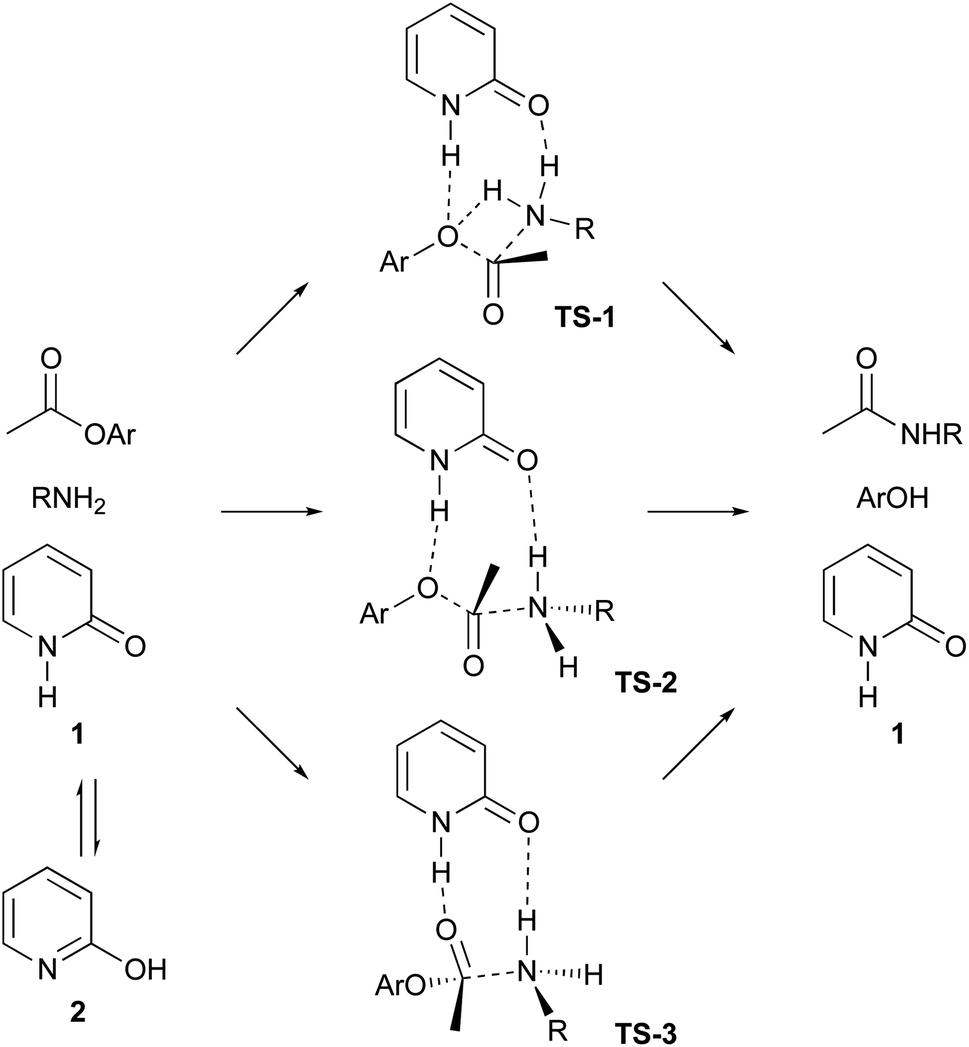 | ||
| Scheme 1 Proposed mechanism of 2-pyridine catalyzed ester aminolysis. | ||
Results and discussion
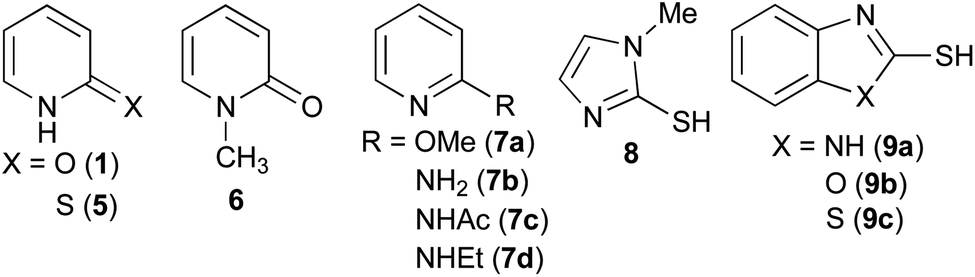
Aminolysis of highly reactive p-nitrophenyl ester 3 with benzylamine (1.2 eq.) in the presence of 1 (20 mol%) in CDCl3 was examined (Table 1, entry 1). After stirring at room temperature for 5 h, 3 was converted to the corresponding amide 4 in 37%. The conversion was calculated by 1H NMR spectroscopy. The reaction without a catalyst achieved only 14% conversion (entry 2). Additionally, N or O-methylated 2-pyridone (6, 7a) revealed lower reactivity compared with 1 (entries 1, 3 and 4). These results support the proposed mechanism described in Scheme 1. Among the screened 2-amino or 2-mercapto-heteroarene catalysts 7b–d, 8 and 9, 2-pyridone (1) showed the highest reactivity (entries 1 and 5–12).17
|
||
|---|---|---|
| Entry | Catalyst | Conversiona |
| a Conversion was calculated by 1H NMR spectroscopy. | ||
| 1 | 2-Pyridone (1) | 37% |
| 2 | None | 14% |
| 3 | N-Methyl 2-pyridone (6) | 25% |
| 4 | 2-Methoxypyridine (7a) | 17% |
| 5 | 2-Mercaptopyridine (5) | 30% |
| 6 | 2-Aminopyridine (7b) | 20% |
| 7 | 2-Acetylaminopyridine (7c) | 20% |
| 8 | 2-Ethylaminopyridine (7d) | 22% |
| 9 | 2-Mercapto-1-methylimidazole (8) | 32% |
| 10 | 2-Mercaptobenzimidazole (9a) | 22% |
| 11 | 2-Mercaptobenzoxazole (9b) | 30% |
| 12 | 2-Mercaptobenzothiazole (9c) | 31% |
 |
||
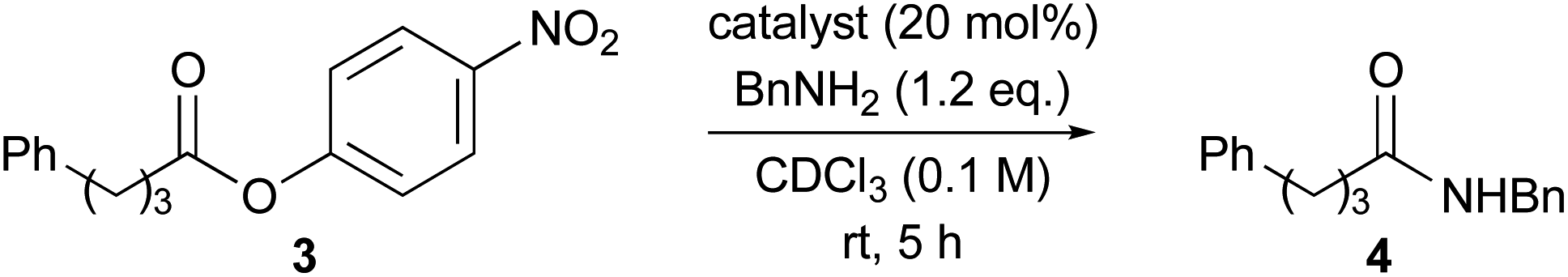
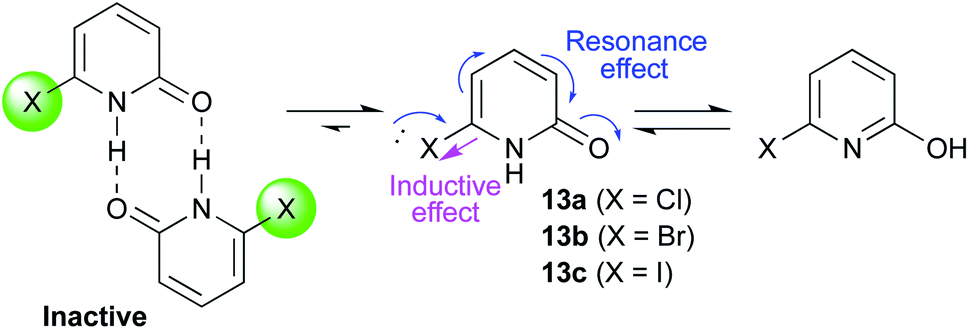
A series of 2-pyridones, including electron-withdrawing and donating substituents at the C3, C4, C5 and C6 positions, were examined as potential catalysts. Selected examples are shown in Table 2. The introduction of electron-withdrawing and donating substituents at the C3 or C4 position did not improve catalytic activity (18–37% conversion, entries 1–6). Additionally, C5-substituted 2-pyridones 12 showed a similar catalytic activity to C4-substituted 2-pyridones 11, but 5-chloro-2-pyridone (12b) revealed slightly higher reactivity compared with 1 (entry 8). A substituent at the C6 position of 2-pyridone was found to be important for catalytic activity. A series of 6-substituted 2-pyridones 13 were examined. Among them, 6-halo-2-pyridone (13a–c) showed the highest catalytic activity for ester aminolysis (entries 10–16). It was assumed that the introduction of electron-withdrawing halogens increased the acidity of the 2-pyridone NH group, which activated ester moiety (Fig. 1).
|
|||
|---|---|---|---|
| Entry | R | Conversiona | |
| a Conversion was calculated by 1H NMR spectroscopy. | |||
| 1 |  |
NO2 (10a) | 23% |
| 2 | Cl (10b) | 27% | |
| 3 | OMe (10c) | 34% | |
| 4 | 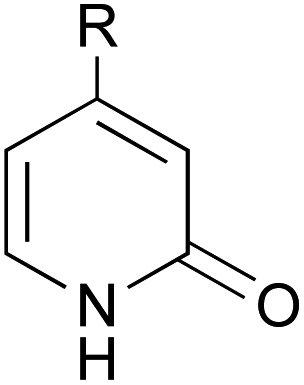 |
CF3 (11a) | 32% |
| 5 | Cl (11b) | 37% | |
| 6 | OMe (11c) | 34% | |
| 7 | 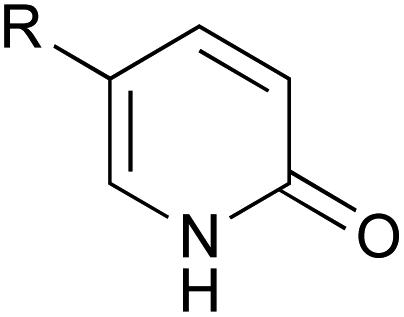 |
NO2 (12a) | 42% |
| 8 | Cl (12b) | 51% | |
| 9 | OMe (12c) | 41% | |
| 10 | 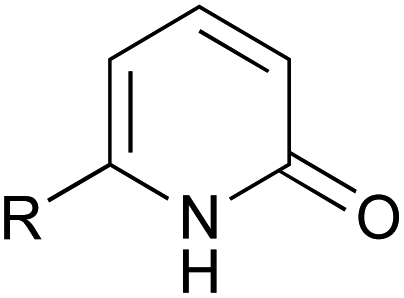 |
Cl (13a) | 81% |
| 11 | Br (13b) | 78% | |
| 12 | I (13c) | 86% | |
| 13 | F (13d) | 31% | |
| 14 | CO2Me (13e) | 50% | |
| 15 | OMe (13f) | 52% | |
| 16 | CH3 (13g) | 34% | |
| 17 | 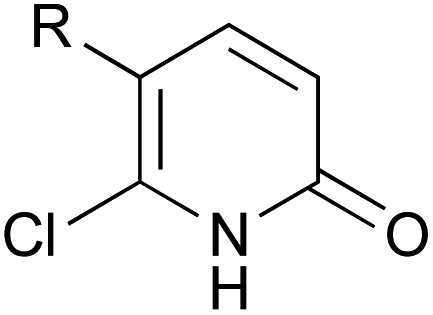 |
Cl (14a) | 58% |
| 18 | NO2 (14b) | 22% | |
 | ||
| Fig. 1 6-Halo-2-pyridone 13a, 13b and 13c. | ||
The basicity of the carbonyl group of the catalyst may also have been increased by the resonance effect of halogens. The steric bulkiness of the C6 substituent may have inhibited the undesired homodimerisation of the catalyst.15a In addition, halogen substituents improved solubility in organic solvents. A certain interaction of halogen atom to the substrate may assist this reaction. However, further introduction of electron-withdrawing groups to the C5 position decreased catalytic activity due to a decrease in the Lewis basicity of pyridone carbonyl (entries 17 and 18). Considering the availability and stability, further investigation was carried out using commercially available 6-chloro-2-pyridone 13a as a catalyst.18,19
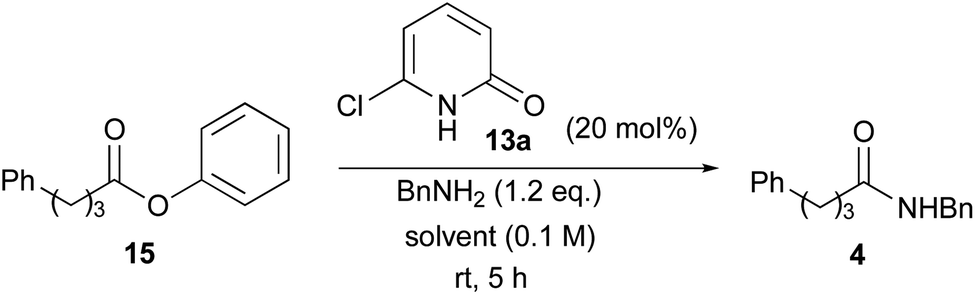
Using 6-chloro-2-pyridone (13a) as a catalyst, various solvents were examined for this reaction. Selected examples are shown in Table 3.17 In this study, relatively less-reactive phenyl ester 15 was used as a substrate. The reactions were carried out in the presence of 20 mol% of 13a at room temperature for 5 h. The reaction proceeded at a 24% rate in CHCl3 (entry 1). The use of diethylether as a solvent showed almost the same results (entry 2). Aromatic solvents, particularly toluene, improved the conversion rate (entries 3–6). A non-polar solvent such as hexane exhibited the highest conversion rate of 74% (entry 7). Conversely, polar solvents such as DMF and t-BuOH were ineffective, and the rate was as low as that in the absence of a catalyst (entries 8 and 9). The reaction rate reduced dramatically without a catalyst in each solvent. These results clearly indicate that the hydrogen bonds between the catalyst 13a and the substrates are crucial for this ester aminolysis.
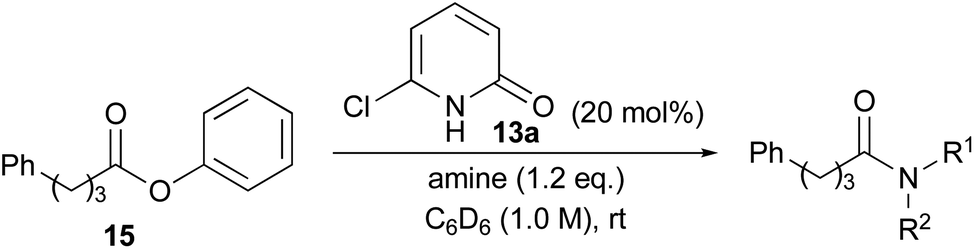
The reactions of a series of amines with phenyl ester 15 in the presence of 20 mol% of 13a were carried out in C6D6, the conversion of which was determined by 1H NMR analysis (Table 4). All reactions were conducted in 1.0 M solution due to the slow reaction in 0.1 M solution (entry 1). Triethylamine was included as an additive when using amine HCl–salt. Aminolysis of phenyl ester 15 with aromatic amine 16, branched amine 17 and secondary amines 18 and 19 was accelerated in the presence of catalyst 13a compared with catalyst-free conditions (entries 2–5). However, ester aminolysis with a highly hindered amine and methanolysis did not proceed at all (entries 6 and 7).
|
||||
|---|---|---|---|---|
| Entry | Amine | Time | Conversiona | Conversiona without catalyst |
| a Conversion was calculated by 1H NMR spectroscopy.b Run in 0.1 M solution.c 1.0 eq. of NEt3 was added.d No reaction. | ||||
| 1b | Aniline (16) | 32 h | 2% | — |
| 2 | 16 | 70 h | 60% | Trace |
| 3 | 1-Phenylethylamine (17) | 49 h | 87% | 14% |
| 4c | MeO(Me)NH·HCl (18) | 48 h | 73% | 20% |
| 5c | H-Pro-OBn·HCl (19) | 47 h | 68% | 22% |
| 6 | t-BuNH2 (20) | 50 h | n.r.d | n.r.d |
| 7 | MeOH | 20 h | n.r.d | n.r.d |
To study the reaction profile, phenyl ester 15 was treated with benzylamine (1.2 eq.) in the presence of 20 mol% 6-chloro-2-pyridone (13a), 6-bromo-2-pyridone (13b) or 6-iodo-2-pyridone (13c) in C6D6 (1.0 M) at room temperature. The progress of this reaction was monitored by 1H NMR spectroscopy, and the amounts of ester 15 and product 4 were quantified to determine the conversion. The time course of conversion is plotted as a dotted line in Fig. 2. It was assumed that the reaction would proceed according to first-order kinetics to each amine (1.2 eq.) and ester (1.0 eq.). Accordingly, kt = (1/(1 − 1.2))ln(1.2 × (1 − x)/(1 × (1.2 − x))). In Fig. 2, time vs. −5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(1.2 × (1 − x))/(1.2 − x) (x = conversion, 0 ≤ x ≤ 1) is plotted as a solid line. As shown in this figure, 6-halo-2-pyridones 13a–c dramatically accelerated the reaction compared with catalyst-free conditions. All 2-pyridones showed similar catalytic activity, but 6-iodo-2-pyridone 13c was slightly higher. These reactions can overall be treated as a pseudo-second-order kinetics.
ln(1.2 × (1 − x))/(1.2 − x) (x = conversion, 0 ≤ x ≤ 1) is plotted as a solid line. As shown in this figure, 6-halo-2-pyridones 13a–c dramatically accelerated the reaction compared with catalyst-free conditions. All 2-pyridones showed similar catalytic activity, but 6-iodo-2-pyridone 13c was slightly higher. These reactions can overall be treated as a pseudo-second-order kinetics.
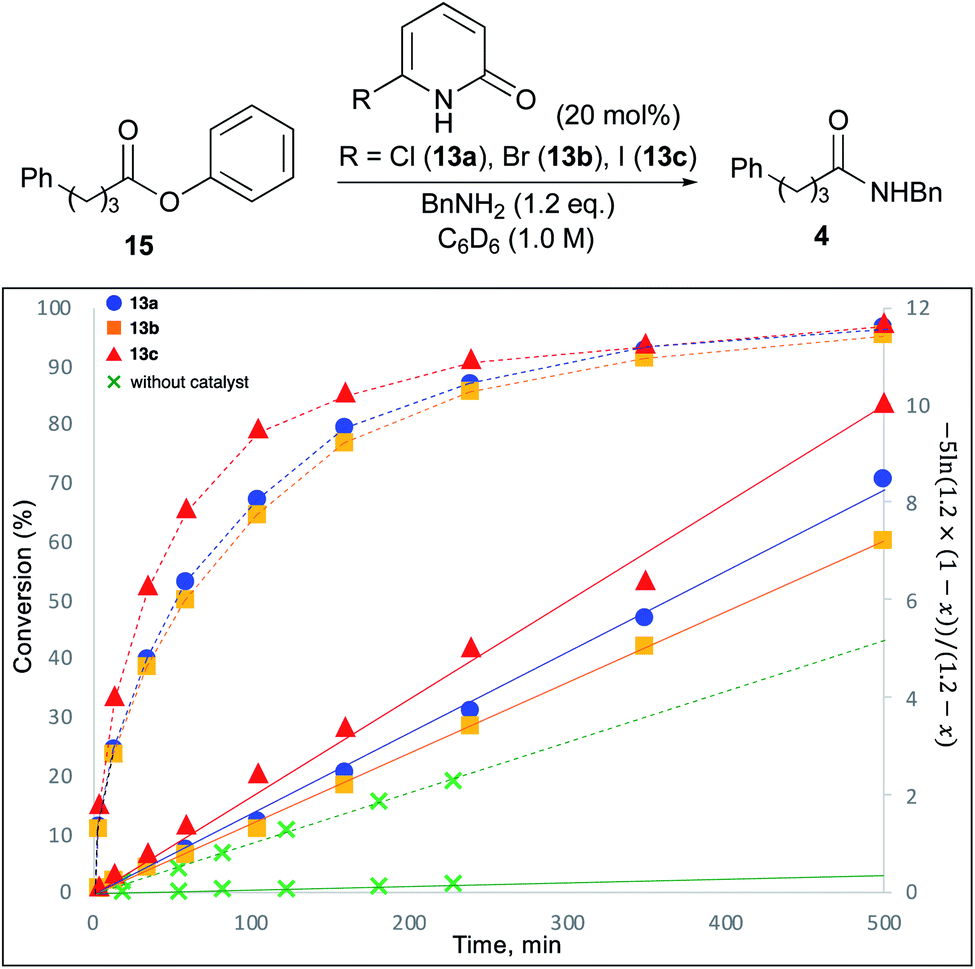 | ||
| Fig. 2 Kinetic study. Time course of conversion was plotted as a dotted line. Time vs. −5ln(1.2 × (1 − x))/(1.2 − x) was plotted as a solid line. | ||
Next, we examined the reactions of less-reactive common esters such as methyl and benzyl esters. Methyl esters 21a, 22 and benzyl ester 21b were successfully reacted with benzylamine (1.2 eq.) in the presence of 13a (20 mol%) in toluene (1.0 M) (Table 5). Although the reaction did not proceed at ambient temperature, methyl ester 21a was consumed after stirring at 110 °C for 72 h. Corresponding amide 4 was isolated at a 99% yield through direct purification by silica gel column chromatography (entry 1). Catalyst 13a was quantitatively recovered. This reaction showed poor conversion (3%) without a catalyst. Benzyl ester 21b was also applicable, and amide 4 was obtained in 98% yield (without a catalyst, 16%; entry 2). Carboxylic acid 21c did not react at all (entry 3). Aminolysis of methyl benzoate (22) proceeded slower compared with that of aliphatic methyl ester 21a. After stirring at 110 °C for 72 h, corresponding amide 23 was obtained in 58% yield with the recovery of 22 (entry 4). It is noted that when the catalyst was changed to 2-pyridone (1), the conversion was significantly reduced to 8%.
|
||||
|---|---|---|---|---|
| Entry | Ester | Conversiona | Isolated yield | Conversiona without catalyst |
| a Conversion was calculated by 1H NMR spectroscopy.b Catalyst was recovered quantitatively.c No reaction.d 2-Pyridone (1) was used as catalyst instead of 13a. | ||||
| 1 | 21a | >99% | 99%b | 3% |
| 2 | 21b | 98% | 98%b | 16% |
| 3 | 21c | n.r.c | n.r.c | — |
| 4 | 22 | 58% | 58%b | 2% (8%)d |
Next, we pursued the aminolysis of amino acid esters, the results of which are summarised in Table 6. Amino acid esters were found to be better substrates for this reaction compared with aliphatic acid esters such as 21a and 21b. Although the reaction was relatively slow, ester aminolysis of Boc-Gly-OBn 24a with benzylamine proceeded even at room temperature, and corresponding amide 27 was obtained in 86% isolated yield after stirring at 40 °C for 24 h (entry 1). The reaction without a catalyst proceeded at a lower conversion (34%). Unexpectedly, sterically less-hindered methyl ester 24b showed lower reactivity than 24a did (65% conversion, entry 2). We envisioned applying this reaction next to prepare a dipeptide. The treatment of L-leucine ethyl ester hydrochloride 26 with 24a in the presence of 13a (20 mol%) and NEt3 at 60 °C for 72 h provided dipeptide 28 in 66% isolated yield with 92% ee (entry 3). Optical purity was determined by chiral HPLC analysis. All other side products such as polypeptides, diketopiperazine and self-condensation products of 26 were not detected. This reaction did not proceed without catalyst 13a. The use of 2-pyridone (1) as a catalyst decreased the conversion to 21%. More hindered secondary amine 18 did not react even at 110 °C (entry 4). Considering the low reactivity of secondary amines, aminolysis of non-protected H-Pro-OBn 19 with benzylamine was conducted (entry 5). As anticipated, the reaction proceeded even at 40 °C to provide corresponding amide 29 in a good isolated yield (87%) while maintaining optical purity (>99% ee). In this reaction, secondary amine groups of the proline residues in substrate 19 and product 29 did not react with benzyl ester. More sterically bulky Boc-L-Leu-OBn 25 was also applicable to this aminolysis. Treatment of 25 with benzylamine at 60 °C for 94 h provided the corresponding amide 30 in 67% yield with almost maintained optical purity (99% ee, entry 6). Raising the temperature to 110 °C improved the conversion, but the optical purity decreased slightly (83% yield, 94% ee, entry 7).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ester | Amine | Temp. | Time | Conversiona | Yield | e.e.b | Conversiona without catalyst |
| a Conversion was calculated by 1H NMR spectroscopy.b Determined by chiral HPLC analysis.c 1.2 eq. of NEt3 was added.d Not detected.e 2-Pyridone (1) was used as catalyst instead of 13a.f No reaction. | ||||||||
| 1 | Boc-Gly-OBn (24a) | BnNH2 | 40 °C | 24 h | 86% | 86% | — | 34% |
| 2 | Boc-Gly-OMe (24b) | BnNH2 | 40 °C | 24 h | 65% | n.d. | — | 13% |
| 3c | 24a | H-L-Leu-OEt·HCl (26) | 60 °C | 72 h | 66% | 66% | 92% | n.d.d (21%)e |
| 4c | 24a | MeO(Me)NH·HCl (18) | 110 °C | 24 h | n.r.f | n.r.f | — | — |
| 5c | H-L-Pro-OBn·HCl (19) | BnNH2 | 40 °C | 24 h | 87% | 87% | >99% | 11% |
| 6 | Boc-L-Leu-OBn (25) | BnNH2 | 60 °C | 94 h | 67% | 67% | 99% | — |
| 7 | 25 | BnNH2 | 110 °C | 24 h | — | 83% | 94% | — |
Based on these results, we show our proposed mechanism in Fig. 3. 2-Pyridone activates ester substrate via a hydrogen bond between proton of N–H in pyridone and carbonyl oxygen, and simultaneously, amine can be activated through a hydrogen bonding to pyridone carbonyl oxygen. This concurrent dual activation also makes the reaction entropically favored. 6-Halogen substitution on 2-pyridone enhances acidity of proton of N–H by negative inductive effect and basicity of carbonyl oxygen can be increased by positive resonance effect. Furthermore, it can be considered that an interaction between ester carbonyl oxygen and empty orbital of halogen atom in pyridone may participate as an additional activation. Besides these considerations, the mechanism based on a lactim tautomer, 2-hydroxypyridine (2), can not be ruled out. More detail discussion must await further study.
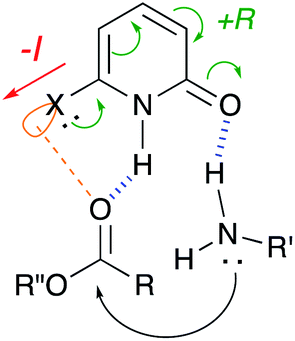 | ||
| Fig. 3 Proposed mechanism. | ||
Conclusions
We found that 6-halo-2-pyridones delivered remarkable catalytic activity for ester aminolysis. The catalyst enabled aminolysis of not only highly reactive aromatic esters but also ordinary methyl and benzyl esters and complete recovery of the catalyst. Biologically important amino acid derivatives apply to this reaction, which did not require any special reagents, strict dryness, anaerobic conditions or extraction and was operationally convenient. We believe that these results may enable a practical approach to catalytic peptide synthesis. Further screening of a highly reactive 2-pyridone catalyst for ester aminolysis and the development of other 2-pyridone catalysed reactions are underway in our laboratory.Experimental section
General procedure for ester aminolysis
A mixture of an ester (0.1 mmol), amine (0.12 mmol) and 6-chloro-2-pyridone (20 mol%) in toluene (0.1 mL) was stirred at an appropriate temperature. The reaction was monitored by TLC. After the completion of the reaction, the mixture was put on a silica gel and purified by column chromatography to provide the corresponding amide. The catalyst 13a could be recovered quantitatively at the purification of silica gel column chromatography (hexane:AcOEt = 10:1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI Grant Numbers 17K08219 and 20K05502. The authors would like to thank MARUZEN-YUSHODO Co., Ltd. for the English language editing.Notes and references
- Selected reviews, see: (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed; (b) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS.
- Selected recent reports, see: (a) W. Muramatsu, T. Hattori and H. Yamamoto, Bull. Chem. Soc. Jpn., 2020, 93, 759 CrossRef CAS; (b) M. Koshizuka, K. Makino and N. Shimada, Org. Lett., 2020, 22, 8658 CrossRef CAS PubMed; (c) M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed; (d) Z. Liu, H. Noda, M. Shibasaki and N. Kumagai, Org. Lett., 2018, 20, 612 CrossRef CAS PubMed; (e) K. Michigami, T. Sakaguchi and Y. Takemoto, ACS Catal., 2020, 10, 683 CrossRef CAS and references cited therein..
- Selected review, see: C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- X. Li, Y. Yuan, C. Kan and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 13225 CrossRef CAS PubMed.
- (a) A. D. Kosal, E. E. Wilson and B. L. Ashfeld, Chem.–Eur. J., 2012, 18, 14444 CrossRef CAS PubMed; (b) A. D. Kosal, E. E. Wilson and B. L. Ashfeld, Angew. Chem., Int. Ed., 2012, 51, 12036 CrossRef CAS PubMed.
- Lewis acidic conditions, see: (a) B. D. Mkhonazi, M. Shandu, R. Tshinavhe, S. B. Simelane and P. T. Moshapo, Molecules, 2020, 25, 1040 CrossRef CAS PubMed; (b) B. C. Ranu and P. A. Dutta, Synth. Commun., 2003, 33, 297 CrossRef CAS; (c) H. Morimoto, R. Fujiwara, Y. Shimizu, K. Morisaki and T. Ohshima, Org. Lett., 2014, 16, 2018 CrossRef CAS PubMed; (d) C. Han, J. P. Lee, E. Lobkovsky and J. A. Porco, J. Am. Chem. Soc., 2005, 127, 10039 CrossRef CAS PubMed; (e) D. C. Lenstra, D. T. Nguyen and J. Mecinović, Tetrahedron, 2015, 71, 5547 CrossRef CAS; (f) D. T. Nguyen, D. C. Lenstra and J. Mecinović, RSC Adv., 2015, 5, 77658 RSC; (g) K. Ishihara, Y. Kuroki, N. Hanaki, S. Ohara and H. Yamamoto, J. Am. Chem. Soc., 1996, 118, 1569 CrossRef CAS; (h) Y. Kuroki, K. Ishihara, N. Hanaki, S. Ohara and H. Yamamoto, Bull. Chem. Soc. Jpn., 1998, 71, 1221 CrossRef CAS; (i) H. Tsuji and H. Yamamoto, J. Am. Chem. Soc., 2016, 138, 14218 CrossRef CAS PubMed.
- Brønsted acidic conditions, see: D. D. Sanz Sharley and J. M. J. Williams, Chem. Commun., 2017, 53, 2020 RSC.
- In the presence of inorganic base, see: (a) B. R. Kim, H.-G. Lee, S.-B. Kang, G. H. Sung, J.-J. Kim, J. K. Park, S.-G. Lee and Y.-J. Yoon, Synthesis, 2012, 44, 42 CrossRef CAS; (b) S. A. Rzhevskiy, A. A. Ageshina, G. A. Chesnokov, P. S. Gribanov, M. A. Topchiy, M. S. Nechaev and A. F. Asachenko, RSC Adv., 2019, 9, 1536 RSC; (c) G. Li and M. Szostak, Nat. Commun., 2018, 9, 4165 CrossRef PubMed; (d) J. D. M. Muñoz, J. Alcázar, A. de la Hoz, Á. Díaz-Ortiz and S. A. Alonso de Diego, Green Chem., 2012, 14, 1335 RSC; (e) T. Ohshima, Y. Hayashi, K. Agura, Y. Fujii, A. Yoshiyama and K. Mashima, Chem. Commun., 2012, 48, 5434 RSC; (f) N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, Chem. Commun., 2015, 51, 9495 RSC.
- In the presence of organic base, see: (a) K. E. Price, C. Larriveé-Aboussafy, B. M. Lillie, R. W. McLaughlin, J. Mustakis, K. W. Hettenbach, J. M. Hawkins and R. Vaidyanathan, Org. Lett., 2009, 11, 2003 CrossRef CAS PubMed; (b) C. Sabot, K. A. Kumar, S. Meunier and C. Mioskowski, Tetrahedron Lett., 2007, 48, 3863 CrossRef CAS; (c) X. Yang and V. B. Birman, Org. Lett., 2009, 11, 1499 CrossRef CAS PubMed; (d) N. Caldwell, C. Jamieson, I. Simpson and T. Tuttle, Org. Lett., 2013, 15, 2506 CrossRef CAS PubMed; (e) N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, ACS Sustainable Chem. Eng., 2013, 1, 1339 CrossRef CAS; (f) N. Caldwell, P. S. Campbell, C. Jamieson, F. Potjewyd, I. Simpson and A. J. B. Watson, J. Org. Chem., 2014, 79, 9347 CrossRef CAS PubMed.
- C,O-activation reactions, see: (a) Y.-S. Bao, B. Zhaorigetu, B. Agula, M. Baiyin and M. Jia, J. Org. Chem., 2014, 79, 803 CrossRef CAS PubMed; (b) S. Shi and M. Szostak, Chem. Commun., 2017, 53, 10584 RSC; (c) A. H. Dardir, P. R. Melvin, R. M. Davis, N. Hazari and M. M. Beromi, J. Org. Chem., 2018, 83, 469 CrossRef CAS PubMed; (d) T. B. Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176 CrossRef; (e) T. B. Halima, J. Masson-Makdissi and S. G. Newman, Angew. Chem., Int. Ed., 2018, 57, 12925 CrossRef PubMed; (f) L. Hie, N. F. F. Nathel, X. Hong, Y. F. Yang, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 2810 CrossRef CAS PubMed; (g) C. Chen, L. Ling, M. Luo and X. Zeng, Bull. Chem. Soc. Jpn., 2021, 94, 762 CrossRef CAS.
- N,H-activation reactions, see: (a) B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682 CrossRef CAS PubMed; (b) Q. Han, X. Xiong and S. Li, Catal. Commun., 2015, 58, 85 CrossRef CAS.
- M. Movassaghi and M. Schmidt, Org. Lett., 2005, 7, 2453 CrossRef CAS PubMed.
- R. D. Gandour, D. A. Walker, A. Nayak and G. R. Newkome, J. Am. Chem. Soc., 1978, 100, 3608 CrossRef CAS.
- (a) H. T. Openshaw and N. Whittaker, J. Chem. Soc. C, 1969, 89 RSC; (b) P. R. Rony, J. Am. Chem. Soc., 1969, 91, 6090 CrossRef CAS.
- (a) L.-H. Wang and H. Zipse, Liebigs Ann., 1996, 1996, 1501 CrossRef; (b) C. B. Fischer, H. Steininger, D. S. Stephenson and H. Zipse, J. Phys. Org. Chem., 2005, 18, 901 CrossRef CAS; (c) X.-Q. Liu, L. Jin, C. K. Kim and Y. Xue, J. Mol. Catal. A: Chem., 2012, 355, 102 CrossRef CAS.
- T. Yamada, T. Hirose, S. Ōmura and T. Sunazuka, Eur. J. Org. Chem., 2015, 296 CrossRef CAS.
- See ESI.†.
- 6-Chloro-2-pyridone (13a) and 6-bromo-2-pyridone (13b) can be purchased from Tokyo Chemical Industry Co., Ltd. for $3 and $70 per gram, respectively. 6-Iodo-2-pyridone (13c) can be prepared from commercially available 2-bromo-6-methoxypyridine in 2 steps. See: Y. Yuan, W. Dong, X. Gao, H. Gao, X. Xie and Z. Zhang, J. Org. Chem., 2018, 83, 2840 CrossRef CAS PubMed.
- 6-Iodo-2-pyridone (13c) is relatively unstable in C6D6 or CDCl3 solution.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04651a |
| This journal is © The Royal Society of Chemistry 2021 |