
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the thermal properties of phase change materials encapsulated in capped carbon nanotubes using molecular dynamics simulations†
Mohsen Abbaspour *a,
Majid Namayandeh Jorabchib,
Hamed Akbarzadeha and
Azra Ebrahimnejada
*a,
Majid Namayandeh Jorabchib,
Hamed Akbarzadeha and
Azra Ebrahimnejada
aDep. of Chemistry, Hakim Sabzevari University, Sabzevar, Iran. E-mail: m.abbaspour@hsu.ac.ir
bDep. of Chemistry, Ferdowsi University of Mashhad, Mashhad, Iran
First published on 14th July 2021
Abstract
Due to the high demand for clean, economic, and recyclable energy, phase change materials (PCMs) have received significant attention in recent years. To improve the performance of PCMs, they are confined in micro- and nano-capsules composed of organic or inorganic materials. In this study, encapsulated phase change material (EPCM) systems were constructed with paraffin molecules as the core material and capped carbon nanotubes (CNTs) as the shell. We investigated the effects of different parameters including CNT diameter, length, and chirality and the length of the alkane molecule chain. We also investigated metal nanocluster-enhanced PCM systems via the addition of Cu, Ag, and Al clusters to the EPCM systems. Different thermodynamic, dynamic, and structural properties including configurational energy, melting range, mean square displacement, self-diffusion coefficient, radial distribution function (RDF), and average end-to-end distance of the confined molecules were examined. We also investigated the effect of metal doping in CNT on the different properties of the confined PCM. The results indicated that a longer CNT has a lower melting point than the normal CNT system. It was also observed that the bigger (30,0) CNT, (14,14) armchair CNT, and icosane systems have higher melting ranges than the normal (25,0) system. The metal cluster systems also have a lower starting melting point than the normal system. Furthermore, it was found that the Al cluster system has the lowest starting melting point among the studied systems.
1. Introduction
PCMs (phase change materials) are materials that absorb (or release) significant latent heat during the melting (or solidifying) process. PCMs are good materials for the storage of thermal energy, and therefore have received much attention in recent years.1–7Proper PCMs must have a suitable phase transition temperature, large specific and latent heat and high thermal conductivity.8 Straight chain n-alkanes (which are also known as paraffins) have high latent heat, low vapor pressure, low supercooling, and high thermal and chemical stability, and therefore have been broadly used as PCMs in experimental and theoretical investigations.9–11 However, paraffins have some disadvantages such as flammability, large volume change during the phase transition, and low thermal conductivity.4 To improve these PCMs and overcome their deficiencies, paraffins are confined into micro and nano capsules usually made from polymer and inorganic materials.12–22 These encapsulated PCMs (also known as EPCMs) exhibit superior properties compared to the usual PCMs, which can improve their energy efficiency as heat transfer materials.4
Due to the high demands for clean, economic, and recyclable energy, EPCMs have been the subject of numerous experimental investigations in recent years.1,3,23–27 For example, Choi et al.23 prepared EPCMs via in situ polymerization. Specifically, they loaded microcapsules with tetradecane as the core and melamine formaldehyde as the shell. They reported that their microcapsules had a high latent heat capacity and durability. Chang et al.24 developed EPCMs using n-octadecane molecules as the core and poly methyl methacrylate–silica as the shell of their microcapsules. They reported that the highest amount of latent heat was achieved using the inorganic/organic ratio of 5% in the capsules. Chao et al.25 synthesized microcapsules using n-octadecane as the core material using an interfacial polycondensation method. Their capsules exhibited the phase transition of octadecane at 29 °C to 30 °C. Recently, Qiu et al.26 fabricated microcapsules using n-octadecane as the core and methylmethacrylate copolymers as the shell of EPCM. They found that the enthalpies of the PCM contents increase by decreasing the length of the side chain of the monomers. Recently, Wang et al.27 synthesized micro-EPCMs consisting of n-octadecane as the core and melamine formaldehyde resin as the shell material. They reported high latent heat and stable performance for the produced micro-EPCMs.
However, although there are numerous experimental investigations on EPCMs, molecular dynamics (MD) simulations are rarely applied in the investigation EPCMs because of their complex structures.1,3,4 Rao et al.4 investigated the melting process of nanocapsulated PCMs using MD simulations. They constructed EPCMs using n-octadecane molecules as the core and SiO2 as the shell. Their results showed that MD simulation is an effective approach to better understand EPCMs. Nie et al.28 investigated the properties of paraffin encapsulated in carbon nanotubes (CNTs) using experimental and MD approaches. They obtained a melting point and latent heat smaller than that of the bulk values. Their MD results showed an ordered distribution of paraffin molecules near the inner wall of the CNT, which disappeared gradually by increasing the temperature. Recently, Keshavarzi and Mansoori and co-workers presented the structures, properties, and phase transitions of confined fluids in carbon nanotubes.29–32
From a wider perspective, it is necessary to study the different properties of EPCMs using MD simulations for expanding both the understanding and applications of phase change materials.33,34 Therefore, in this study, our aim was to investigate the thermal properties of octadecane and icosane molecules encapsulated in different CNTs using MD simulations. The configurational energy, self-diffusion coefficient, and radial distribution function (RDF) of the different systems were also computed. We investigated the effects of different parameters in this study, including the length, diameter, and chirality effects of the CNTs and also the effect of the length of the paraffin molecule. To improve the thermal conductivity of the systems, we also examined the effect of the addition of different metal nanoclusters (such as Cu, Ag, and Al) to the octadecane molecules encapsulated in the CNTs.
2. Simulation details
In this study, the EPCM system was constructed with 17 octadecane molecules as the core material and a graphene-capped (25,0) CNT with the length of 42.68 Å as the shell. The capping of CNTs with graphene surfaces has been experimentally investigated in previous studies.35 The capped CNTs with the confined molecules were put in the center of the simulation box with the dimensions of 100 × 100 × 100 Å3. Firstly, we put 17 octadecane molecules with an ordered configuration in the middle of a CNT. Then, to anneal the molecules, we heated the systems from 200 K to 1000 K, and then cooled them from 1000 K to 200 K at the temperature interval of 50 K. The final structure of the cooling process at 200 K was adopted as the initial configuration for the different simulations.All the simulations were carried out in the NVT ensemble using the Nosé–Hoover thermostat and DL_POLY software.36 The Ewald summation technique was used for the electrostatic interactions. We used 12 Å as the cutoff distance. Periodic boundary conditions were also employed in three directions. The MD simulations were performed for 2 ns (1 ns for the equilibration step followed by 1 ns of production for computing the properties), which is comparable to that of previous MD simulations4,8,37 on confined water molecules between graphene plates. We kept the CNTs fixed in these simulations. The OPLS AA force field38,39 was adopted for the PCM (alkane) molecules and the Lennard-Jones model with the Lorentz–Berthelot combination rules40,41 was employed for the interactions between the PCM and the CNT.
To examine the effect of the CNT length, we used the (25,0) CNT with a length of 64.86 Å. To study the effect of the CNT diameter, we used the (30,0) CNT with a length of 42.68 Å. To investigate the effect of the chirality of the CNT, the (14,14) CNT with a similar diameter (19.45 Å) and length (42.68 Å) was used. To study the effect of the chain length of the PCM, we used 17 icosane molecules in the (25,0) CNT.
Metal nanoclusters have also been used with different PCMs experimentally.42,43 For example, Qian et al.42 experimentally investigated organic PCMs with Cu nanoparticles. They used silver nanoparticles decorated on diatomite with polyethylene glycol (PEG) and found that the presence of metal nanoparticles decreased the melting and freezing periods. They also concluded that the heat transfer is enhanced through the thermal conductivity improvement by the metal nanoparticles. Jafarian et al.43 experimentally examined paraffin wax (as a PCM) with Cu nanoparticles. They concluded that the concentration, size, and type of metal nanoparticle have an important effect on the thermal conductivity of PCMs. They found that the thermal conductivity of the PCM increased with an increase in the nanoparticle concentration. It also increased with a decrease in the nanoparticle size.
To examine the effect of the presence of metal nanoclusters, we confined 32-atom nanoclusters of Cu, Ag, and Al (with the initial fcc structure) with 17 octadecane molecules in the (25,0) CNT. Previous simulations on the melting of small metal nanoclusters also used the initial fcc structures.44,45 We also ran some test simulations using different initial structures of the metal clusters (fcc, icosahedral, and annealed structures) and found that the initial structure has a small effect on the results. The snapshots of these test simulation results are presented at initially and after 0.7 ns of simulation in Fig. S1 in the ESI.†
The quantum Sutton–Chen model was used for the metal–metal interactions and their parameters are presented in Table S1 in the ESI.†46,47 The metal–CNT and metal–PCM interactions were modeled using the LJ potential. Finally, we investigated the thermal properties of the PCM molecules confined in the (25,0) CNT doped with Al atoms. Recently, we showed that the metal doping of CNTs can affect the different properties of the confined molecules.48
3. Results and discussion
3.1 The normal EPCM system
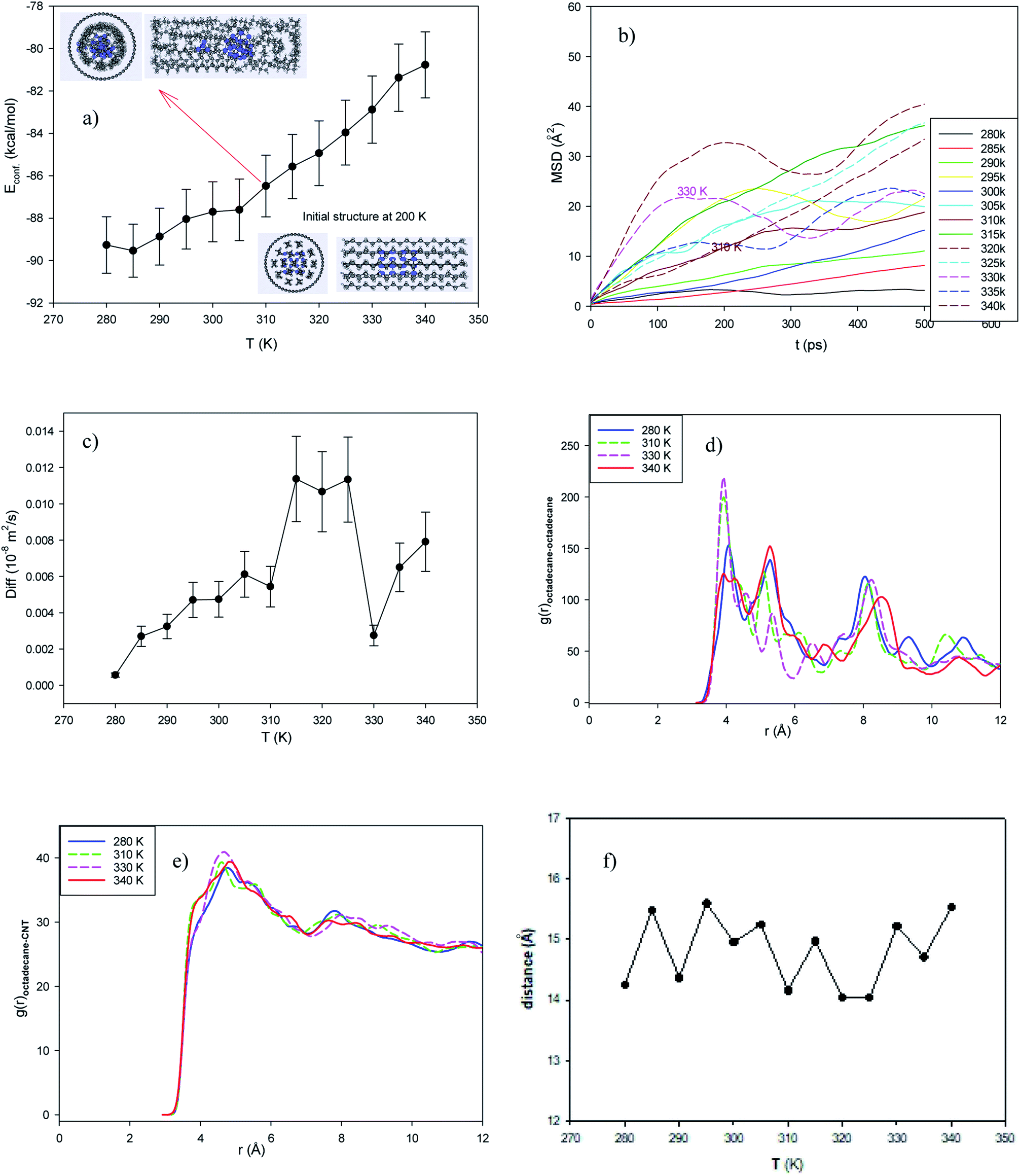
In the normal EPCM system, 17 octadecane molecules were placed in the middle of the capped (25,0) CNT with a length of 42.68 Å. The configurational energy of the system from 280 to 350 K is presented in Fig. 1a. The configurational energy is the result of the forcefield effect on the system and the sum of the different intermolecular and intramolecular energies such as the van der Waals energy and coulombic energy.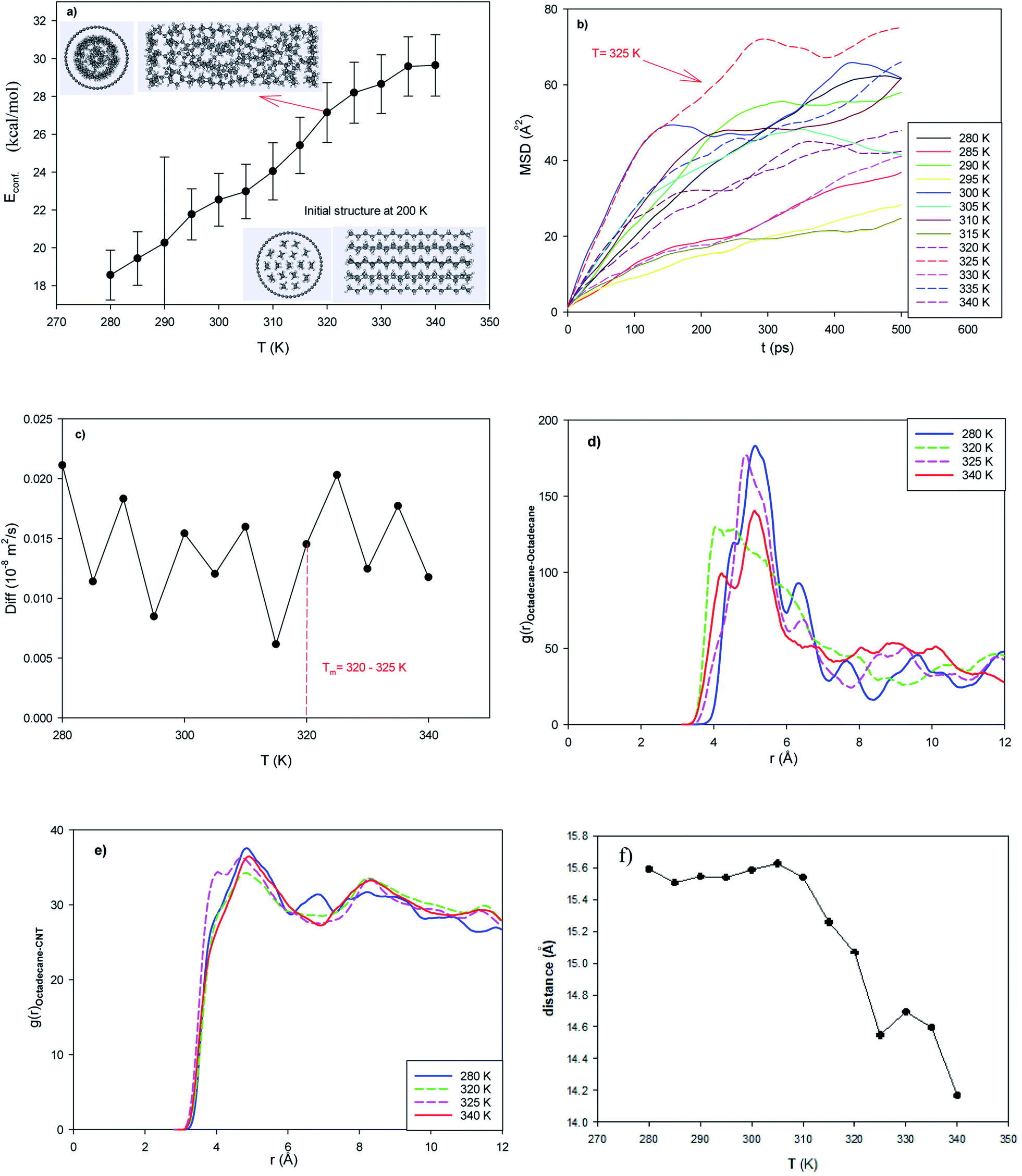 | ||
| Fig. 1 Normal system consisting of 17 octadecane molecules in the capped (25,0) CNT with a length of 42.68 Å. (a) Configurational energy, (b) mean square displacement (MSD), (c) self-diffusion coefficient, (d) octadecane–octadecane radial distribution function (RDF), (e) octadecane–CNT RDF, and (f) average end-to-end distance. | ||
As shown in this figure, the energy of the confined octadecane molecules increases with an increase in temperature. Also, sudden changes in the configurational energy can be distinguished, which may indicate a phase transition process from the solid phase to the liquid state. However, the configurational energy curve cannot represent the phase transition range alone. According to the inserted snapshots of the system in Fig. 1a, the confined molecules have an ordered structure at 200 K and this ordered configuration disappears at 320 K. The confined molecules also form a two-layer structure in the CNT after melting.
The experimental melting temperature of bulk octadecane is 301 K.29,39 This result shows that the encapsulation of the octadecane molecules in the CNT increases their melting temperature. This result is in agreement with simulation result of Akbarzadeh et al.49 on the melting of ionic liquids (ILs) in carbon nanotubes, where they showed that the confinement of IL molecules in CNTs increased their melting temperature and thermal stability. Yafei et al.50 also experimentally investigated encapsulated systems containing octadecane molecules as the core and polystyrene as the shell. They concluded that the microencapsulation of octadecane increases its melting range.
To investigate the melting range and to examine the dynamics of the confined octadecane molecules in the CNT, the self-diffusion coefficients were also calculated from the mean square displacement (MSD) plots using the following equation, where they show linear behaviors:51
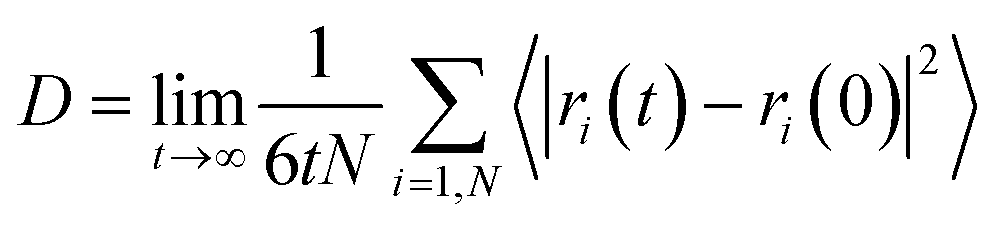 | (1) |
As shown in this figure, the slope of the MSD plots exhibits a sharp change and increases in the temperature range of 320 to 325 K (see the MSD line at 325 K). The sharp increase in the dynamics of the confined molecules at the melting range can be better distinguished in the self-diffusion plot shown in Fig. 1c, which indicates a first-order phase transition. Our simulated self-diffusion coefficients are in good agreement with the previous simulation results of Rao et al.52 Although our simulated self-diffusion coefficients are in the same order of the experimental values53 of bulk octadecane, they are somewhat smaller. For example, the experimental self-diffusion of bulk octadecane at 323 K is 0.046 × 10−8 (m2 s−1),53 which is greater than our calculated value for the confined octadecane at this temperature (0.018 × 10−8 (m2 s−1)). This result means that the encapsulation of the octadecane molecules decreases their dynamics. The same result was obtained by Akbarzadeh and Abbaspour54 in the simulation of ionic liquids confined in different nanotubes. They also concluded that the self-diffusion of the confined ionic liquid is less than that of the bulk ionic liquid.
Our simulated self-diffusion coefficients are greater than that reported by Rao et al.52 They simulated the self-diffusion of octadecane molecules encapsulated in an SiO2 shell. This difference is due to the stronger interactions of the Si atoms with the octadecane molecules than the CNT atoms. Specifically, confining the paraffin molecules in the CNT leads to faster dynamics of the molecules compared to the SiO2 nanocapsules. According to Fig. 1c, some fluctuations in the diffusion coefficient can be observed. This phenomenon may be due to the smaller accessible volume for the confined alkane molecules, which causes the diffusion value to fluctuate. In fact, when an alkane molecule starts to melt, the chains of the neighboring molecules hinder the twisting of the molecule. After melting, twisting occurs, and the diffusion coefficient increases and the length of the alkane chain decreases (Fig. 1f). This phenomenon was not observed in the longer nanotube with a larger accessible volume to the alkane molecules (Fig. 2c). We will discuss this phenomenon in the next section.
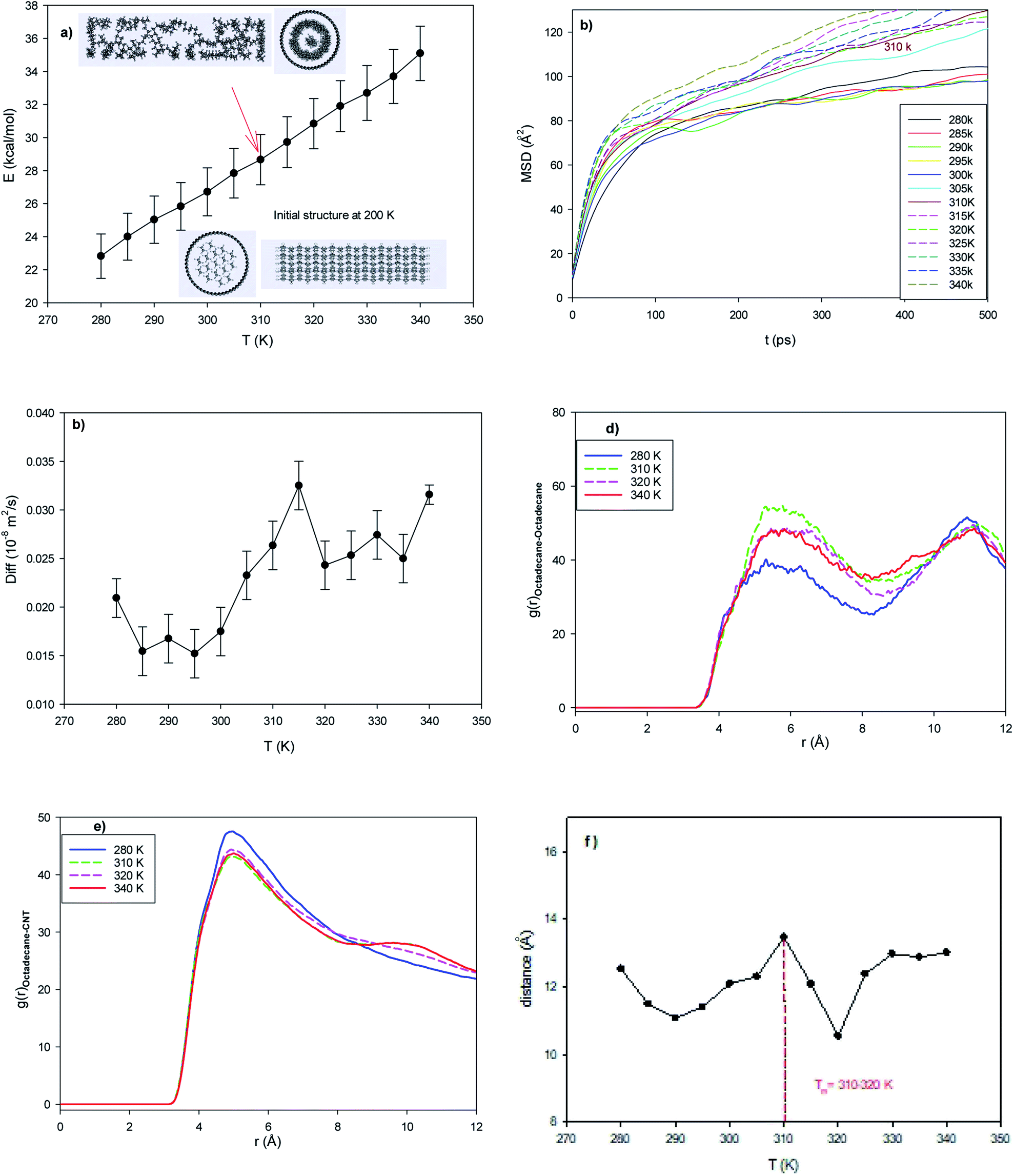 | ||
| Fig. 2 Longer CNT system consisting of 17 octadecane molecules in the capped (25,0) CNT with a length of 64.86 Å. (a) Configurational energy, (b) MSD, (c) self-diffusion coefficient, (d) octadecane–octadecane RDF, (e) octadecane–CNT RDF, and (f) average end-to-end distance. | ||
To investigate the structural properties of the confined octadecane molecules, the radial distribution function RDF or (g(r)) was calculated using the following formula:51
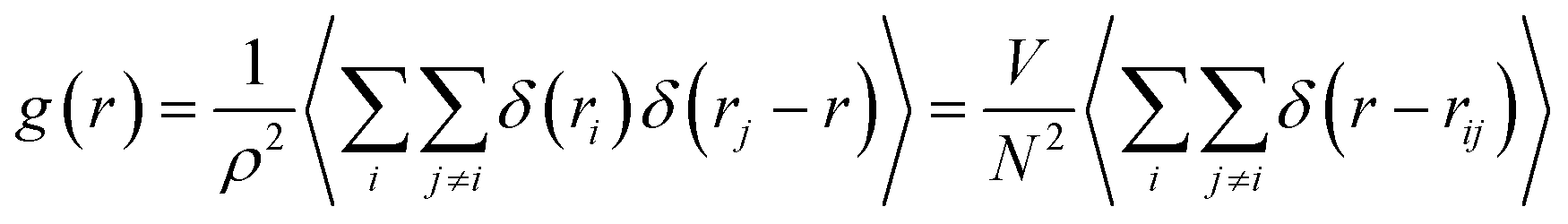 | (2) |
The octadecane molecules may exhibit different configurations. The trans configuration (in which the carbon atoms are in a plane) and the gauche configuration are the two common structures of alkanes. The end-to-end distance is the longest distance when all sections of a molecule are in the trans configuration (solid state). We also present the average end-to-end distance of the confined octadecane molecules at different temperatures in Fig. 1f. As shown in this figure, the end-to-end distance decreases with an increase in temperature, which obeys the thermodynamic theory that the order parameter decreases with the melting process. This is due to the fact that the alkane molecules gain more kinetic energy after melting and tend to twist and orient to the gauche configuration and roll in the nanotube, and therefore the end-to-end distance decreases. Therefore, the end-to-end distance can be considered a thermodynamic order parameter to monitor the melting of long chain alkanes.52 Fig. 1f also shows a sudden change in the end-to-end distance in the melting range, which supports the energy and diffusion results.
3.2 The effect of CNT length
To investigate the effect of the length of the CNT, 17 octadecane molecules were positioned in the middle of the capped (25,0) CNT with a length of 64.86 Å. According to Fig. 2a, the configurational energy increases with an increase in temperature, but the melting range is not obvious. The snapshots in this figure also show that the confined molecules are in the melted state at 310 K. This result shows that an increase in the CNT length decreases the melting temperature. In agreement with the configurational energy results in Fig. 2a, the MSD curves show an increase in the slope of the curves from 310 K, which indicates the melting process (Fig. 2b). The jump in the self-diffusion coefficients of the confined octadecane molecules obviously exhibits the melting process in the melting range of around 310–320 K in Fig. 2c. It can also be found that the self-diffusion of the confined molecules in the longer CNT is greater than that in the normal system. This is due to the increase in the accessible volume of the confined molecules in the longer CNT than the shorter CNT, which leads to the faster melting of the alkane molecules. We also present snapshots of the octadecane molecules in both the normal and long CNTs at the same temperature (T = 305 K) in Fig. S2 in the ESI.† As shown in this figure, there is more accessible volume to the confined molecules in the longer CNT. The octadecane molecules are rolled more completely in the longer CNT and are in the melted state, whereas the octadecane molecules are more compacted and in the solid state in the normal CNT at the same temperature.The center of mass RDFs of the octadecane molecules at different temperatures are presented in Fig. 2d. As shown in this figure, the octadecane–octadecane RDF peaks increase in the melting range. This is due to the increase in the alkane–alkane interactions in the melting range, where the confined molecules tend to roll in the inner CNT wall (Fig. S2†). The RDFs of the center of mass of the octadecane molecules with the CNT wall are also presented in Fig. 2e. According to this figure, the RDF peaks suddenly change in the melting range and the RDF peaks become slightly smaller, which is due to the weakening of the molecule–wall interactions by the increase in temperature. It is also shown that the octadecane–octadecane RDFs are smaller, whereas the octadecane–CNT wall RDFs are higher than the corresponding peaks of the normal system. These results show that the confined octadecane molecules have less interactions with each other, but have stronger interactions with the CNT wall. This is another reason for the decrease in the melting point of the confined molecules with an increase in the CNT length.
We also present the average end-to-end distance of the confined octadecane molecules in the longer CNT at different temperatures in Fig. 2f. According to this figure, a sharp decrease in the end-to-end distance can be observed at around the melting range (310–320 K). It is also shown that the end-to-end distance of the confined molecules in the longer CNT is smaller than that in the normal CNT after the melting range. This is due to the greater accessible volume for the confined molecules in the longer CNT, in which the octadecane molecules can roll more completely in the CNT in the melted state.
3.3 The effect of CNT diameter
To examine the effect of the diameter of the CNT, 17 octadecane molecules were placed in the middle of the capped (30,0) CNT with a length of 42.68 Å. The configurational energy of the system is presented in Fig. S3a.† The melting temperature range cannot be distinguished in this figure. However, according to the inserted snapshots, the confined molecules are in the liquid (melted) state at 310 K and the confined octadecane molecules form one layer in the CNT due to the increase in the CNT diameter.To distinguish the melting range, the MSD plots for the confined octadecane molecules in the (30,0) CNT at different temperatures are presented in Fig. S3b.† As shown in the MSD plots, the behavior and slope of the curves sharply increase from 310 K to 340 K, which indicates the melting process. The change in the self-diffusion coefficients of the confined molecules also indicates the melting process from 310–340 K, as shown in Fig. S3c.† In comparison with the normal system, the self-diffusion of the confined molecules increases with an increase in the CNT diameter, which is due to the increase in the accessible volume for the confined molecules to move. The self-diffusion of the confined molecules in the (30,0) CNT is also greater than that in the longer (25,0) CNT.
To investigate the structural properties of the confined octadecane molecules, the center of mass RDFs of the octadecane molecules and the octadecane–CNT at different temperatures are presented in Fig. S3d and e,† respectively. As shown in these figures, the RDF peaks become smaller with an increase in temperature and the sharp changes correspond to the melting range (from 310–340 K). We also present the average end-to-end distance of the confined octadecane molecules in the (30,0) CNT at the different temperatures in Fig. S3f.† However, the melting temperature cannot be distinguished using the end-to-end distance.
3.4 The effect of CNT chirality
To examine the effect of chirality, we positioned 17 octadecane molecules in the middle of the capped (14,14) CNT with a diameter of 19.45 Å and length of 42.68 Å. According to Fig. S4a in the ESI,† a small jump can be observed at 320 K. According to the snapshot, the confined molecules are in the melted state at 320 K. The MSD curves also show an increase in the slope of the curves from 320 to 340 K, which indicate the melting process (Fig. S4b†). A jump in the self-diffusion coefficients from 320 to 340 K can also be observed in Fig. S4c.† In comparison with the results of the normal zigzag (25,0) CNT, the octadecane molecules in the armchair (14,14) CNT have almost similar but slightly more interactions, and thus they have a higher melting temperature range. It should be noted that the type of molecule-carbon wall interaction is the same for the armchair or zigzag CNT but the different configurations of the carbon atoms in the CNT wall result in different interactions and a small difference in the melting range. As shown in Fig. S4c,† the self-diffusion of the confined molecules in the armchair CNT is smaller, which also indicates the stronger interactions in this system than the zigzag CNT. As shown in Fig. S4d and e,† the RDFs decrease with an increase in temperature, but they do not help to distinguish the melting range. We also presented the average end-to-end distance of the confined octadecane molecules in the armchair (25,0) CNT at different temperatures in Fig. S4f.† As shown in this figure, there is a sharp decrease in the end-to-end distance, which indicates the melting range initiates from 320 K.3.5 The effect of alkane chain length
To examine the effect of the alkane chain length, we put 17 icosane molecules in the middle of the capped (25,0) CNT with a length of 42.68 Å. According to the jumps in the configurational energy and the inserted snapshot in Fig. S5a in the ESI,† the melting temperature range is 310 to 335 K. Comparing the results for the octadecane molecules, the increase in alkane chain slightly increases the melting temperature. The increase in the MSD curves and the self-diffusion values also show that the melting range initiates from 310 K (Fig. S5b and c†). The changes in the behaviors of the icosane–icosane and icosane–CNT RDFs also confirm that the melting range is around 310–335 K (Fig. S5d and e†). The icosane–icosane RDFs decrease with an increase in temperature, whereas the first peaks of the icosane–CNT RDFs increase with the temperature. We also present the average end-to-end distance of the confined icosane molecules in the (25,0) CNT at different temperatures in Fig. S5f.† According to this figure, the melting point exists at around 325 K, which is in agreement with the other results (310–335 K).3.6 The effect of the addition of metal nanoclusters
 | ||
| Fig. 3 Cu cluster system consisting of a copper cluster with 17 octadecane molecules in the capped (25,0) CNT with a length of 42.68 Å. (a) Configurational energy, (b) MSD, (c) self-diffusion coefficient, (d) octadecane–octadecane RDF, (e) octadecane–CNT RDF, and (f) average end-to-end distance. | ||
According to the sharp increase in the configurational energy values, the phase transition process from the solid phase to the liquid state can be observed from 310 K to 330 K. As shown in the snapshot of the system in Fig. 3a, the ordered configuration starts to disappear from 310 K. The starting melting temperature of the nanocluster-enhanced system is also lower than that of the normal system (320 K). In addition to the increase in thermal conductivity, the decrease in the melting temperature is another result of the presence of metallic clusters in the EPCM systems. These results are in good agreement with the previous experimental results.55 It should also be noted that the presence of metal nanoparticles increases the melting range compared to that of the normal system from 325 K to 330 K.
The slope of the MSD plots of the confined molecules exhibits a sharp increase from 310 K to 330 K, as shown in Fig. 3b. The sharp increase in the self-diffusion of the confined molecules in Fig. 3c also indicates a first-order phase transition in the melting range of 310–330 K. In comparison with the normal system without the clusters, it can be found that the presence of the metal nanocluster decreases the self-diffusion value due to the stronger interactions between the octadecane molecules and the metal atoms.
According to Fig. 3d and e, the octadecane–octadecane and octadecane–CNT RDFs of the confined molecules in the cluster-enhanced system show important changes in the melting range. As shown in these figures, the octadecane–octadecane and octadecane–CNT RDFs show a sharp increase in the melting range, which represents the strengthening of the molecule–molecule and molecule–CNT interactions in the melting range. However, the octadecane–octadecane interaction decreases at a higher temperature (T = 340 K). We also present the average end-to-end distance of the confined octadecane molecules in the Cu-EPCM system at different temperatures in Fig. 3f. The melting temperature cannot be distinguished using the end-to-end distance, which is due to the presence of the metal cluster. The end-to-end distance does not seem to decrease significantly for the metal-encapsulated CNT systems in the melting range. This is due to the fact that the alkane–nanocluster interaction prevents the alkane molecule from twisting (or roll), and therefore, its length does not decrease after the melting range. However, the next sections show that the average end-to-end distance decreases in the melting range for the nanocluster systems with weaker interactions than Cu.
The MSD plots of the confined molecules and the self-diffusion values also exhibit a sharp increase at 310 K and 325 K in Fig. S6b and c,† respectively. The octadecane–octadecane and octadecane–CNT RDFs in Fig. S6d and e† also confirm the change in the behavior of the confined molecules in the melting range. We also present the average end-to-end distance of the confined molecules in the (25,0) CNT at different temperatures in Fig. S6f.† According to this figure, the sharp decrease in the end-to-end distance also confirms the melting range from 310 to 325 K, which is consistent with the other results.
According to Fig. S7d and e,† the octadecane–octadecane and octadecane–CNT RDFs also confirm the change in the behavior of the confined molecules in the melting range. The octadecane–octadecane interaction also increases in the melting range, whereas it decreases at a higher temperature (340 K). We also present the average end-to-end distance of the confined molecules in the (25,0) CNT at different temperatures in Fig. S7f.† This figure indicates the sharp changes in the end-to-end distance in the melting range of around 300 to 340 K.
3.7 The effect of Al doping in CNT
In this system, 32 Al atoms were doped in the (25,0) CNT randomly. Then, 17 octadecane molecules were positioned in the middle of the capped metal doped-CNT. According to the configurational energy and corresponding snapshots in Fig. 4a, the temperature range for the phase transition is from 310 K to 335 K, which is less than the melting point of the octadecane molecules in the normal zigzag CNT. The MSD plots and the self-diffusion values also confirm the melting range in Fig. 4b and c, respectively (however, the dynamic results show the starting melting point of 305 K). The self-diffusion of the confined molecules in the metal-doped CNT is lower than that in the normal CNT, which is due to the stronger interactions between the confined molecules and the Al-doped atoms. According to Fig. 4d and e, the octadecane–octadecane and octadecane–CNT RDFs of the confined molecules in the cluster-enhanced system show important changes in the melting range. According to these figures, the octadecane–octadecane interaction decreases with an increase in temperature, whereas the octadecane–CNT interaction increases with an increase in temperature. We also present the average end-to-end distance of the confined octadecane molecules in the Al-doped system at different temperatures in Fig. 4f. As shown in this figure, the end-to-end distance decreases around the melting range, confirming the other results.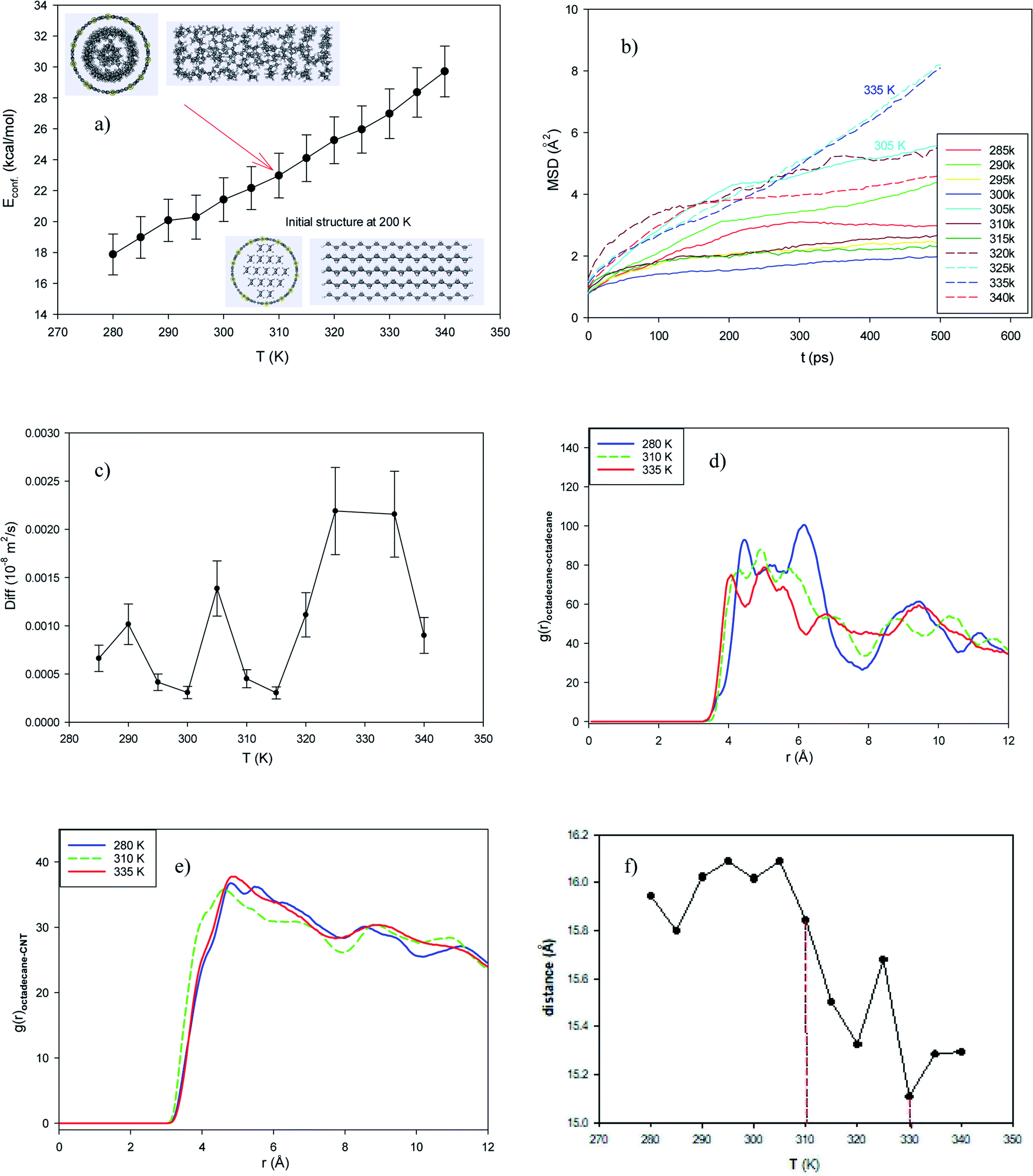 | ||
| Fig. 4 Al-doped system consisting of 17 octadecane molecules in the Al-doped CNT. (a) Configurational energy, (b) MSD, (c) self-diffusion coefficient, (d) octadecane–octadecane RDF, (e) octadecane–CNT RDF, and (f) average end-to-end distance. | ||
3.8 Comparison of the cluster structures in different systems
To examine the structures of the metal clusters in the different systems, the octadecane–cluster and cluster–cluster RDFs are presented at different temperatures in Fig. S8.† The following trend can be observed for the metal–metal RDFs: Cu–Cu > Ag–Ag > Al–Al, which is in good agreement with the order of the interaction potential of the Sutton–Chen model (parameter ε in Table S1†).46,47 Due to the same reason, the snapshots of the metal clusters in Fig. 5 show that the silver and aluminium clusters (unlike the copper cluster) are distributed along the CNT. Although the interaction potential of Cu is stronger than that of Ag and Al, the Ag and Al cluster systems exhibit higher cluster–octadecane RDFs than the Cu metal system. This is due to the fact that the silver and aluminium clusters (unlike the copper cluster) are distributed between the octadecane molecules along the CNT, and therefore the octadecane–metal interactions in the Ag and Al systems are greater than that in the Cu system. It is also shown that the octadecane–metal RDFs decrease with an increase in temperature, which indicates the weakening of the alkane–metal interactions with an increase in temperature.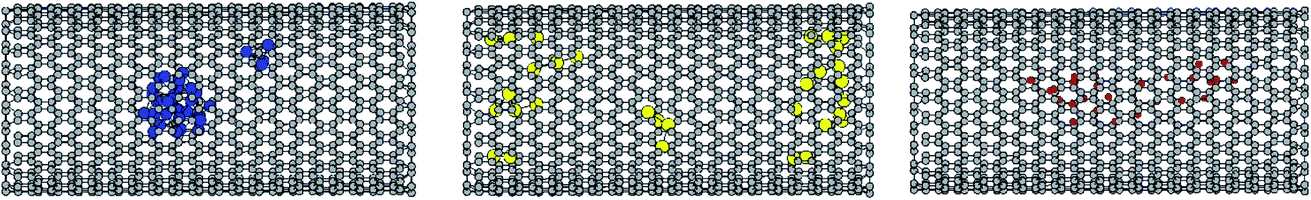 | ||
| Fig. 5 Snapshots of the metal clusters in the different systems at their melting ranges. The Cu, Al, and Ag atoms are in blue, yellow, and red, respectively. | ||
3.9 Comparison of the melting ranges of the different systems
According to the obtained results from the different nanosystems, it is shown that the determination of the melting point of the encapsulated alkane molecules is not an easy process and cannot be distinguished using only the energy curves because they show fluctuations in their properties and exhibit a range of melting temperatures. The MSD, self-diffusion, and RDF curves should be also considered to determine the melting range more accurately. Recently, Rao et al.52 investigated the melting process of nanocapsulated PCMs using MD simulations. They constructed the EPCMs using n-octadecane molecules as the core and SiO2 as the shell. However, they did not report the exact melting point and only studied the melting range of the confined octadecane molecules by observing the change in the MSD and diffusion values. Previous experimental and MD simulations on bulk octadecane have also reported melting ranges and not melting points.56,57The melting ranges of the different systems are compared in Fig. 6. Due to the larger accessible volume for the confined octadecane molecules, the longer (25,0) CNT has a lower melting point than that of the normal (25,0) CNT system. It is also shown that the (30,0) CNT, armchair CNT, and icosane systems have higher melting ranges than that of the normal (25,0) system. It is also shown that the metal cluster systems have a lower starting point of melting than that of the normal system. This result is in good agreement with the experimental results of Alomair et al.55 They showed that the presence of nanoparticles accelerates the melting process of phase change materials. It is observed that the Al cluster system has the lowest starting melting point among the studied systems.
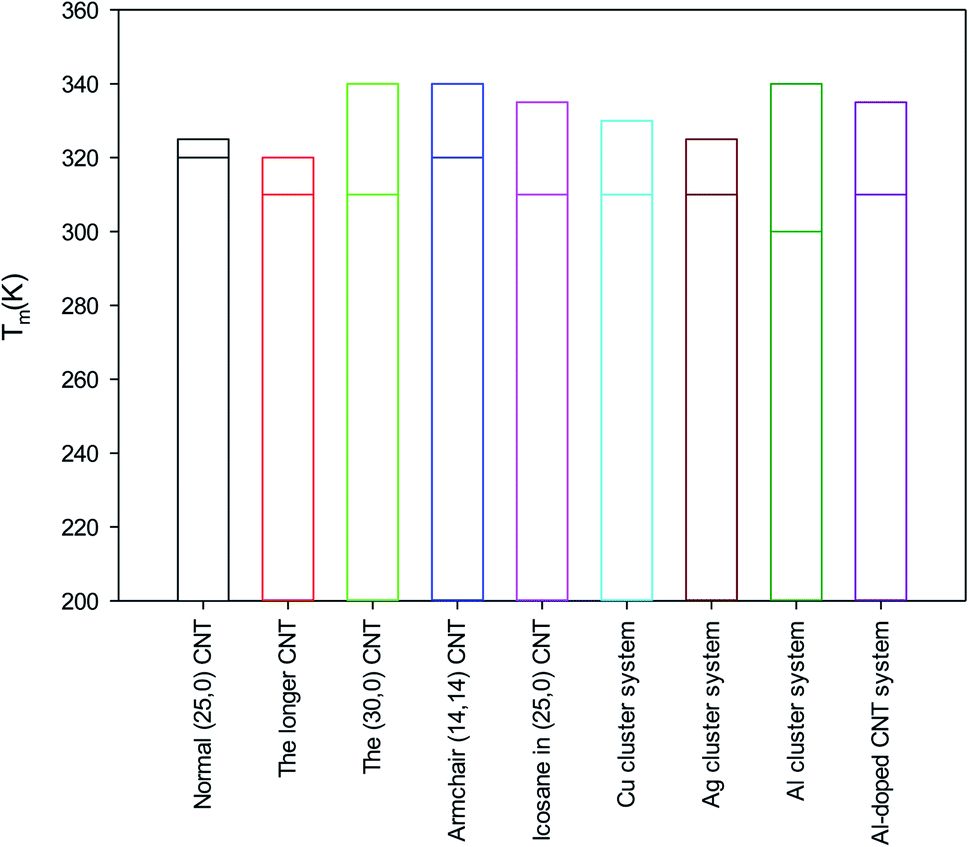 | ||
| Fig. 6 Comparison of the melting ranges for the different systems in this work. | ||
4. Concluding remarks
In this research, EPCM systems were constructed with 17 alkane molecules as the core material and a graphene-capped CNT as the shell. We investigated the effects of different parameters including the CNT diameter, length, and chirality and the length of the alkane chain. We also investigated metal nanocluster-enhanced PCM systems using Cu, Ag, and Al clusters. The following important results were obtained:(1) The energy of the metal nanocluster-enhanced systems is lower (more negative) than that of the other systems, which is due to the stronger metal interactions.
(2) Due to the larger accessible volume for the confined octadecane molecules, the longer (25,0) CNT has a lower melting point than that of the normal (25,0) CNT system. It was also shown that the (30,0) CNT, armchair CNT, and icosane systems have higher melting ranges than that of the normal (25,0) system.
(3) Our dynamics investigations using MSD curves and self-diffusion coefficients confirmed the melting ranges obtained using the configurational energy curves and also indicated that an increase in the CNT diameter and length increases the diffusion coefficient. The presence of metal clusters also decreases the diffusion values due to the stronger interactions with the confined molecules. The self-diffusion of the confined molecules in the metal-doped CNT is also smaller than that of the normal CNT.
(4) According to our structural investigations, the octadecane–octadecane and octadecane–CNT RDFs of the confined molecules indicate important changes in the melting range, which exhibit significant changes in their interactions in the melting range.
(5) Although the interaction potential of Cu is stronger than that of Ag and Al, the Ag and Al cluster systems exhibit higher cluster–octadecane RDFs than the Cu metal system. This is due to the fact that the silver and aluminium clusters (unlike the copper cluster) are distributed between the octadecane molecules along the CNT, and therefore the octadecane-metal interactions in the Ag and Al systems are greater than that in the Cu system.
(6) The metal cluster systems have a smaller starting melting point than that of the normal system. It was shown that the Al cluster system has the lowest starting melting point among all the studied systems.
(7) The end-to-end distance of the confined molecules decreases with an increase in temperature, which obeys the thermodynamic theory that the order parameter decreases with the melting process. The sharp decrease in the average end-to-end distance of the confined molecules around the melting temperature also confirmed the melting range obtained using the energy, MSD, diffusion, and RDF results.
Conflicts of interest
There are no conflicts to declare.References
- M. Zhang, C. Wang, A. Luo, Z. Liu and X. Zhang, Molecular dynamics simulation on thermophysics of paraffin/EVA/graphene nanocomposites as phase change materials, Appl. Therm. Eng., 2020, 166, 114639 CrossRef CAS.
- S. Zhang, L. Pu, L. Xu, R. Liu and Y. Li, Melting performance analysis of phase change materials in different finned thermal energy storage, Appl. Therm. Eng., 2020, 115425 CrossRef.
- T. Xiong, L. Zheng and K. W. Shah, Nano-enhanced phase change materials (NePCMs): A review of numerical simulations, Appl. Therm. Eng., 2020, 115492 CrossRef CAS.
- Z. H. Rao, S. Wang, M. Wu, Y. Zhang and F. Li, Molecular dynamics simulations of melting behavior of alkane as phase change materials slurry, Energy Convers. Manage., 2011, 64, 152–156 CrossRef.
- J.-F. Su, X.-Y. Wang, S.-B. Wang, Y.-H. Zhao and Z. Huang, Fabrication and properties of microencapsulated-paraffin/gypsum-matrix building materials for thermal energy storage, Energy Convers. Manage., 2012, 55, 101–107 CrossRef CAS.
- Z. H. Rao and S. F. Wang, A review of power battery thermal energy management, Renewable Sustainable Energy Rev., 2011, 15(9), 4554–4571 CrossRef CAS.
- Z. H. Rao, S. F. Wang and G. Q. Zhang, Simulation and experiment of thermal energy management with phase change material for ageing LiFePO(4) power battery, Energy Convers Manage, 2011, 52(12), 3408–3414 CrossRef CAS.
- Z. Rao, S. Wang and F. Peng, Self diffusion and heat capacity of n-alkanes based phase change materials: a molecular dynamics study, Int. J. Heat Mass Transfer, 2013, 64, 581–589 CrossRef CAS.
- A. Sari, Form-stable paraffin/high density polyethylene composites as solid–liquid phase change material for thermal energy storage: preparation and thermal properties, Energy Convers. Manage., 2004, 45(13–14), 2033–2042 CrossRef CAS.
- B. He, V. Martin and F. Setterwall, Phase transition temperature ranges and storage density of paraffin wax phase change materials, Energy, 2004, 29(11), 1785–1804 CrossRef CAS.
- A. Sari and A. Karaipekli, Thermal conductivity and latent heat thermal energy storage characteristics of paraffin/expanded graphite composite as phase change material, Appl. Therm. Eng., 2007, 27(8–9), 1271–1277 CrossRef CAS.
- M. N. A. Hawlader, M. S. Uddin and M. M. Khin, Microencapsulated PCM thermal-energy storage system, Appl. Energy, 2003, 74(1–2), 195–202 CrossRef CAS.
- Y. Fang, S. Kuang, X. Gao and Z. Zhang, Preparation and characterization of novel nanoencapsulated phase change materials, Energy Convers. Manage., 2008, 49(12), 3704–3707 CrossRef CAS.
- G. Fang, H. Li, F. Yang, X. Liu and S. Wu, Preparation and characterization of nanoencapsulated n-tetradecane as phase change material for thermal energy storage, Chem. Eng. J., 2009, 153(1–3), 217–221 CrossRef CAS.
- S. Sinha-Ray, R. P. Sahu and A. L. Yarin, Nano-encapsulated smart tunable phase change materials, Soft Matter, 2011, 7(19), 8823–8827 RSC.
- C. Y. Zhao and G. H. Zhang, Review on microencapsulated phase change materials (MEPCMs): fabrication, characterization and applications, Renew. Sustain. Energy Rev., 2011, 15(8), 3813–3832 CrossRef CAS.
- M. Karkri, M. Lachheb, Z. Nógellová, B. Boh, B. Sumiga, M. A. AlMaadeed, A. Fethi and I. Krupa, Thermal properties of phase-change materials based on high-density polyethylene filled with micro-encapsulated paraffin wax for thermal energy storage, Energy and Buildings, 2015, 88, 144–152 CrossRef.
- V. V. Tyagi, S. C. Kaushik, S. K. Tyagi and T. Akiyama, Development of phase change materials based microencapsulated technology for buildings: a review, Renew Sustain Energy Rev, 2011, 15(2), 1373–1391 CrossRef CAS.
- M. A. Rady, A. S. Huzayyin, E. Arquis, P. Monneyron, C. Lebot and E. Palomo, Study of heat and mass transfer in a dehumidifying desiccant bed with macroencapsulated phase change materials, Renewable Energy, 2009, 34(3), 718–726 CrossRef CAS.
- C. Alkan, A. Sari and A. Karaipekli, Preparation, thermal properties and thermal reliability of microencapsulated n-eicosane as novel phase change material for thermal energy storage, Energy Convers. Manage., 2011, 52(1), 687–692 CrossRef CAS.
- D. Zhang, M. G. Li, Y. Zhang and Y. H. Xu, Effect of different amounts of surfactant on characteristics of nanoencapsulated phase-change materials, Polym. Bull., 2011, 67(3), 541–552 CrossRef.
- Z.-H. Chen, F. Yu, X.-R. Zeng and Z.-G. Zhang, Preparation, characterization and thermal properties of nanocapsules containing phase change material ndodecanol by miniemulsion polymerization with polymerizable emulsifier, Appl. Energy, 2012, 91(1), 7–12 CrossRef CAS.
- J. K. Choi, J. G. Lee, J. H. Kim and H. S. Yang, Preparation of microcapsules containing phase change materials as heat transfer media by in situ polymerization, J. Ind. Eng. Chem., 2001, 7(6), 358–362 CAS.
- C. C. Chang, Y. L. Tsai, J. J. Chiu and H. Chen, Preparation of phase change materials microcapsules by using PMMA network-silica hybrid shell via sol-gel process, J. Appl. Polym. Sci., 2009, 112(3), 1850–1857 CrossRef CAS.
- J. S. Cho, A. Kwon and C. G. Cho, Microencapsulation of octadecane as a phase-change material by interfacial polymerization in an emulsion system, Colloid Polym. Sci., 2002, 280(3), 260–266 CrossRef CAS.
- X. Qiu, W. Li, G. Song, X. Chu and G. Tang, Microencapsulated n-octadecane with different methylmethacrylate-based copolymer shells as phase change materials for thermal energy storage, Energy, 2012, 46(1), 188–199 CrossRef CAS.
- Y. Wang, Z. Liu, X. Niu and X. Ling, Preparation, Characterization, and Thermal Properties of Microencapsulated Phase Change Material for Low-Temperature Thermal Energy Storage, Energy Fuels, 2019, 33(2), 1631–1636 CrossRef CAS.
- C. Nie, X. Tong, S. Wu, S. Gong and D. Peng, Paraffin confined in carbon nanotubes as nano-encapsulated phase change materials: experimental and molecular dynamics studies, RSC Adv., 2015, 5(113), 92812–92817 RSC.
- S. Himran, A. Suwono and G. A. Mansoori, Characterization of alkanes and paraffin waxes for application as phase change energy storage medium, Energy Sources, 1994, 16(1), 117–128 CrossRef CAS.
- T. Keshavarzi, R. Sohrabi and G. A. Mansoori, An analytic model for nano confined fluids phase-transition: applications for confined fluids in nanotube and nanoslit, J. Comput. Theor. Nanosci., 2006, 3(1), 134–141 CAS.
- G. A. Mansoori and S. A. Rice, Confined fluids: Structure, properties and phase behavior, in Advances in Chemical Physics, John Wiley & Sons, 2015, vol. 156, pp. 197–294 Search PubMed.
- G. A. Mansoori and T. E. Keshavarzi, Phase Transitions in Small/Nano Systems, in Phytonanotechnology, ed. M. Ghorbanpour and S. H. Wani, Academic Press, 2019, ch. 1, adv Search PubMed.
- E. Vardoulakis, D. Karamanis, M. N. Assimakopoulos and G. Mihalakakou, Solar cooling with aluminium pillared clays, Sol. Energy Mater. Sol. Cells, 2011, 95(8), 2363–2370 CrossRef CAS.
- D. Karamanis and E. Vardoulakis, Application of zeolitic materials prepared from fly ash to water vapor adsorption for solar cooling, Appl. Energy, 2012, 97, 334–339 CrossRef CAS.
- X. T. Tran, M. Hussain and H. T. Kim, Facile and fast synthesis of a reduced graphene oxide/carbon nanotube/iron/silver hybrid and its enhanced performance in catalytic reduction of 4–nitrophenol, Solid State Sci., 2020, 100, 106107 CrossRef CAS.
- I. T. Todorov, W. Smith, K. Trachenko and M. T. Dove, DL_POLY_3: new dimensions in molecular dynamics simulations via massive parallelism, J. Mater. Chem., 2006, 16, 1611 RSC.
- Z. Rao, S. Wang, M. Wu, Y. Zhang and F. Li, Molecular dynamics simulations of melting behavior of alkane as phase change materials slurry, Energy Convers. Manage., 2012, 64, 152–156 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- S. W. Siu, K. Pluhackova and R. A. Böckmann, Optimization of the OPLS-AA force field for long hydrocarbons, J. Chem. Theory Comput., 2012, 8(4), 1459–1470 CrossRef CAS.
- M. Neek-Amal, R. Asgari and M. R. Tabar, The formation of atomic nanoclusters on graphene sheets, Nanotechnology, 2009, 20, 135602 CrossRef CAS PubMed.
- K. Malek and M. Sahimi, Molecular dynamics simulations of adsorption and diffusion of gases in silicon-carbide nanotubes, J. Chem. Phys., 2010, 132, 014310 CrossRef.
- T. Qian, J. Li, X. Min, W. Guan, Y. Deng and L. Ning, Enhanced thermal conductivity of PEG/diatomite shape-stabilized phase change materials with Ag nanoparticles for thermal energy storage, J. Mater. Chem. A, 2015, 3(16), 8526–8536 RSC.
- M. Jafarian, M. Omid, M. Khanali and M. MokhtariMotameniShirvan, Thermal Conductivity Enhancement of Phase Change Material for Thermal Energy Storage Using Nanotechnology, Iran. J. Anim. Biosyst., 2019, 50(2), 319–329 Search PubMed.
- H. Akbarzadeh and H. Yaghoubi, Molecular dynamics simulations of silver nanocluster supported on carbon nanotube, J. Colloid Interface Sci., 2014, 418, 178–184 CrossRef CAS PubMed.
- H. Akbarzadeh, H. Yaghoubi, A. N. Shamkhali and F. Taherkhani, CO adsorption on Ag nanoclusters supported on carbon nanotube: a molecular dynamics study, J. Phys. Chem. C, 2014, 118(17), 9187–9195 CrossRef CAS.
- T. Çagin, Y. Qi, H. Li, Y. Kimura, H. Ikeda, W. L. Johnson and W. A. Goddard III, The quantum Sutton-Chen many-body potential for properties of fcc metals, MRS Symposium Ser, 1999, 554, 43 CrossRef.
- P. Puri and V. Yang, Molecular Dynamics Study of Melting of Nano Aluminum Particles, in 45th AIAA Aerospace Sciences Meeting and Exhibit, 2007, p. 1429 Search PubMed.
- M. Abbaspour, M. N. Jorabchi, H. Akbarzadeh, S. Salemi and R. Ebrahimi, Molecular dynamics simulation of anticancer drug delivery from carbon nanotube using metal nanowires, J. Comput. Chem., 2019, 40, 2179 CAS.
- H. Akbarzadeh, M. Abbaspour, S. Salemi and S. Abdollahzadeh, Investigation of the melting of ionic liquid emimPF6 confined inside carbon nanotubes using molecular dynamics simulations, RSC Adv., 2015, 5(5), 3868–3874 RSC.
- A. Yafei, J. Yong, S. Jing and W. Deqing, Microencapsulation of n-hexadecane as phase change material by suspension polymerization, e-Polym., 2007, 7(1), 1–9 Search PubMed.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquid, Oxford, Clarendon, 1997 Search PubMed.
- Z. Rao, S. Wang and F. Peng, Self diffusion of the nano-encapsulated phase change materials: a molecular dynamics study, Appl. Energy, 2012, 100, 303–308 CrossRef CAS.
- D. C. Douglass and D. W. McCall, Diffusion in paraffin hydrocarbons, J. Phys. Chem. A, 1958, 62(9), 1102–1107 CrossRef CAS.
- H. Akbarzadeh and M. Abbaspour, Effects of diameter and chirality on structural and dynamical behavior of [EMIM][PF6] encapsulated in carbon nanotube: a molecular dynamics study, J. Mol. Liq., 2015, 212, 423–429 CrossRef CAS.
- M. A. Alomair, Y. A. Alomair, H. A. Abdullah, S. Mahmud and S. Tasnim, Nanoparticle enhanced phase change material in latent heat thermal energy storage system: an experimental study, in Proc. Int. Conf. Energy Harvest. Storage, Transf, 2017, pp. 1–5 Search PubMed.
- A. Hassan, M. Shakeel Laghari and Y. Rashid, Micro-encapsulated phase change materials: a review of encapsulation, safety and thermal characteristics, Sustainability, 2016, 8(10), 1046 CrossRef.
- R. Zhong-Hao, L. Xin-Jian, Z. Rui-Kai, L. Xiang, W. Chang-Xing, W. Hao-Dong and L. Yi-Min, A comparative study on the self diffusion of n-octadecane with crystal and amorphous structure by molecular dynamics simulation, Chin. Phys. Lett., 2014, 31(1), 010501 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02033a |
| This journal is © The Royal Society of Chemistry 2021 |