
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing photoluminescence mechanisms of single CsPbBr3/Cs4PbBr6 core/shell perovskite nanocrystals†
Huafeng Ding‡
a,
Yansu Shan‡a,
Jizhou Wang‡b,
Qinfeng Xu *a,
Jing Hana,
Mengmeng Jiaoa,
Kunjian Caoa,
Mingliang Liua,
Haifeng Mu*a,
Shufang Zhanga and
Chuanlu Yang*a
*a,
Jing Hana,
Mengmeng Jiaoa,
Kunjian Caoa,
Mingliang Liua,
Haifeng Mu*a,
Shufang Zhanga and
Chuanlu Yang*a
aDepartment of Physics and Optoelectronic Engineering, Ludong University, Yantai 264025, People's Republic of China. E-mail: xuqf5678@163.com; hai-fengmu@163.com; yangchuanlu@126.com
bLanzhou Institute of Space Technology and Physics, Lanzhou 730000, People's Republic of China
First published on 13th September 2021
Abstract
CsPbBr3 nanocrystals (NCs) encapsulated by Cs4PbBr6 has attracted extensive attention due to good stability and high photoluminescence (PL) emission efficiency. However, the origin of photoluminescence (PL) emission from CsPbBr3/Cs4PbBr6 composite materials has been controversial. In this work, we prepare CsPbBr3/Cs4PbBr6 core/shell nanoparticles and firstly study the mechanism of its photoluminescence (PL) at the single-particle level. Based on photoluminescence (PL) intensity trajectories and photon antibunching measurements, we have found that photoluminescence (PL) intensity trajectories of individual CsPbBr3/Cs4PbBr6 core/shell NCs vary from the uniform longer periods to multiple-step intensity behaviors with increasing excitation level. Meanwhile, second-order photon correlation functions exhibit single photon emission behaviors especially at lower excitation levels. However, the PL intensity trajectories of individual Cs4PbBr6 NCs demonstrate apparent “burst-like” behaviors with very high values of g2(0) at any excitation power. Therefore, the distinguishable emission statistics help us to elucidate whether the photoluminescence (PL) emission of CsPbBr3/Cs4PbBr6 core/shell NCs stems from band-edge exciton recombination of CsPbBr3 NCs or intrinsic Br vacancy states of Cs4PbBr6 NCs. These findings provide key information about the origin of emission in CsPbBr3/Cs4PbBr6 core/shell nanoparticles, which improves their utilization in the further optoelectronic applications.
1. Introduction
All-inorganic perovskite nanocrystals (NCs) have received extensive attention in solar cell and light-emitting devices due to their facile synthesis1–3 and excellent photophysical properties such as high photoluminescence quantum yields (PLQYs),4 narrow emission width,5,6 and tunable emission features that cover the full visible range.7,8 As a result, they have shown promising applications in optoelectronics, including photovoltaics,9,10 lasing,11,12 light-emitting diodes13–15 and photodetectors.16,17 However, the photoluminescence quantum yields (PLQYs) of CsPbX3 NCs are greatly reduced when they are in the form of agglomerates compared to well-dispersed colloidal NCs.18 At present, an effective approach to solve these problems is to modify the surface structures of CsPbX3 NCs and alleviate the stability.19,20 CsPbX3 perovskite NCs coated with heterostructure shell have been reported, such as CsPbX3/ZnS core/shell NCs,21 CsPbBr3/SiO2 and CsPbBr3/Ta2O5.22 Recently, it is unsurprising that Cs4PbBr6 lattice spacing could match the cubic CsPbBr3 lattice constants and this match satisfies the CsPbBr3 NCs inside the Cs4PbBr6 matrix, with enhanced passivation of the surfaces without additional strain on the NCs. Therefore, CsPbBr3 coated with the same chemical constituents Cs4PbBr6 has been proven to be an effective way to maintain the highly efficient photoluminescence quantum yields (PLQYs) and simultaneously improve the stabilities.23,24Despite the synthesis of CsPbBr3 perovskite NCs encapsulated by Cs4PbBr6 material enables high emission efficiency and excellent stability, the photoluminescence origin of CsPbBr3/Cs4PbBr6 core/shell perovskite NCs have attracted a lot of attention as well as intensive debates. The debates on the origin of bright photoluminescence emission involve two opposing opinions. Due to the extreme similarity of its photoluminescence emission with that of CsPbBr3 nanocrystals, it is believed encapsulated CsPbBr3 nanocrystals are responsible for the highly efficient photoluminescence.25–30 However, some researchers have attributed the photoluminescence emission to Br defects in the Cs4PbBr6 and regarding the photoluminescence emission as an intrinsic property of Cs4PbBr6.31–36 To fully exploit the origin of their PL emission, it is vital to deeply gain the important information about the interaction with the encapsulating media and complex PL dynamics process. Therefore, studies of the PL properties at the single-particle level has been proved to be a very effective method for elucidate its PL origins.
In this report, we present a simple solution-processed chemical method to synthesize CsPbBr3/Cs4PbBr6 core/shell NCs and investigate the PL intensity trajectories from individual CsPbBr3/Cs4PbBr6 core/shell NCs and Cs4PbBr6 NCs by time-correlated single photon counting (TCSPC). We analyse PL lifetimes and second-order correlation functions g2(τ) at different excitation levels in order to explain the origin of their PL emission.
2. Experimental section
The CsPbBr3/Cs4PbBr6 core/shell perovskite nanocrystals were synthesized through the hot-injection method with detailed processes presented in ESI.† The resulting CsPbBr3 NCs solution was lowered to room temperature and 0.1 mmol (or 0.2 mmol) ZnBr2 was added to the flash, degassed at 50 °C for 20 min under vacuum. Afterward the temperature was raised to 70 °C under a nitrogen atmosphere, Cs-oleate with different amount (0.203 mmol corresponding to a shell thickness of 1.5 nm) was quickly injected. The solution was lowered to room temperature using a water bath after 3 min. The solution turned bright green again. To collect the NCs, the crude solution was then centrifuged at 8000 rpm for 5 min. After centrifugation, the supernatant was discarded and the CsPbBr3/Cs4PbBr6 core/shell NCs were prepared.For the single-dot measurements, CsPbBr3/Cs4PbBr6 core/shell NCs solution diluted in toluene has been mixed with PMMA (5wt%) and spin-coated onto a glass coverslip at the speed of 3000 rpm. The QDs density was kept below 0.1 μm−2 to allow us to probe single QD with detailed processes presented in ESI.†
3. Results and discussion
The CsPbBr3/Cs4PbBr6 core/shell perovskite nanocrystals were synthesized through a hot-injection method with detailed processes provided in experimental section (see ESI†). Fig. 1a indicates the TEM image of CsPbBr3/Cs4PbBr6 core/shell perovskite NCs. The TEM image of CsPbBr3/Cs4PbBr6 core/shell perovskite nanocrystals have a cubic shape and the core CsPbBr3 are coated by Cs4PbBr6 shell (inset in Fig. 1a). | ||
| Fig. 1 (a) TEM images of CsPbBr3/Cs4PbBr6 core/shell perovskite NCs and corresponding magnified individual core/shell NC (top right corner); (b) XRD patterns of Cs4PbBr6 NCs, CsPbBr3/Cs4PbBr6 core/shell NCs and CsPbBr3 NCs; (c) absorption and PL spectra of Cs4PbBr6, CsPbBr3/Cs4PbBr6 core/shell NCs and CsPbBr3 NCs; (d) photoluminescence excitation (PLE) spectra of Cs4PbBr6, CsPbBr3/Cs4PbBr6 core/shell NCs and CsPbBr3 NCs, the monitored wavelength for PLE is 520 nm and the excitation wavelength for PL is 405 nm. | ||
To further confirm the formation of CsPbBr3/Cs4PbBr6 core/shell NCs, the X-ray diffraction (XRD) spectroscopy was performed as shown in Fig. 1b. The CsPbBr3/Cs4PbBr6 core/shell NCs display a combination between the cubic CsPbBr3 phase and hexagonal Cs4PbBr6 phase. According to the results from X-ray diffraction spectroscopy (XRD), the main peaks should correspond to the cubic crystal, indicating that the CsPbBr3 still maintain their initial crystal phase. Meanwhile, the minor peaks can be ascribed to hexagonal phase of Cs4PbBr6, revealing the formation of Cs4PbBr6 shell. Therefore, it implies that the XRD of CsPbBr3/Cs4PbBr6 core/shell NCs is different from the simple mixtures of CsPbBr3 and Cs4PbBr6 in bulk.
The absorption and PL spectra of the CsPbBr3 NCs, CsPbBr3/Cs4PbBr6 core/shell NCs and Cs4PbBr6 NCs are presented in Fig. 1c. After the Cs4PbBr6 shell encapsulation, it was found that the additional characteristic peaks were located at 315 nm, which can be attributed to the absorption from Cs4PbBr6, suggesting the formation of Cs4PbBr6 shell structures. The PL peak of Cs4PbBr6 perovskite NCs displays an obvious red shift from 515 nm to 520 nm. Fig. 1d shows the Photoluminescence excitation (PLE) spectra of CsPbBr3/Cs4PbBr6 core/shell NCs emission at 515 nm. Compared with CsPbBr3/Cs4PbBr6 core/shell NCs, the Cs4PbBr6 exhibits a obvious sharp dip at the wavelength of 310 nm, where light is fully absorbed by the Cs4PbBr6. Therefore, it also demonstrate that CsPbBr3/Cs4PbBr6 core/shell NCs is different from Cs4PbBr6 NCs.
In order to get deep insights into the carrier dynamics induced by the core/shell structure, we investigate time-resolved photoluminescence (TRPL) decay behavior of CsPbBr3/Cs4PbBr6 core/shell NCs as shown in Fig. 2a. The average lifetime of CsPbBr3/Cs4PbBr6 core/shell NCs is 7.1 ns, which is much shorter than average lifetime 9.6 ns of pure CsPbBr3 NCs. This could be attribute to the thicker shell structures of Cs4PbBr6 that reduce the probability of carriers being captured by the surface traps due to the enhanced overlap of wave functions. According to analysis on absorption spectra, the CsPbBr3 core (2.4 eV, 515 nm) was coated by Cs4PbBr6 shell with large band gap (4.0 eV, 310 nm). Therefore, the photogenerated electron and hole will be confined in the core regions. The shell can passivate the surface states of the core and reduce nonradiative recombination pathways. After CsPbBr3 encapsulated with Cs4PbBr6 shell, the PLQYs was improved from 83.6% to 96.8% corresponding to a 13% improvement after shell encapsulation. Under the same excitation experimental conditions, the PL peak of CsPbBr3/Cs4PbBr6 core/shell NCs remains almost unaltered at 515 nm, showing a robust resistance to anion exchange reaction. Under continuous excitation by 405 nm laser, CsPbBr3/Cs4PbBr6 core/shell NCs show a much better stability than colloidal CsPbBr3 NCs. CsPbBr3/Cs4PbBr6 core/shell NCs retains about 77% emission intensity after illumination for 10 days and CsPbBr3 NCs retains less than 45% as shown in Fig. 2b, indicating improved PL stability in air compared to the pure CsPbBr3 NCs.
 | ||
| Fig. 2 (a) Time-resolved photoluminescence (TRPL) decays of CsPbBr3 NCs and CsPbBr3/Cs4PbBr6 core/shell NCs; (b) PLQYs of Cs4PbBr6/CsPbBr3 core/shell NCs and CsPbBr3 NCs exposed to air for different days under ambient conditions. | ||
Fluorescence blinking has been extensively investigated in semiconductor nanostructures, providing unique insight into their photo-excited carrier dynamics. Here, we firstly explored blinking trajectories of individual CsPbBr3/Cs4PbBr6 core/shell NCs with different excitation level, respectively. Fig. 3a and c present the PL intensity trajectories with obvious on states and gray states under the low excitation level of 〈N〉 = 0.1 and 〈N〉 = 0.3, respectively. Fig. 3b and d show single quantum emitters with low values g2(0)<0.5 corresponding to low excitation level of 〈N〉 = 0.1 and 〈N〉 = 0.3, respectively. However, the blinking behavior of the CsPbBr3/Cs4PbBr6 NCs visibly changed at high excitation level of 〈N〉 = 1.0. It is clearly seen that blinking trajectories is no longer represented by two-state emission but is multiple-step intensity emission as shown in Fig. 3e. PL lifetimes extracted from different intensity levels are composed of the fast (approximate 1 to 2 ns) and slow (approximate 9 to 10 ns) component with varying amplitudes as shown in Fig. 3f. Longer lifetime components predominate in the higher intensity bursts and the shorter ones in the low intensity intervals correspond to the on and gray states, respectively. The PL lifetime of on state obviously demonstrates the near band edge excitonic transitions. On the contrary, the PL lifetime of the gray state is much shorter and it indicates that the emission is likely coming from a charged excitonic state. These results correspond well with previously measured PL lifetimes for individual CsPbBr3 NCs.37,38
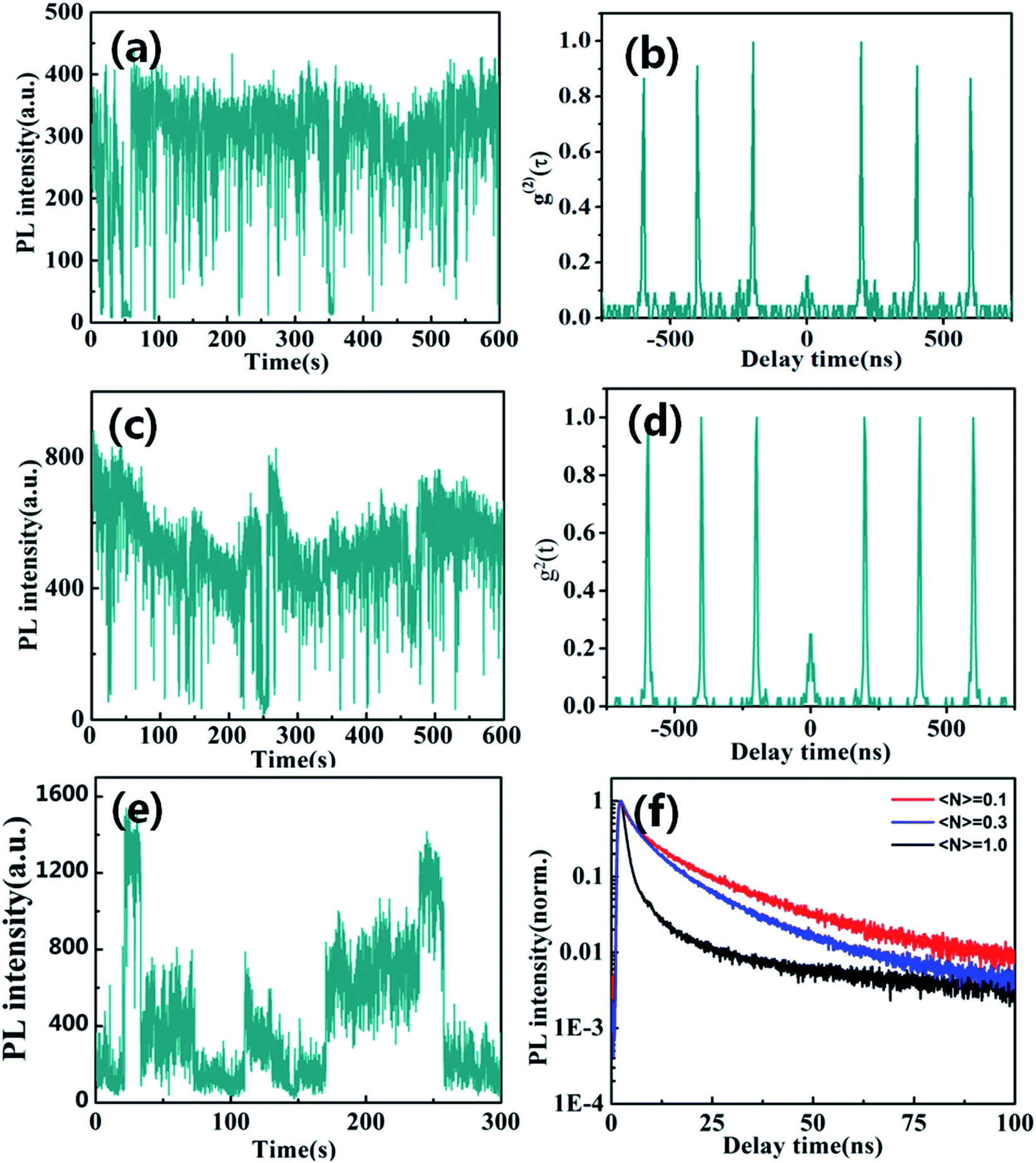 | ||
| Fig. 3 Blinking trajectories of individual Cs4PbBr6 NC at low (a) ∼〈N〉 = 0.1 and high excitation level (c) ∼〈N〉 = 0.5, (d) ∼〈N〉 = 1.0, respectively. Antibunching functions g2(t) at low (b) ∼〈N〉 = 0.1 and high (e) ∼〈N〉 = 1 excitation levels. (f) Corresponding PL lifetimes at different excitation levels. | ||
When individual CsPbBr3/Cs4PbBr6 core/shell NCs was excited with 〈N〉 = 1.3 higher excitation levels for longer times, the blinking trajectories demonstrate a very peculiar emission behavior as shown in Fig. 4a. It starts at the stable intensity level with 1000 to 1200 counts, typical for two state emission. After 100 seconds, the PL intensity starts to exhibit large jumps to the 400 to 1200 counts level. The lifetimes extracted from different intensity bursts demonstrate dramatically similar lifetimes and the zero delay time second-order correlation function g2(0) is approximate ∼1 as shown in Fig. 4b. Such behavior is very similar to that of Cs4PbBr6 NCs. These results thus confirm that the high efficiency PL emission of the CsPbBr3/Cs4PbBr6 core/shell NCs is from the core of CsPbBr3 NCs at lower excitation levels. However, it is clearly seen that the blinking behaviors of many CsPbBr3/Cs4PbBr6 core/shell NCs initially demonstrate two-state emission and then are burst-like behaviors at higher excitation level. Thus, we can suggest that laser-induced structural reorganization in CsPbBr3/Cs4PbBr6 core/shell NCs may lead to the formation of PL-active Br vacancy states at higher excitation level. Hence, these results confirm that CsPbBr3 NCs encapsulated by Cs4PbBr6 shell structures exhibit different PL characteristics in comparison to pure CsPbBr3 QDs.
 | ||
| Fig. 4 (a) Blinking trajectories and (b) antibunching functions g2(t) of individual CsPbBr3/Cs4PbBr6 core/shell NC at high excitation level (〈N〉 = 1.3). | ||
Compared with the above CsPbBr3/Cs4PbBr6 core/shell NCs, Fig. 5a presents the PL intensity trajectory of individual Cs4PbBr6 NCs at the 〈N〉 = 0.1 excitation level. The PL intensity trajectory shows obvious noisiness behaviors that attribute to charge carrier recombination from the trap states located on the interior and surface of the Cs4PbBr6 NCs.38 Because Br vacancies can act as a trap state and the trapping of electrons recombinate with holes, making the exciton recombination centers more efficient for green emission in Cs4PbBr6 NCs. We found that the majority of individual Cs4PbBr6 NCs still do not exhibit single photon properties with high g2(0) values at low excitation levels 〈N〉 = 0.1 as shown in Fig. 5b. The PL lifetimes are fitted with bi-exponential functions with a short lifetimes of 2–3 ns and a long lifetimes of 11–13 ns as shown in Fig. 5f. The short and long components can be ascribed to the excitons located on the surface and interior of the Cs4PbBr6 NCs, respectively. Previous studies have shown that CsPbBr3 NCs exhibit shorter PL lifetime compared to the Cs4PbBr6 NCs due to the presence of different deactivation pathways.39,40
 | ||
| Fig. 5 Blinking trajectories of individual Cs4PbBr6 NC at low (a) ∼〈N〉 = 0.1 and high excitation level (c) ∼〈N〉 = 0.5, (d) ∼〈N〉 = 1.0, respectively. Antibunching functions g2(t) at low (b) ∼〈N〉 = 0.1 and high (e) ∼〈N〉 = 1 excitation levels. (f) Corresponding PL lifetimes at different excitation levels. | ||
As the excitation power further increases, the blinking behavior of Cs4PbBr6 NC visibly changes as shown in Fig. 5c and d. We observe a multitude intensity spikes that closely resemble the typical blinking behavior of individual molecular emitters. The PL intensity superlinearly increase due to the activation of more emissive centers in the Cs4PbBr6 NCs. Moreover, PL lifetimes extracted from different intensity bursts in the blinking trace are very close, suggesting their similar origins. Therefore, it has been proposed that intrinsic defect states within the band gap may serve as radiative recombination centers for the observed emission of Cs4PbBr6 NCs.41,42
4. Conclusions
In summary, CsPbBr3/Cs4PbBr6 core/shell NCs were prepared and their PL properties were investigated at individual particle level. These results demonstrate that the Cs4PbBr6 shell with large band gap is helpful to confine the exciton and reach high PLQYs and high stability. The PL emission of CsPbBr3/Cs4PbBr6 core/shell structures could be attributed to direct band-edge electron–hole radiative recombination in the CsPbBr3 NCs, while the green emission (∼2.4 eV) in Cs4PbBr6 originates from the exciton recombination at Br vacancy instead of a direct band-to-band transition. Therefore, the different carrier dynamics studies allow us to distinguish the origin of green emission between Cs4PbBr6 NCs and CsPbBr3 NCs. The fundamental knowledge about interaction of excitons with interfaces in CsPbBr3/Cs4PbBr6 core/shell NCs will provide new physical insights into the design and optimization of novel optoelectronics devices.Author contributions
Q. F. Xu, S. F. Zhang, C. L. Yang conceived and planed the project. Y H. F. Ding, Y. S. Shan performed and analyzed the spectroscopy experiments. J. Z. Wang, J. Han, M. M. Jiao, K. J. Cao, H. F. Mu, M. L. Liu synthesized the materials. Q. F. Xu wrote the paper and discussed with other authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the financial support of the Natural Science Foundation of China (No. 61905106), Shandong Province Natural Science Foundation (No. ZR2019MF057 and ZR2019MA066), Science and Technology Program of Shandong Province (J18KA222) and the Taishan Scholars Project of Shandong Province (tsqn201812098 and ts201511055).References
- T. Zhang, M. I. Dar, G. Li, F. Xu, N. Guo, M. Grätzel and Y. Zhao, Sci. Adv., 2017, 3, e1700841 CrossRef PubMed.
- J. Y. Sun, F. T. Rabouw, X. F. Yang, X. Y. Huang, X. P. Jing, S. Ye and Q. Y. Zhang, Adv. Funct. Mater., 2017, 27, 1704371 CrossRef.
- X. Li, F. Cao, D. Yu, J. Chen, Z. Sun, Y. Shen, Y. Zhu, L. Wang, Y. Wei, Y. Wu and H. Zeng, Small, 2017, 13, 1603996 CrossRef PubMed.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- A. Waleed, M. M. Tavakoli, L. Gu, Z. Wang, D. Zhang, A. Manikandan, Q. Zhang, R. Zhang, Y.-L. Chueh and Z. Fan, Nano Lett., 2017, 17, 523–530 CrossRef CAS PubMed.
- Z. K. Liu, Y. Bekenstein, X. C. Ye, S. C. Nguyen, J. Swabeck, D. D. Zhang, S. T. Lee, P. D. Yang, W. L. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS PubMed.
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao, X. Yi, G. Zhu, H. Lv, L. Ma, T. Chen, Z. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2017, 139, 2852 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Y. Wang, Z. Chen, F. Deschler, X. Sun, T.-M. Lu, E. A. Wertz, J.-M. Hu and J. Shi, ACS Nano, 2017, 11, 3355–3364 CrossRef CAS PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- W. Zhang, L. Peng, J. Liu, A. Tang, J.-S. Hu, J. Yao and Y. S. Zhao, Adv. Mater., 2016, 28, 4040–4046 CrossRef CAS PubMed.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- X. Li, D. Yu, J. Chen, Y. Wang, F. Cao, Y. Wei, Y. Wu, L. Wang, Y. Zhu, Z. Sun, J. Ji, Y. Shen, H. Sun and H. Zeng, ACS Nano, 2017, 11, 2015–2023 CrossRef CAS PubMed.
- H. Lu, W. Tian, F. Cao, Y. Ma, B. Gu and L. Li, Adv. Funct. Mater., 2016, 26, 1296–1302 CrossRef CAS.
- Y. Kim, E. Yassitepe, O. Voznyy, R. Comin, G. Walters, X. Gong, P. Kanjanaboos, A. F. Nogueira and E. H. Sargent, ACS Appl. Mater. Interfaces, 2015, 7, 25007–25013 CrossRef CAS PubMed.
- D. Chen, G. Fang and X. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 40477–40487 CrossRef CAS PubMed.
- S. Ye, M. H. Yu, W. Yan, J. Song and J. L. Qu, J. Mater. Chem. C, 2017, 5, 8187–8193 RSC.
- W. Chen, J. Hao, W. Hu, Z. Zang, X. Tang, L. Fang, T. Niu and M. Zhou, Small, 2017, 13, 1604085 CrossRef PubMed.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. L. Zheng, Z. Y. Yang and E. H. Sargent, Adv. Mater., 2017, 29, 6 CrossRef PubMed.
- Y. M. Chen, Y. Zhou, Q. Zhao, J. Y. Zhang, J. P. Ma, T. T. Xuan, S. Q. Guo, Z. J. Yong, J. Wang, Y. Kuroiwa, C. Moriyoshi and H. T. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 15905–15912 CrossRef CAS PubMed.
- Q. A. Akkerman, A. L. Abdelhady and L. Manna, J. Phys. Chem. Lett., 2018, 9, 2326–2337 CrossRef CAS PubMed.
- D. Han, H. L. Shi, W. M. Ming, C. K. Zhou, B. W. Ma, B. Saparov, Y. Z. Ma, S. Y. Chen and M. H. Du, J. Mater. Chem. C, 2018, 6, 6398–6405 RSC.
- Z. K. Liu, Y. Bekenstein, X. C. Ye, S. C. Nguyen, J. Swabeck, D. D. Zhang, S. T. Lee, P. D. Yang, W. L. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS PubMed.
- C. de Weerd, J. H. Lin, L. Gomez, Y. Fujiwara, K. Suenaga and T. Gregorkiewicz, J. Phys. Chem., 2017, 121, 19490–19496 Search PubMed.
- L. Yang, D. M. Li, C. Wang, W. Yao, H. Wang and K. X. Huang, J. Nanopart. Res., 2017, 19, 13 CrossRef.
- J. M. Bao and V. G. Hadjiev, Nano-Micro Lett., 2019, 11, 18 CrossRef PubMed.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 840–845 CrossRef CAS.
- J. Yin, H. Yang, K. Song, A. M. El-Zohry, Y. Han, O. M. Bakr, J. L. Bredas and O. F. Mohammed, J. Phys. Chem. Lett., 2018, 9, 5490–5495 CrossRef CAS PubMed.
- J. Yin, Y. H. Zhang, A. Bruno, C. Soci, O. M. Bakr, J. L. Bredas and O. F. Mohammed, ACS Energy Lett., 2017, 2, 2805–2811 CrossRef CAS.
- J. H. Cha, J. H. Han, W. Yin, C. Park, Y. Park, T. K. Ahn, J. H. Cho and D. Y. Jung, J. Phys. Chem. Lett., 2017, 8, 565–570 CrossRef CAS PubMed.
- S. Seth and A. Samanta, J. Phys. Chem. Lett., 2017, 8, 4461–4467 CrossRef CAS PubMed.
- X. Chen, D. Q. Chen, J. N. Li, G. L. Fang, H. C. Sheng and J. S. Zhong, Dalton Trans., 2018, 47, 5670–5678 RSC.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924 CrossRef CAS PubMed.
- Y. Zhang, T. Guo, H. Yang, R. Bose, L. Liu, J. Yin, Y. Han, O. M. Bakr, O. F. Mohammed and A. V. Malko, Nat. Commun., 2019, 10, 2930 CrossRef PubMed.
- A. A. Cordones and S. R. Leone, Chem. Soc. Rev., 2013, 42, 3209 RSC.
- N. Yarita, H. Tahara, T. Ihara, T. Kawawaki, R. Sato, M. Saruyama, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 1413 CrossRef CAS PubMed.
- Y. Zhang, T. Guo, H. Yang, R. Bose, L. Liu, J. Yin, Y. Han, O. M. Bakr, O. F. Mohammed and A. V. Malko, Nat. Commun., 2019, 10, 2930 CrossRef PubMed.
- J. Yin, P. Maity, M. De Bastiani, I. Dursun, O. M. Bakr, J. L. Bredas and O. F. Mohammed, Sci. Adv., 2017, 3, 1701793 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04981j |
| ‡ H. F. Ding, Y. S. Shan and J. Z. Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |