
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkali-modified heterogeneous Pd-catalyzed synthesis of acids, amides and esters from aryl halides using formic acid as the CO precursor†
Charles O. Oseghale,
Oluwatayo Racheal Onisuru,
Dele Peter Fapojuwo,
Batsile M. Mogudi,
Pule Petrus Molokoane,
Nomathamsanqa Prudence Maqunga and
Reinout Meijboom *
*
Research Center for Synthesis and Catalysis, Department of Chemical Sciences, University of Johannesburg, PO Box 524, Auckland Park 2006, Johannesburg, South Africa. E-mail: rmeijboom@uj.ac.za; Fax: +27 11 559 2819; Tel: +27 72 894 0293
First published on 5th August 2021
Abstract
To establish an environmentally friendly green chemical process, we minimized and resolved a significant proportion of waste and hazards associated with conventional organic acids and molecular gases, such as carbon monoxide (CO). Herein, we report a facile and milder reaction procedure, using low temperatures/pressures and shorter reaction time for the carboxyl- and carbonylation of diverse arrays of aryl halides over a newly developed cationic Lewis-acid promoted Pd/Co3O4 catalyst. Furthermore, the reaction proceeded in the absence of acid co-catalysts, and anhydrides for CO release. Catalyst reusability was achieved via scalable, safer, and practical reactions that provided moderate to high yields, paving the way for developing a novel environmentally benign method for synthesizing carboxylic acids, amides, and esters.
Introduction
Carbonylation reactions involving transition metals have proven to be the most convenient and versatile method for synthesizing carbonyl-containing compounds such as anhydrides, esters, amides, aldehydes, alcohols, ketones, and acids,1–6 which have found applications in a variety of biologically active compounds, pharmaceuticals, and natural products. As a result, the carbonylation process has become an indispensable building block and a highly effective technique for synthesizing fine and bulk chemicals in academia and industry. However, carbonylation reactions have not been extensively used in a laboratory setting for chemical synthesis. This is due to the handling of extremely flammable and toxic CO gas, which works under higher pressure and requires sophisticated stainless-steel autoclave reactors. To address this issue on a laboratory scale, much emphasis has been directed towards the use of CO surrogates.7,8–10 Numerous surrogates, including aldehydes,11 formic acids,7,12–15 and formates,16,17 have been used in carbonylation processes catalyzed by transition metals. Among these CO substitutes, formic acid (HCOOH) is the most practical surrogate since it can be readily obtained via CO2 hydrogenation18,19 and bio-waste fermentation.20,21 However, the ambiguous breakdown route of HCOOH to H2 and CO2 or CO and H2O remains a common issue with the effective use of formic acid as a surrogate.The decomposition of formic acid to H2 and CO2 has been successfully achieved using a variety of homogeneous catalysts,22,23 whereas the decomposition of formic acid to CO and H2O is currently being investigated owing to the difficulty encountered in its transformation at lower temperatures. Although formic acid dehydration in the presence of strong mineral acids, such as sulfuric acid, may produce CO gas preferentially. The acid is thought to act as an activator for the CO release but has compatibility issues when applied to other carbonylation reactions owing to the process's harsh reaction conditions. Similarly, acetic anhydride,19,24,25 triethylamine,7 and others have been reportedly employed as activators for selective CO release. However, acetic anhydride requires a stoichiometric amount to promote CO production, which generates waste and complicates the work. As a result, the quest for more environmentally friendly methods to perform these reactions in situ in solution persists.
Developing a stable and reusable heterogeneous catalyst to perform carbonylation reactions under these conditions is highly desirable. Although homogeneous catalysis remains vital in academia and chemical industrial processes, it is estimated to account for more than 20% of catalytic reactions in the chemical industry today.26–28 Carbonylation reactions catalyzed by transition metals have developed into a critical tool in organic synthesis. These reactions have been carried out using a variety of catalytically active metals. The palladium metal-catalyzed addition of CO to alkynes and alkenes in the presence of a suitable nucleophile has garnered significant interest in recent years for its potential use in synthesizing a wide variety of essential chemical products. Palladium-supported phosphine functionalized porous polymers and various palladium–phosphine complexes have been extensively described.29–31 However, these synthetic protocols often require the use of non-recyclable palladium precursors, a longer reaction time, and phosphine ligands, as well as harsh reaction conditions. In addition, these protocols sometimes intricate the carbonylation of the unprotected nitrogen in the phosphine ligands. Moisture-sensitive phosphine ligands and non-recyclable palladium precursors pose significant challenges in terms of catalyst recovery, structural flexibility, application, accessibility, leaching, and reusability.32,33 This necessitates the development of highly reusable, economically viable, and environmentally benign carbonylation catalysts. Recently, researchers have developed a method for immobilizing clusters or metallic compounds (palladium complexes) on solid supports such as organic polymers,34,35 Fe3O4,36 SBA-15,37,38 MOF-5,39 ZIF-8 (ref. 40) that is sustainable, green, easy, and cost-effective. The heterogenization of homogeneous catalysts has been extensively researched recently to address sustainability concerns about recyclability, besides designing new ligands to enrich the homogeneous catalysts.
As part of our ongoing work on heterogeneous catalyzed carbonylation reactions, we became motivated to fill the gap by designing a new palladium heterogeneous catalyst for these reactions. The catalytic system utilizes the synergistic combination of alkali metals as ion-promoters on the palladium supported on Co3O4. The spinel mesoporous Co3O4 is versatile and constitutes an interesting class of nanomaterials for noble metal support in vast catalytic reactions due to the intrinsic catalytic activity they exhibit.41–44 Notably, incorporating dopants such as alkali metals into catalysts has developed into an interesting and active field in catalysis. We recently reported that alkali metals might induce a phenomenon and improve the overall catalyst activity.2,41,45 Various studies have documented the increased catalytic activity of these alkali-promoted catalysts due to their electronic and structural effects on these nanomaterials.46–48 It was discovered in this study that doping with alkali metals stabilized and induced the active dispersed palladium species on reducible Co3O4, thus minimizing active site leaching, acting as a potential “in-built” cationic Lewis acid co-catalyst, and improving the catalyst's binding properties.
Furthermore, between the generated CO molecules, the newly designed catalyst, and the substrates or products that may limit the substrate's scope, reaction design must account for the undesirable chemistry associated with these reactions. As a result, performing carbonylation reactions on a laboratory scale with various substrates has thus far remained elusive. The study reports a highly reusable catalyst and an operationally simple strategy for the stoichiometric carbonylation reaction under milder conditions (Scheme 1). Notably, this new system exhibited superior activity for the reactions in the absence of acid co-catalysts and anhydrides (activators). The critical success may be attributed to CO generated in situ in the solution containing the ion-promoted Pd-catalyst. We anticipated that this catalytic system would improve the rates of carboxy- and carbonylation reactions by utilizing formic acid as a CO precursor. Additionally, we demonstrated that this green method might be used to effectively synthesize many biologically active amide compounds. Notably, this facile protocol serves as a critical tool for drug development programs and discovery.
 | ||
| Scheme 1 Alkali-cation promoted Pd/Co3O4-catalyzed carboxy- and carbonylation of aryl halides. | ||
Experimental section
Chemicals
Apart from the calcium (98%) and potassium (>99%) nitrate salts purchased from Associated Chemical Enterprises, all other chemicals used in this work were of analytical grade and purchased from Sigma-Aldrich. The reactions were performed using an autoclave and a drilled aluminum block with securely capped 20 mL dram-vials at different times. In addition, high purity solvents from commercial suppliers were used without the need for further purification.Synthesis of the catalysts
The deposition–precipitation synthetic technique was used to synthesize undoped Pd/Co3O4 and alkali metal (x = Cs, Na, Li, Ca and K) doped Pd/Co3O4 catalysts with the same alkali metal/Pd mole ratio (2% mol/mol).49 Typically, 1 g of the as-prepared Co3O4 (ref. 2) was added to an aqueous Pd(OAc)2 solution and stirred at room temperature (RT) for 2 h. Urea![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd salt at a (2:1) molar ratio was added to the reaction mixture with heating and stirring for 1 h at 80 °C. After cooling to room temperature, freshly prepared NaBH4 was added to the reaction mixture at a 10:1 molar excess and agitated for a further 4 h at RT. The alkali metal was then added to the mixture and stirred for 1 h. The precipitate obtained was washed with 100 mL of excess distilled water, dried, and calcined for 3 h at 350 °C. Prior to conducting the activity tests, the catalysts were reduced in H2 for 20 min at 350 °C and labeled as Co3O4 (X1) and 3% Pd/Co3O4 (X2). The doped equivalents were denoted as x–Pd/Co3O4 or Pd/Co3O4–x; 3% Pd/Co3O4–Ca (X3), 3% Pd/Co3O4–Na (X4), 3% Pd/Co3O4–K (X5), 3% Pd/Co3O4–Cs (X6), 3% Pd/Co3O4–Li (X*), 1% Pd/Co3O4–Li (X7) and 5% Pd/Co3O4–Li (X8). The % mole of the alkali metal cations was maintained at 2 wt%.
:Pd salt at a (2:1) molar ratio was added to the reaction mixture with heating and stirring for 1 h at 80 °C. After cooling to room temperature, freshly prepared NaBH4 was added to the reaction mixture at a 10:1 molar excess and agitated for a further 4 h at RT. The alkali metal was then added to the mixture and stirred for 1 h. The precipitate obtained was washed with 100 mL of excess distilled water, dried, and calcined for 3 h at 350 °C. Prior to conducting the activity tests, the catalysts were reduced in H2 for 20 min at 350 °C and labeled as Co3O4 (X1) and 3% Pd/Co3O4 (X2). The doped equivalents were denoted as x–Pd/Co3O4 or Pd/Co3O4–x; 3% Pd/Co3O4–Ca (X3), 3% Pd/Co3O4–Na (X4), 3% Pd/Co3O4–K (X5), 3% Pd/Co3O4–Cs (X6), 3% Pd/Co3O4–Li (X*), 1% Pd/Co3O4–Li (X7) and 5% Pd/Co3O4–Li (X8). The % mole of the alkali metal cations was maintained at 2 wt%.
Catalyst characterization
Various characterization techniques, including ICP-OES, FT-IR, p-XRD, TEM, SEM, TPR, and TPD, were used to characterize the as-prepared nanomaterials. The metal loading (wt%) capacity of the catalyst was determined using the Spectro Arcos ICP-OES instrument. A thermogravimetric analyzer (PerkinElmer STA 6000) was used to perform the thermogravimetric analyses (TGA) under air atmosphere (20–1000 °C) at a flow and heating ramping rates of 20 mL min−1 and 10 °C min−1, respectively. The surface morphologies of the synthesized nanomaterials were studied using a Vega 3 Tescan LMH Scanning Electron Microscope (SEM) equipped with EDX and operating at a high voltage of 20 kV. The 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were acquired using a Bruker-500 MHz spectrometer, with values given relative to tetramethylsilane (0.0) as the internal standard. The N2-sorption studies were conducted using a Micromeritics ASAP 2460 device. Prior to the experiments, the samples were degassed at 90 °C under N2 atmosphere and vacuum for 14 h and 2 h, respectively, to remove physisorbed moisture from the nanomaterials. The surface area was calculated using the Brunauer–Emmett–Teller (BET) technique. Powder X-ray diffraction (p-XRD) analyses were performed using a Rigaku MiniFlex-600 instrument operating with Cu K radiation (= 1.5406). Both low (0.5–4.0°) and wide (20–70°) 2θ angles diffraction patterns were measured at a step rate of 0.1° min−1. Fourier transform infrared (FT-IR) spectroscopy of the materials was performed using a Bruker FT-IR Alpha spectrometer (KBr pellets in 4000 to 500 cm−1). On a Micromeritics AutoChem II chemisorption analyzer, the hydrogen temperature-programmed reduction (H2-TPR) was conducted. About 25 mg of the catalyst was loaded into a quartz tube reactor and pretreated for 1 h at 200 °C under helium/argon flow. After cooling, the materials were probed by passing 10% H2 (Ar) at a flow rate of 50 mL min−1 from ambient temperature to 600 °C (10 °C min−1). The ammonia temperature-programmed desorption (NH3-TPD) tests were performed on the Micromeritics AutoChem II equipment to identify the acidic sites on the catalyst. Approximately (0.20–0.30 g) was loaded into a tube reactor containing quartz wool and further probed by passing 10% NH3 at a temperature range of 20–700 °C using helium as the probed gas. The flow and heating rates were maintained at 25 mL min−1 and 3 °C min−1 for both experiments, respectively. The morphology of the catalyst was captured using a Transmission Electron Microscope (TEM) (JEOL Jem-2100F) operated at a 200 kV accelerating voltage. A total of 0.2 mg of the catalyst was measured and ultrasonicated in 1 mL ethanol for 30 min before being deposited on the carbon-coated TEM-Cu grids.Catalytic test for carbonylation of the aryl halides
Typically, the reaction tube equipped with a stirring bar was charged with a 0.3 g catalyst (previously reduced at 350 °C for 20 min in H2), 4 mmol substrate, 3 mL solvent, 1 eq. Et3N B1, 1 mmol (200 L) decane (internal standard), 3 mL HCOOH, and 3 mL nucleophile (for alkoxy- and aminocarbonylation). For 8–24 hours, the mixture was heated to 90–130 °C at a stirring speed of 550 rpm. Upon cooling to RT, 50 mL of water was added to separate the organic phase using a separating funnel. Ethyl acetate was used to extract the aqueous layer (3 × 25 mL). The combined layer was washed with a 50 mL brine solution and then dried over magnesium sulfate. The residual solvent was evaporated using a rotary evaporator, and the product was purified by flash column chromatography on silica gel with hexane/ethyl acetate eluents at various intervals. The products were quantified using a Shimadzu GC-2010 equipped with a flame ionization detector (FID) and N2 as the carrier gas; the FID and injection temperatures were adjusted to 370 and 200 °C, respectively. The products were confirmed and compared using GC-MS, 1H, and 13C NMR instruments. Simple filtration, followed by washing and drying in a vacuum, was used to test the catalyst's reusability.Results and discussions
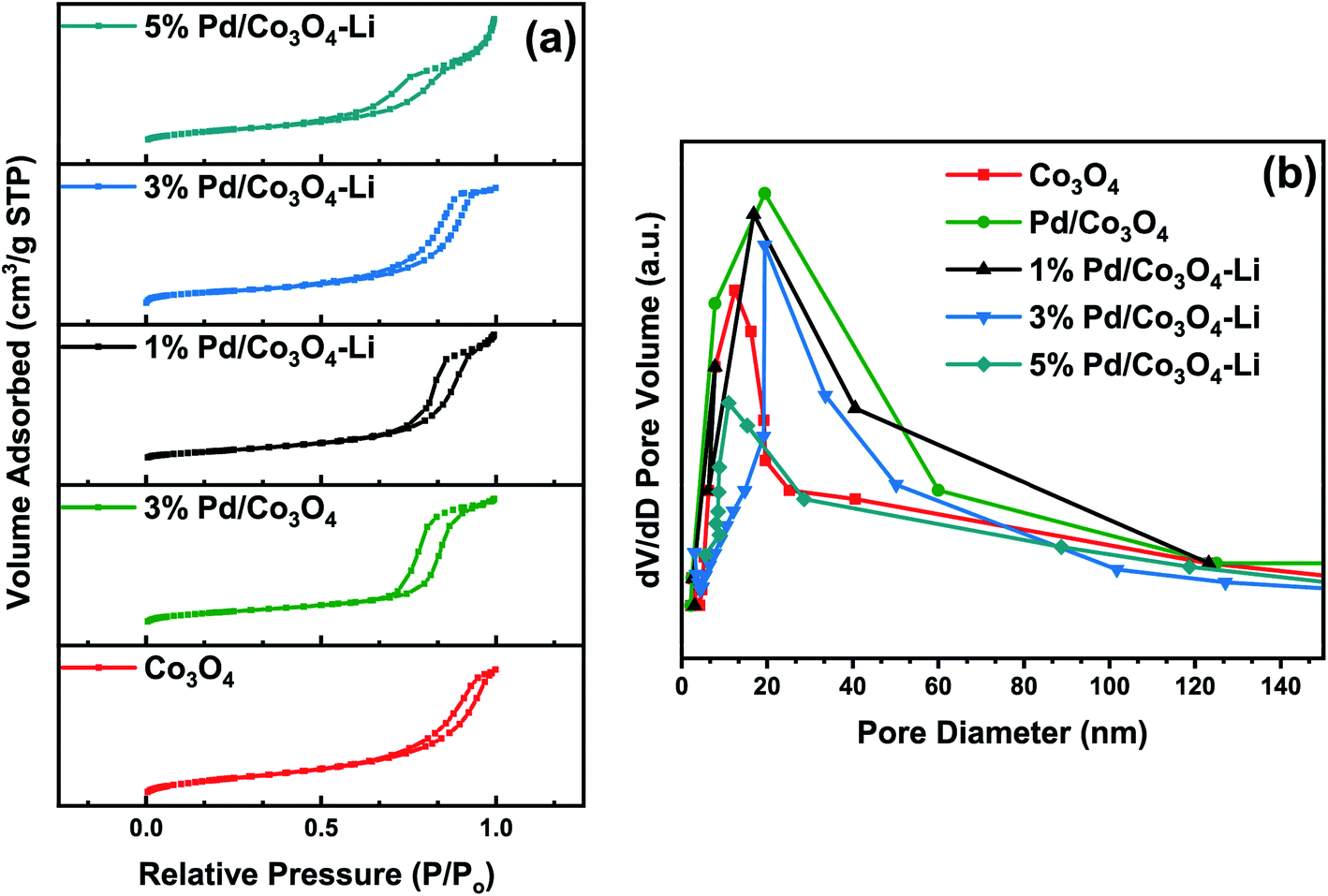
In this study, the newly designed alkali-promoted Pd catalyst was employed as an efficient catalyst for the carboxy- and carbonylation reactions of diverse arrays of aryl halides. Under milder reaction conditions and utilizing formic acid as a CO surrogate source, the carbonylation processes were carried out with excellent conversions, selectivities, and yields to their respective desired products. Most importantly, eliminating the use of acid co-catalyst and sacrificial acetic anhydrides needed for the CO release. In general, this type of carbonylation process is difficult to perform without external additives (organic acids and anhydrides) since these additives are often required to obtain reasonable reactivities.7,24 Similarly, homogeneous catalytic systems have been extensively used to carry out these reactions, but with catalyst recovery and reusability problems. As a result, the design of a more versatile heterogeneous catalyst capable of performing these reactions with excellent product yields is highly desired. In this work, we report the first heterogeneous Pd-based catalyst, promoted by alkali metal ions (Cs, Na, K, Li, and Ca) for the carboxy- and carbonylation of aryl halides. Although the PdNPs were the active species for the reaction, however, we found out that the alkali cations in the catalysts had a remarkable promotional effect on the catalytic system by acting as a promoter and an in built cationic Lewis acid co-catalyst. Additionally, the activity was linked with the NH3-TPD analysis of each catalyst, showing that the lithium-promoted catalyst with the highest amount of acidic sites was the most active for the reactions (Fig. 4). With the results, we concluded that there was a strong bonding interaction between the alkali cations, palladium nanoparticles, and the mesoporous Co3O4 support.Structural characterization of the catalysts
 | ||
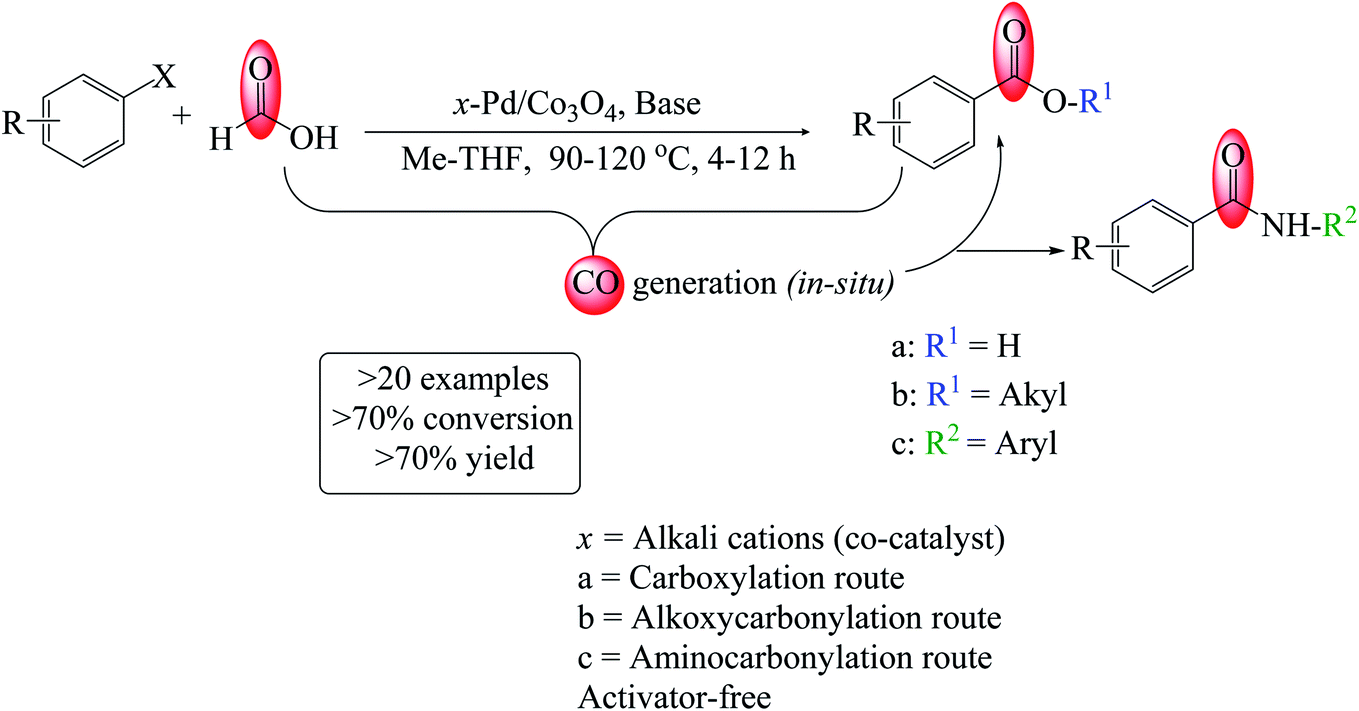
| Fig. 1 (a) Wide-angle X-ray diffractograms of A (Co3O4), B (3% Pd/Co3O4), C (1% Pd/Co3O4–Li), D (3% Pd/Co3O4–Li), and E (5% Pd/Co3O4–Li) and (b) low-angle p-XRD patterns of the nanomaterials. ♦ – Co3O4-phase (PDF no. 73-1701) and ♣ – PdO-phase (PDF no. 85-0713). | ||
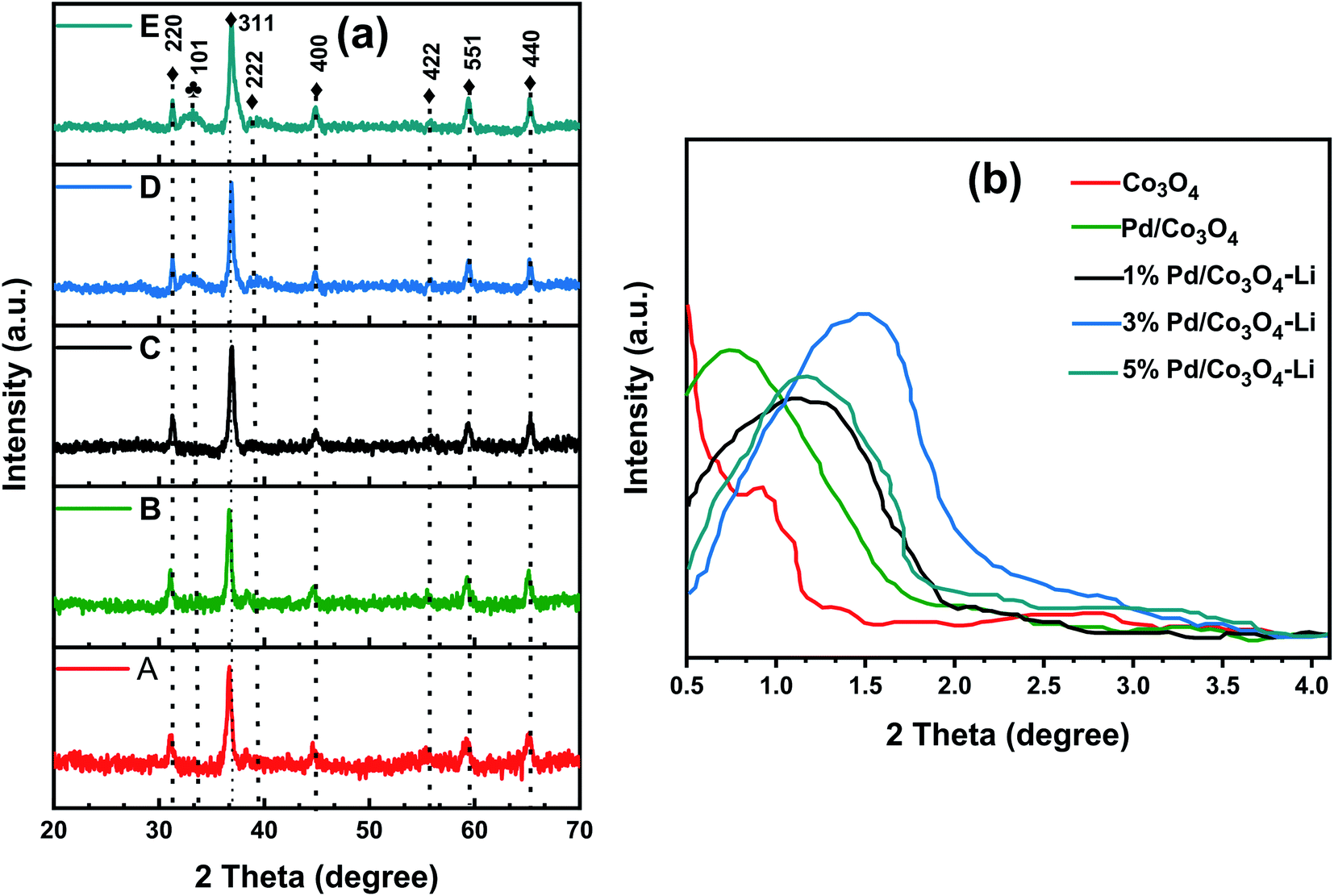
| Catalysts | SABETa (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | Crystallite sizesb (nm) |
|---|---|---|---|---|
| a Determined by N2 BET.b Calculated using Scherrer's equation. | ||||
| Co3O4 | 63 | 0.357 | 12.8 | 7.4 |
| 3% Pd/Co3O4 | 58 | 0.392 | 19.2 | 11.6 |
| 1% Pd/Co3O4–Li | 51 | 0.378 | 16.8 | 14.2 |
| 3% Pd/Co3O4–Li | 43 | 0.371 | 15.7 | 16.8 |
| 5% Pd/Co3O4–Li | 29 | 0.244 | 10.6 | 22.4 |
Additionally, the PdO particles grew in size with increased intensities for the 3 and 5% Pd loadings. However, with a lower Pd loading (1 wt%), no PdO peaks were seen, indicating that the particles were too tiny to be detected by the XRD machine. The peaks corresponding to the Pd metal were not very visible in the spectra. The XRD analysis revealed that the crystallinity of Co3O4 was preserved, suggesting that the PdNPs and alkali cations were successfully loaded onto its matrix.
 | ||
| Fig. 2 (a) and (b) SEM images of Co3O4 and 5% Pd/Co3O4–Li, respectively. TEM images and particle size distribution chart (inset) for (c) 1% Pd/Co3O4–Li, (d) 5% Pd/Co3O4–Li and (e) HR-TEM image magnification of 5% Pd/Co3O4–Li (inset). The nanoparticles are indicated in green circles. | ||
 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption curves and (b) BJH pore size distributions for the catalysts. | ||
 | ||
| Fig. 4 NH3-TPD profiles of the catalysts. All Pd loadings were set at 3 wt%. | ||
Following that, we conducted the H2-TPR measurements to determine the nanomaterials' reducibility (Fig. 5). Two reduction peaks at 236 °C and 264 °C, assigned to Co3+ → Co2+ and Co2+ → Co, respectively, were observed for the mesoporous Co3O4. The addition of palladium and an alkali cation to Co3O4 facilitated the catalyst's reduction behavior, consistent with previous reports.58–60 The Pd loadings substantially influenced the shift to lower reduction temperatures; these peaks at lower temperatures were attributed to PdO reductions, and the peak positions are highly dependent on the PdO dispersion on the support.61 Although the low-temperature peaks may not be entirely due to PdO reduction, because catalysts have been shown to display a reduction of Co3+ → Co2+ at temperatures as low as 180–200 °C, apparently due to the H2 spillover effect.61–63 However, the lower reduction peak in the range of 151–196 °C for the 3% X* and 5% X8 catalysts may be attributed to PdO reduction, as this was also evident from the XRD spectra. It is worth noting that the trend toward increasing catalytic activity of the catalyst may also be linked to the nanomaterial's low reduction temperatures.
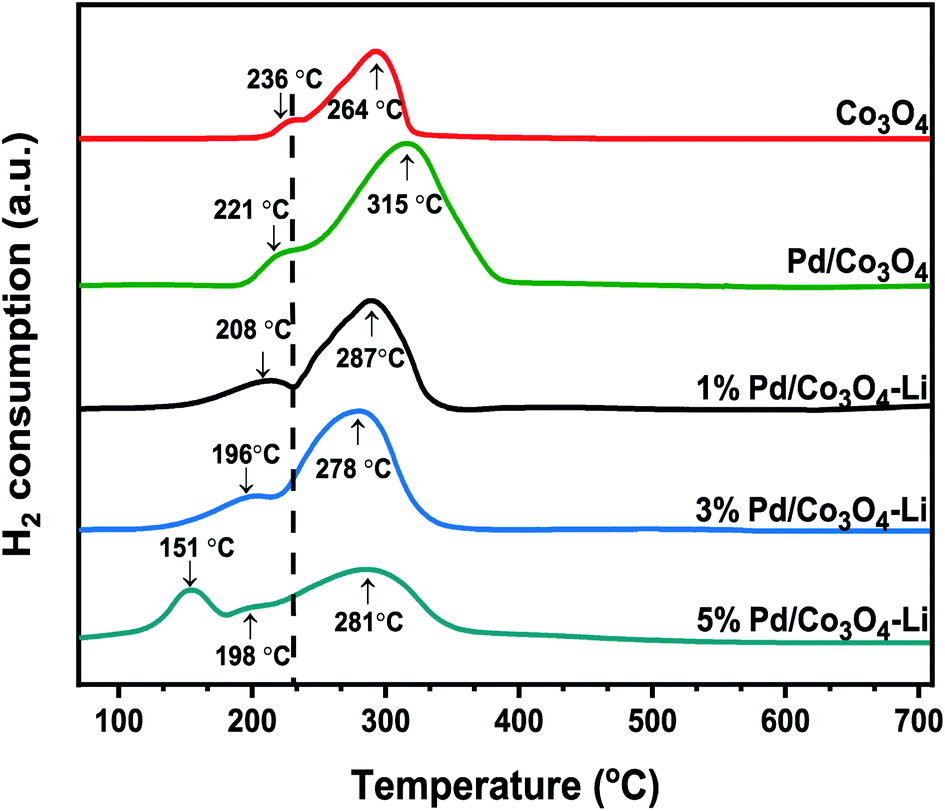 | ||
| Fig. 5 H2-TPR profiles of the catalysts. | ||
Catalytic reactions
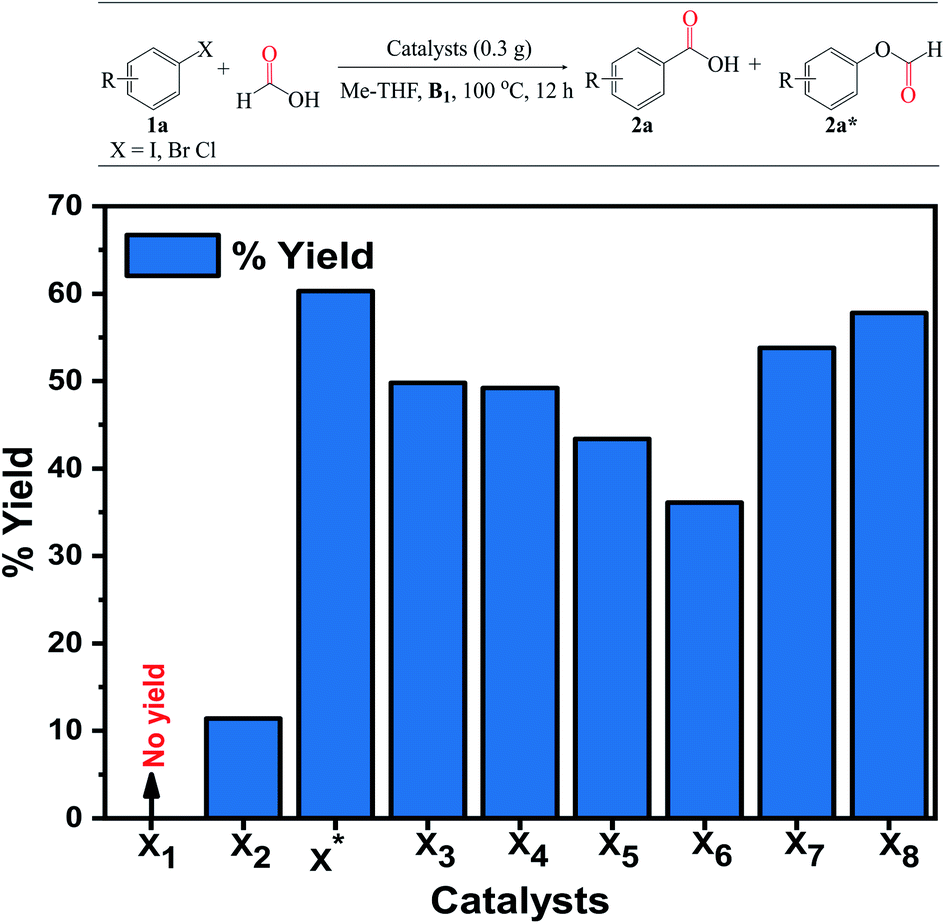 | ||
| Fig. 6 Catalyst screening. Reaction conditions: catalyst (0.3 g), 1a (4 mmol), HCOOH:Me-THF 3:3 (mL), B1 (1 equiv.), 100 °C, 12 h. Isolated yield and selectivity (%) determined by GC-FID analysis. | ||
| Entry | Cat. X* (g) | Time (h) | Temp. (°C) | Solvents | Nucleophiles | Conv.b (%) | Sel.b (%) | Yieldb (%) |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 3% X* (0.1–0.5 g), 1a (4 mmol), HCOOH:nucleophiles:Me-THF 3:3:3 (mL), B1 (1 equiv.).b Conversion, selectivity and isolated yields determined by GC-FID.c (X*, 0.5 g) with K2CO3 (1 equiv.). Entries 1–12 (carboxylation). Entries 13–18 (alkoxycarbonylation). |
||||||||
| 1 | 0.3 | 12 | 100 | Me-THF | — | 75.1 | 80.4 | 60.3 |
| 2 | 0.1 | 12 | 100 | Me-THF | — | 66.3 | 71.3 | 47.4 |
| 3 | 0.5 | 12 | 100 | Me-THF | — | 79.9 | 76.8 | 61.7 |
| 4 | 0.5 | 8 | 100 | Me-THF | — | 52.8 | 70.4 | 36.8 |
| 5 | 0.5 | 16 | 100 | Me-THF | — | 81.8 | 79.6 | 66.2 |
| 6 | 0.5 | 12 | 110 | Me-THF | — | 85.5 | 90.2 | 76.2 |
| 7 | 0.5 | 12 | 120 | Me-THF | — | 93.6 | 74.7 | 71.1 |
| 8 | 0.5 | 12 | 70 | Me-THF | — | <20 | — | <20 |
| 9 | 0.5 | 12 | 110 | Acetonitrile | — | 70.7 | 65.3 | 45.9 |
| 10 | 0.5 | 12 | 110 | 1,4-Dioxane | — | 90.5 | 67.1 | 59.8 |
| 11 | 0.5 | 12 | 110 | Toluene | — | 85.5 | 70.3 | 59.5 |
| 12c | 0.5 | 12 | 110 | Me-THF | — | 80.6 | 38.2 | 30.6 |
| 13 | 0.5 | 12 | 110 | — | Methanol | 85.3 | 57.2 | 48.7 |
| 14 | 0.5 | 12 | 110 | — | Ethanol | 75.2 | 51.9 | 39.1 |
| 15 | 0.5 | 12 | 110 | — | Propanol | 68.4 | 52.4 | 35.9 |
| 16 | 0.5 | 12 | 110 | — | Butanol | 75.3 | 44.2 | 33.2 |
| 17 | 0.5 | 12 | 110 | — | tert-Butanol | 60.9 | 38.5 | 23.5 |
| 18 | 0.5 | 12 | 110 | — | Cl-butanol | 69.3 | 50.5 | 35.1 |
Additionally, to demonstrate the versatility of this novel catalyst under the optimized reaction conditions, we validated the possibility of using different nucleophiles to investigate these alkyl group's effects on the alcohol for the alkoxycarbonylation reaction. Notably, methanol was the most efficient nucleophile compared to the others, though, no significant difference in product yields and selectivities (Table 2, entries 13–18). The observation thus implies that an increase in the alkyl chain length on the alcohols may substantially reduce yields owing to the formation of additional side products. The yield of 2a dropped to 23.5% when tert-butanol was used, presumably due to steric hindrance from the bulk methyl substituents attached to the alcohol molecule (Table 2, entry 17). Interestingly, the yield of the chloro-substituted butanol significantly increased compared to its unsubstituted counterpart, demonstrating the effect of the electron-withdrawing group on the nucleophile (Table 2, entry 18).
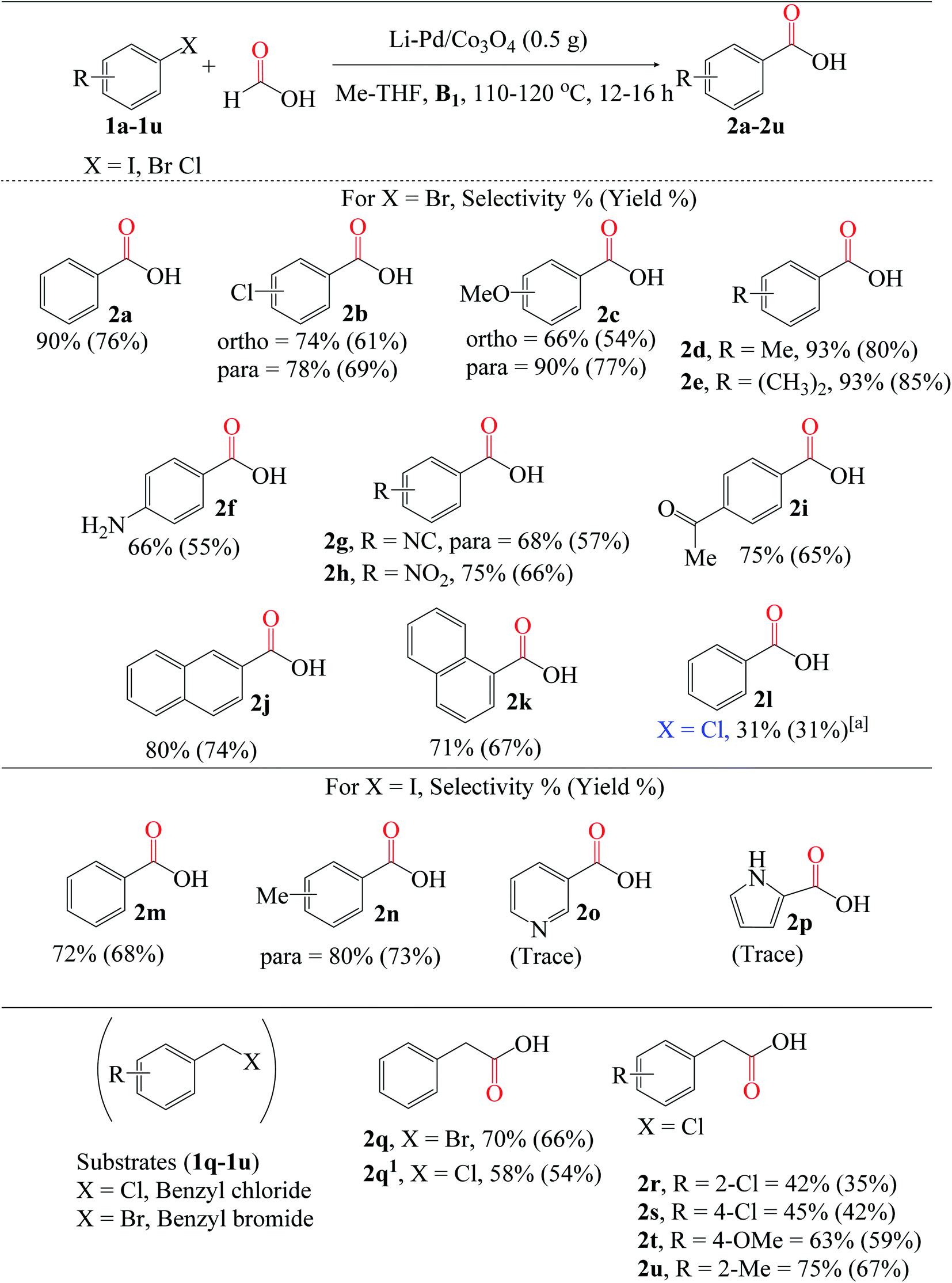 | ||
| Fig. 7 Reaction conditions: 3% X* (0.5 g), substrates (4 mmol), HCOOH:Me-THF 3:3 (mL), B1 (1 equiv.). Conversion and isolated yields determined by GC-FID analysis. aReaction conditions: 5% X* (1.0 g), 1l (4 mmol), HCOOH:Me-THF 6:6 (mL), B1 (1 equiv.), 130 °C, 24 h. | ||
Besides aryl iodides and bromides, benzyl halides (BnCl and BnBr) can also be converted to carboxylic acids. Comparing the reactivities of 1q (BnBr) and 1q1 (BnCl), the former yielded much more 2q than the latter (2q1), at 66% and 54%, respectively; this is directly related to and consistent with the relative stabilities of the C(sp3)–X bonds. Regardless of the substitution position, moderate yields of 2r (2-Cl) and 2s (4-Cl) were obtained using chloro-substituted benzyl chloride (1r and 1s). Similarly, electron-donating groups (OMe and Me) attached to the benzyl chloride substrates 1t and 1u were effectively carboxylated to 2t (59%) and 2u (67%), respectively.
Additionally, we broadened the substrate scope for amino- and alkoxycarbonylation of aryl halides (Fig. 8). Since these reactions are relatively the same, we also utilized the optimized reaction conditions stated above in Fig. 7. The aminocarbonylation of p-chlorobromobenzene 1b was catalyzed by X* using benzylamine as the nucleophile. Electron-donating group substrates, such as 1c (MeO) and 1d (Me), were successfully utilized to obtain the desired amides (3c and 3d). Also, the chlorosubstituted 1b substrate was tolerated. Next, we explored the versatility of the catalyst for the alkoxycarbonylation reaction. The chlorosubstituted bromobenzene 1b was successfully alkoxycarbonylated to their corresponding ester 4b in moderate yield. Interestingly, excellent yields of 4c–4h were obtained using substrates substituted with electron-donating groups such as 1c (OMe), 1d (Me), 1e (CH3)2, and 1f (NH2), as well as electron-withdrawing groups such as 1g (NC) and 1h (NO2). Lastly, it is worth noting that the alkoxycarbonylation of 1d was completed in a shorter reaction time than the aminocarbonylation reaction, demonstrating the substrate's high reactivity for the alkoxycarbonylation reaction.
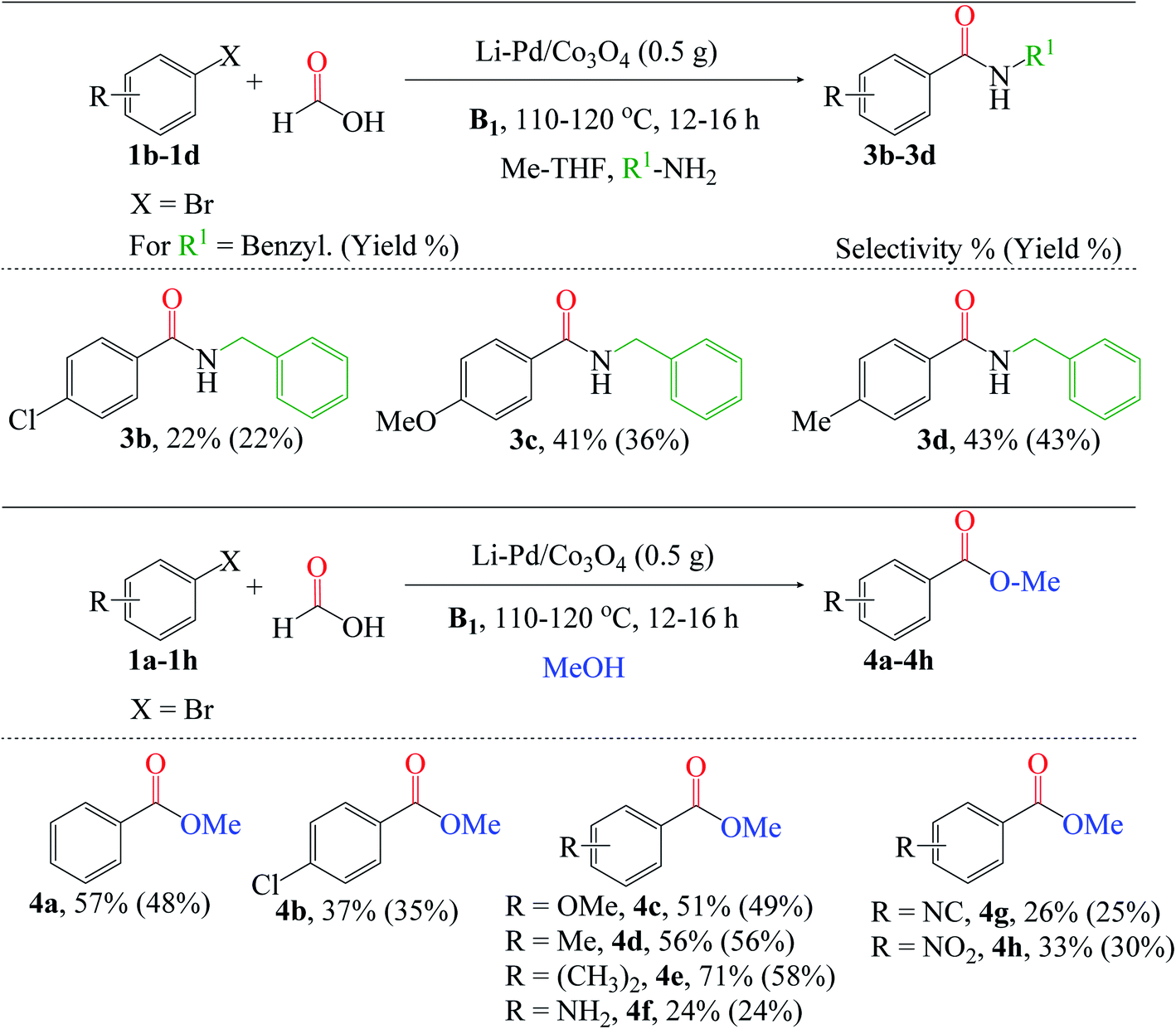 | ||
| Fig. 8 Reaction conditions: 3% X* (0.5 g), substrates (4 mmol), HCOOH:MeOH:R1-NH2:Me-THF 3:3:3:3 (mL), B1 (1 equiv.). Conversion and isolated yields were determined by GC-FID analysis. | ||
Kinetic and GC-TCD studies
The kinetic analysis was carried out on the substrates 1a (bromobenzene) and 1l (chlorobenzene) (Fig. 9). Carboxylation of 1a was much faster than 1l at a 8 h reaction time, with conversion and yield to the target product 2a reaching 73% and 51%, respectively. Conversion and yield of the 1l substrate were both <5% under identical reaction conditions. When the time was extended to 20 h, the conversion of 1a reached almost 100%, while the complete conversion of 1l took around 24–30 h. The kinetic study implies that a shorter time (low pressure) is required for carboxylation of the 1a, while a longer time (higher pressure/temperature) is needed for carboxylation of the 1l due to the stronger C(sp3)–Cl bonds in the 1l molecule (Fig. 9).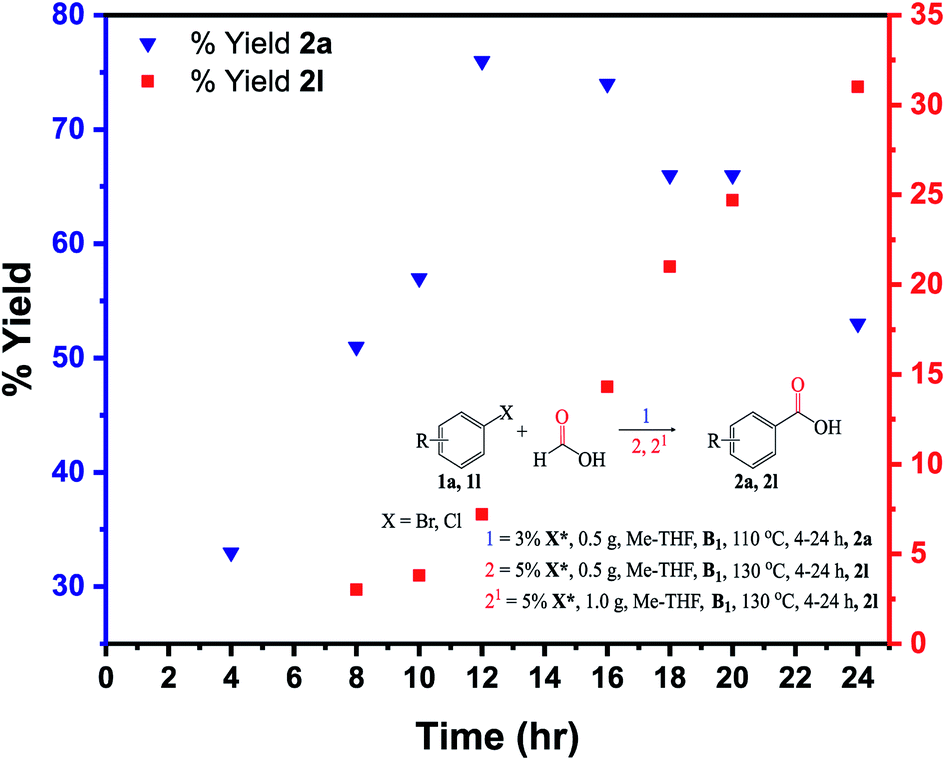 | ||
| Fig. 9 Reaction kinetics for the 1a and 1l substrates: isolated yields determined by GC-FID analysis. Under the same reaction conditions (110 °C, 8 h), the 1l substrate recorded a yield of 2l at <5%. At 5% X* (1.0 g), 130 °C, 24 h, the 2l yield reached >30%. | ||
To gain a better insight into the formic acid decomposition of this new system, a GC-TCD analysis was performed (Fig. S5†). Several literature works have reported the use of acid anhydrides and other activators to facilitate the CO release from formic acid.19,65,66 Despite the moderate to good yields obtained with these additives, product contamination has been associated with this protocol due to the anhydride's slow decomposition and severe reaction conditions, resulting in a longer time required to stabilize the catalytic system. Additionally, it is well established that the same acid anhydrides are incompatible with aminocarbonylation reactions due to the formylating agents (acetic formic anhydride) formed in solution during the CO release process.67 This necessitates the development of safer procedures for this type of reaction. Typically, an autoclave equipped with a stirring bar was charged with X* (0.5 g), HCOOH (3 mL), Me-THF (3 mL), and B1 (1 eq.) and vigorously stirred at 550 rpm for 16 h (110–130 °C). The reaction was halted and allowed to cool to ambient temperature before being analyzed on the GC-TCD machine.
Interestingly, the formic acid decomposed into a mixture of CO and CO2, with the highest amount/area obtained for the CO, suggesting that this system can readily be used in lieu of acid anhydrides for the in situ generation of CO from formic acid. Although, a minute amount of H2 gas was also detected in the spectra after 3.436 retention time. It is worth noting that the new catalyst serves a dual purpose: it facilitated the decomposition of the HCOOH and triggers the substrates' C(sp3)–X bonds, allowing for clean carbonylative transformations.
Additionally, an open-system experiment was conducted to prove the in situ generation of CO (Fig. S6†). The 2a yield decreased substantially under this “open-system condition” compared to the conventional procedure described before, probably due to the reduced gas pressure released throughout the process. As a result, the experiment verifies that the generated gas is CO (see Fig. S6 in the ESI†).
Reusability and heterogeneity test
The reusability of the Pd-based heterogeneous catalyst was tested with the model reaction (carboxylation) using our benchmark substrate 1a, under the optimized reaction conditions of 110 °C, 12 h (Fig. 10). Centrifugation at 450 rpm for 30 min was used to separate the catalyst, followed by simple filtration and reactivation in an oven at 350 °C for 30 min before reuse. As shown in Fig. 10, the 2a yield decreased in the second, third, and fourth cycles but significantly rose in the fifth and sixth cycles, reaching 82 and 85%, respectively, indicating its stability. Additionally, our initial test reveals that the unpromoted catalyst exhibited significant leaching of the active Pd in the reaction mixture. The increased stability of the Li-promoted catalyst over its unpromoted counterpart may be attributed to the alkali metal in the catalyst, which minimized leaching of the active Pd species, improved the catalyst's binding properties, and enhanced the catalytic activity for the reactions (Fig. 10). It is worth noting that the reusability of thi novel catalyst significantly improves the viability of our catalytic system in comparison to the homogeneous systems.68–72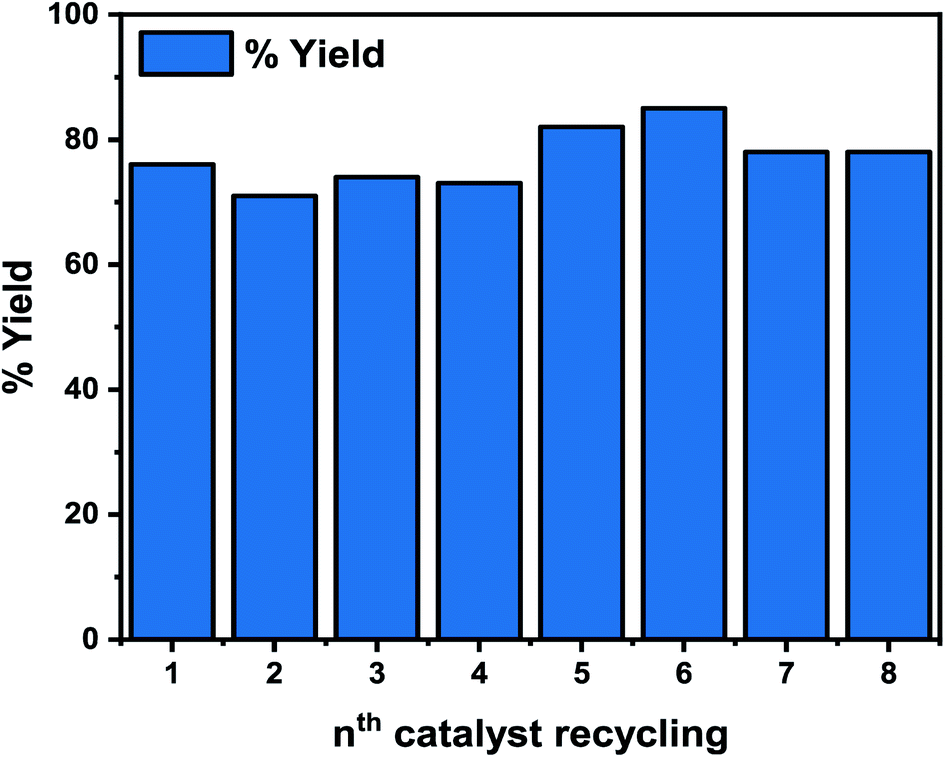 | ||
| Fig. 10 Catalyst reusability. Reaction conditions: 110 °C, 12 h. | ||
Next, the heterogeneity test was performed by first filtering the composite catalyst from the reaction mixture, and after that, the filtrate was reintroduced into the reaction tube under the reaction conditions of 110 °C and 8 h. Interestingly, no change in conversion was observed after 8 h of reaction time (Fig. S7†), suggesting that the active Pd species that catalyzed the reaction was not leached into the mixture. In addition, the ICP-OES analysis of the filtrate was performed after the 8th run, with no possible bleeding of the Pd species.
Conclusion
The carboxy- and carbonylation reactions were achieved with the newly designed heterogeneous Pd catalyst and synthetic protocol, which in situ generates CO from formic acid. Although the system was more favorable for carboxylation reaction, acceptable product yields were also obtained for alkoxy- and aminocarbonylation processes. Without the need for acid co-catalysts and acetic anhydrides, the reactions proceeded smoothly with this new catalyst under mild reaction conditions and at a shorter reaction time. Besides the catalytic system's superior selectivity and unprecedented reactivity, another more exciting feature was observed in the catalyst's practical reusability. We believe that the reported results adequately demonstrate the synthetic utility of this green carbonylation protocol and therefore establish it as a critical tool for development in academia, the chemical industry, and drug delivery applications.Conflicts of interest
There are no conflicts of interest to declare.References
- T. Fukuyama, T. Totoki and I. Ryu, Green Chem., 2014, 16, 2042–2050 RSC.
- C. O. Oseghale, B. M. Mogudi, C. A. Akinnawo and R. Meijboom, Appl. Catal., A, 2020, 117735 CrossRef CAS.
- X. Qi, R. Zhou, H.-J. Ai and X.-F. Wu, J. Catal., 2020, 381, 215–221 CrossRef CAS.
- S. Zhang, H. Neumann and M. Beller, Chem. Soc. Rev., 2020, 49, 3187–3210 RSC.
- C. Zhu, J. Liu, M.-B. Li and J.-E. Bäckvall, Chem. Soc. Rev., 2020, 49, 341–353 RSC.
- S. P. Chavan and B. M. Bhanage, Eur. J. Org Chem., 2015, 2015, 2405–2410 CrossRef CAS.
- C. Veryser, S. Van Mileghem, B. Egle, P. Gilles and W. M. De Borggraeve, React. Chem. Eng., 2016, 1, 142–146 RSC.
- R. Sang, C. Schneider, R. Razzaq, H. Neumann, R. Jackstell and M. Beller, Org. Chem. Front., 2020, 7, 3681–3685 RSC.
- J. H. Choi, H. Do Kim, J. Y. Kang, T. Jeong, P. Ghosh and I. S. Kim, Bull. Korean Chem. Soc., 2021, 42, 542–547 CrossRef CAS.
- R. Geitner, A. Gurinov, T. Huang, S. Kupfer, S. Graefe and B. M. Weckhuysen, Angew. Chem., 2021, 133, 3464–3469 CrossRef.
- J. Cao, Z.-J. Zheng, Z. Xu and L.-W. Xu, Coord. Chem. Rev., 2017, 336, 43–53 CrossRef CAS.
- L. Liu, X.-C. Chen, S.-Q. Yang, Y.-Q. Yao, Y. Lu and Y. Liu, J. Catal., 2021, 394, 406–415 CrossRef CAS.
- N. Hussain, A. K. Chhalodia, A. Ahmed and D. Mukherjee, ChemistrySelect, 2020, 5, 11272–11290 CrossRef CAS.
- M.-C. Fu, in Studies on Green Synthetic Reactions Based on Formic Acid from Biomass, Springer, 2020, pp. 1–26 Search PubMed.
- L. Wang, H. Neumann and M. Beller, Angew. Chem., 2018, 130, 7026–7030 CrossRef.
- P. Gautam and B. M. Bhanage, Catal. Sci. Technol., 2015, 5, 4663–4702 RSC.
- T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 5370–5373 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- R. Sang, P. Kucmierczyk, K. Dong, R. Franke, H. Neumann, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2018, 140, 5217–5223 CrossRef CAS.
- F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji and H. Enomoto, Green Chem., 2008, 10, 612–615 RSC.
- R. Wölfel, N. Taccardi, A. Bösmann and P. Wasserscheid, Green Chem., 2011, 13, 2759–2763 RSC.
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
- Y. Manaka, W.-H. Wang, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2014, 4, 34–37 RSC.
- X.-D. Lang, F. You, X. He, Y.-C. Yu and L.-N. He, Green Chem., 2019, 21, 509–514 RSC.
- M. V Khedkar, S. R. Khan, T. L. Lambat, R. G. Chaudhary and A. A. Abdala, Curr. Org. Chem., 2020, 24, 2588–2600 CrossRef.
- J. Hagen, Industrial catalysis: a practical approach, John Wiley & Sons, 2015 Search PubMed.
- L. Wang, W. Sun and C. Liu, Chin. J. Chem., 2018, 36, 353–362 CrossRef CAS.
- J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514–1517 CrossRef CAS PubMed.
- B. Urbán and R. Skoda-Földes, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 302–306 CrossRef.
- B. Urbán, P. Szabó, D. Srankó, G. Sáfrán, L. Kollár and R. Skoda-Földes, J. Mol. Catal., 2018, 445, 195–205 CrossRef.
- Y. Lei, L. Wu, X. Zhang, H. Mei, Y. Gu and G. Li, J. Mol. Catal. Chem., 2015, 398, 164–169 CrossRef CAS.
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, Angew. Chem. Int. Ed., 2018, 57, 15316–15329 CrossRef CAS.
- F. Chen, X. Jiang, L. Zhang, R. Lang and B. Qiao, Chin. J. Catal., 2018, 39, 893–898 CrossRef CAS.
- A. Mansour and M. Portnoy, J. Mol. Catal. Chem., 2006, 250, 40–43 CrossRef CAS.
- Z. S. Qureshi, S. A. Revankar, M. V Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148–153 CrossRef CAS.
- A. S. Prasad and B. Satyanarayana, J. Mol. Catal. Chem., 2013, 370, 205–209 CrossRef.
- M. Genelot, N. Villandier, A. Bendjeriou, P. Jaithong, L. Djakovitch and V. Dufaud, Catal. Sci. Technol., 2012, 2, 1886–1893 RSC.
- M. Genelot, V. Dufaud and L. Djakovitch, Adv. Synth. Catal., 2013, 355, 2604–2616 CrossRef CAS.
- T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805–1807 RSC.
- T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406–1410 CrossRef CAS.
- C. O. Oseghale, B. M. Mogudi, O. R. Onisuru, C. A. Akinnawo, D. P. Fapojuwo and R. Meijboom, ChemCatChem, 2021, 13, 1311–1316 CrossRef CAS.
- Z. Jiang, X. Feng, J. Deng, C. He, M. Douthwaite, Y. Yu, J. Liu, Z. Hao and Z. Zhao, Adv. Funct. Mater., 2019, 29, 1902041 CrossRef.
- Y. Wang, H. Arandiyan, J. Scott, H. Dai and R. Amal, Adv. Sustainable Syst., 2018, 2, 1700119 CrossRef.
- X. Hu, Z. Zhang, Y. Zhang, L. Sun, H. Tian and X. Yang, Eur. J. Inorg. Chem., 2019, 2019, 2933–2939 CrossRef CAS.
- B. M. Mogudi, P. Ncube, N. Bingwa, N. Mawila, S. Mathebula and R. Meijboom, Appl. Catal., B, 2017, 218, 240–248 CrossRef CAS.
- Y. Li, C. Zhang, H. He, J. Zhang and M. Chen, Catal. Sci. Technol., 2016, 6, 2289–2295 RSC.
- S. Biswas, A. S. Poyraz, Y. Meng, C.-H. Kuo, C. Guild, H. Tripp and S. L. Suib, Appl. Catal., B, 2015, 165, 731–741 CrossRef CAS.
- T. Grewe and H. Tüysüz, J. Mater. Chem. A, 2016, 4, 3007–3017 RSC.
- A. Aguilar-Tapia, L. Delannoy, C. Louis, C. W. Han, V. Ortalan and R. Zanella, J. Catal., 2016, 344, 515–523 CrossRef CAS.
- W. Zhou, Y. Guan, D. Wang, X. Zhang, D. Liu, H. Jiang, J. Wang, X. Liu, H. Liu and S. Chen, Chem.–Asian J., 2014, 9, 1648–1654 CrossRef CAS.
- S. Li, Y. Zhang, J. Shi, G. Zhu, Y. Xie, Z. Li, R. Wang and H. Zhu, Nanomaterials, 2020, 10, 31 CrossRef CAS.
- M. N. Pahalagedara, L. R. Pahalagedara, C.-H. Kuo, S. Dharmarathna and S. L. Suib, Langmuir, 2014, 30, 8228–8237 CrossRef CAS.
- Y. Liu, J.-F. Chen and Y. Zhang, RSC Adv., 2015, 5, 29002–29007 RSC.
- S. Zhuang, Y. Liu, S. Zeng, J. Lv, X. Chen and J. Zhang, J. Sol-Gel Sci. Technol., 2016, 77, 109–118 CrossRef CAS.
- S. Sun, H. Li and Z. J. Xu, Joule, 2018, 2, 1024–1027 CrossRef.
- C. Gregor, M. Hermanek, D. Jancik, J. Pechousek, J. Filip, J. Hrbac and R. Zboril, Eur. J. Inorg. Chem., 2010, 16, 2343–2351 CrossRef.
- J. Dou, Z. Bao and F. Yu, ChemCatChem, 2018, 10, 250–258 CrossRef CAS.
- Y. Wang, C. Zhang, F. Liu and H. He, Appl. Catal., B, 2013, 142, 72–79 CrossRef.
- Z. Chen, S. Wang, Y. Ding, L. Zhang, L. Lv, M. Wang and S. Wang, Appl. Catal., A, 2017, 532, 95–104 CrossRef CAS.
- N. Yang, S. Ni, Y. Sun and Y. Zhu, J. Mol. Catal., 2018, 452, 28–35 CrossRef CAS.
- J.-P. Dacquin, C. Dujardin and P. Granger, J. Catal., 2008, 253, 37–49 CrossRef CAS.
- B. Liu, B. Ouyang, Y. Zhang, K. Lv, Q. Li, Y. Ding and J. Li, J. Catal., 2018, 366, 91–97 CrossRef CAS.
- B. Ouyang, S. Xiong, Y. Zhang, B. Liu and J. Li, Appl. Catal., A, 2017, 543, 189–195 CrossRef CAS.
- A. Brennführer, H. Neumann, S. Klaus, T. Riermeier, J. Almena and M. Beller, Tetrahedron, 2007, 63, 6252–6258 CrossRef.
- X. Qi, H. Li and X. Wu, Chem.–Asian J., 2016, 11, 2453–2457 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Lett., 2003, 5, 4269–4272 CrossRef CAS PubMed.
- W. Stevens and A. Van Es, Recl. Trav. Chim. Pays-Bas, 1964, 83, 863–872 CrossRef CAS.
- B. Bartal, G. Mikle, L. Kollár and P. Pongrácz, J. Mol. Catal., 2019, 467, 143–149 CrossRef CAS.
- C. Li, X. Qi and X. Wu, ChemistrySelect, 2016, 1, 1702–1704 CrossRef CAS.
- F. Wu, J. Peng, L. Meng, X. Qi and X. Wu, ChemCatChem, 2017, 9, 3121–3124 CrossRef CAS.
- A. Ahmed, N. Hussain, M. Bhardwaj, A. K. Chhalodia, A. Kumar and D. Mukherjee, RSC Adv., 2019, 9, 22227–22231 RSC.
- X. Qi, C. Li and X. Wu, Chem.–Eur. J., 2016, 22, 5835–5838 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05177f |
| This journal is © The Royal Society of Chemistry 2021 |