
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCofactor-assisted three-way DNA junction-driven strand displacement†
Yufeng Jiaa and
Yingxin Hu *b
*b
aSchool of Economics and Management, Shijiazhuang Tiedao University, Shijiazhuang 050043, P. R. China
bCollege of Information Science and Technology, Shijiazhuang Tiedao University, Shijiazhuang 050043, P. R. China. E-mail: huyingxin@mail.stdu.edu.cn
First published on 10th September 2021
Abstract
Toehold-mediated strand displacement is widely used to construct and operate DNA nanodevices. Cooperative regulation of strand displacement with diverse factors is pivotal in the design and construction of functional and dynamic devices. Herein, a cofactor-assisted three-way DNA junction-driven strand displacement strategy was reported, which could tune the reaction kinetics by the collaboration of DNA and other types of stimulus. This strategy is responsive to various inputs by incorporation of the specific sequence into the three-way junction structure. Specifically, the cooperation of multiple factors changes the conformation of the specific domain and promotes the reaction. To demonstrate the strategy, adenosine triphosphate (ATP), HG2+, and pH were used as cofactors to modulate the displacement reaction. The electrophoresis and fluorescence experiments showed that the cooperative regulation of the strand displacement reaction could be achieved by diverse factors using this strategy. The proposed strategy provides design flexibility for dynamic DNA devices and may have potential in biosensing and biocomputing.
Introduction
DNA is an excellent material in the area of nanotechnology owing to the precise Watson–Crick base pairing rule, programmable interaction, and low synthetic cost.1 DNA has been employed to fabricate functional nanodevices such as circuits,2–5 sensors,6–9 and molecular machines.10–13 The dynamic operations of most nanodevices are driven by toehold-mediated DNA strand displacement reactions.14,15 The strand displacement is a strand-exchange reaction in which an invading strand displaces a short strand from an initial duplex. Hybridization of the invading strand is initiated from the toehold region, typically a 4–8 nt single-stranded DNA.In typical strand displacement, the toehold and branch migration domains are adjacent to each other,16 which makes flexible and fine adjustment of the displacement reaction difficult and hinders scaling up DNA networks. To overcome the limitations of the proximal toehold, many regulative methods for the control of strand displacement reactions have been proposed. To control the reaction kinetics, a remote toehold mechanism that introduced a spacer between the toehold and displacement domain was reported.17 Other approaches that introduced mismatches or transient recognition interaction or the water-soluble FeII4L4 tetrahedral cage were also proposed to tune the displacement kinetics.18–20 To build more flexible dynamic systems, three and four-way DNA junction-driven strand displacement was proposed, which distributed the toehold domain and branch migration domain on two different strands.21–23 Other approaches such as allosteric toehold, combinatorial toehold, and wedge-like DNA tool were also investigated.24–27 In addition, to broaden the application scope of the strand displacement reaction, small molecules, metal ions, proteins, antibodies, and other environmental stimuli such as pH were also employed to modulate the displacement reaction.23,28–36 For example, a responsive “hidden toehold” that utilized ATP to regulate DNA strand displacement reaction was proposed.32 The metallic-toehold was reported to control strand displacement through variation of the concentration of Hg2+ ions or metal–ligand complexation.28,37 DNA tetraplexes such as G-quadruplexes or i-motifs were also used to tune the displacement reaction.29 Binding-induced strand displacement and antibody-templated strand exchange have also been applied to biosensing or molecular circuit.16,30,34,38 Moreover, fuzzy DNA strand displacement or CRISPR-mediated strand displacement with toehold-free DNA that utilized enzymes were proposed to construct DNA networks.39,40
One of the basic requirements for smart nanodevices is the ability to respond to a variety of external stimuli.41,42 The above strategies for regulating strand displacement are mainly based on DNA or a certain factor, which restricts the development of multi-responsive systems. Therefore, novel strategies would be needed to modulate the strand displacement reaction with multiple inputs and stimuli. Three-way junction (TWJ)-driven strand displacement could provide more choices to build sensing platforms or dynamic nanodevices.23 It would be interesting to introduce various stimuli such as small molecules, metal ions, and pH into the structure as an extra regulatory factor to modulate the reaction kinetics.
In this work, a cofactor-assisted TWJ-driven strand displacement strategy was developed, which could tune the strand displacement kinetics by the cooperation of DNA and stimulus such as ATP, Hg2+, or pH. The strategy achieved multiple stimuli-responsiveness by introducing the aptamer sequence into the TWJ structure and the DNA input. Without the target, the mismatch in the TWJ substrate and DNA input slowed down or blocked the displacement reaction. Whereas, upon addition of the target, the binding of it with the aptamer altered the conformation and then facilitated the reaction. The strategy was confirmed by native polyacrylamide gel electrophoresis and fluorescence assay. This strategy provides additional design flexibility for dynamic DNA devices and has potentials in biosensing and biocomputing.
Experimental
Materials and reagents
All DNA strands (ESI Table S1†) were ordered from Sangon Biotech Co., Ltd (Shanghai, China). Unmodified DNA strands were purified by PAGE (polyacrylamide gel electrophoresis), and modified DNA strands with fluorophore and quenchers were purified by HPLC (high-performance liquid chromatography). ATP2Na was obtained from Sigma-Aldrich Co. LLC. Mercury(II) nitrate [Hg(NO3)2] was purchased from Wanshan Mineral Products (Tongren, Guizhou, China). The DNA oligonucleotides for the ATP reaction were dissolved in Tris–HCl (20 mM, pH 8.3) buffer A containing 5 mM MgCl2 and 300 mM NaCl. The DNA oligonucleotides for mercury reaction were dissolved in buffer B consisting of 50 mM Tris–HOAc, 100 mM NaOAc, 5 mM Mg (OAc)2, and 30 mM KNO3 at pH 7.4. The DNA oligonucleotides for the pH-induced reaction were dissolved in buffer C consisting of 50 mM Tris, 100 mM NaCl, and 5 mM MgCl2. The concentration of DNA strands was calculated using extinction coefficients (λ = 260 nm) by a Quawell Q6000. All other chemicals were of analytical grade and used without further purification.The preparation of the DNA complex
Various DNA complexes were formed by mixing corresponding single strands with a designated concentration in the reaction buffer. The mixtures were heated to 85 °C for 5 minutes and then slowly cooled to 25 °C for over 2 hours.Gel electrophoresis analysis
The samples (2 μM) were mixed with 6× loading buffer (Thermo Fisher) and subjected to electrophoresis analysis on a 15% polyacrylamide gel. The analysis was performed in 1× TAE buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA, pH 8.0) supplemented with 12.5 mM MgCl2 at 90 V for 1–2 hours at 4 °C. After EB (Sigma-Aldrich) staining, gels were imaged using MiniGel (Beijing Sage Creation Science Co, Ltd).Fluorescent experiments
All experiments were carried out at 25 °C in the reaction buffer using real-time fluorescence PCR (Xi'an TianLong Science and Technology Co., Ltd). In a typical reaction, the total volume of the solution was 100 μL for detection. The FAM fluorescence signal was detected at 492 nm excitation and 518 nm emission at 2 min intervals. Here, fluorescence data were normalized to make the initial fluorescent signal value correspond to zero.Results and discussion
Design of cofactor-assisted TWJ-driven strand displacement
As illustrated in Fig. 1a, the strand exchange reaction between stable DNA duplex A/R and invading strand I2 is relatively slow due to a lack of toehold. To regulate the reaction, an extra strand I1 is introduced to partly hybridize with strand A and R respectively and thus a TWJ structure is formed (Fig. 1c). In this way, the newly formed structure possesses both toehold domain and binding domain which are essential for displacement reaction. To achieve responsiveness to an external stimulus such as Hg2+, ATP, or pH, a specific domain (grey) such as T–T mismatches or higher-order structure (G-quadruplex or i-motif) is inserted between the toehold domain and the binding domain. Without the participation of external stimuli, the mismatch caused by the specific domain blocks the binding and slows down the reaction. However, once the specific domain-relevant stimulus is involved in the reaction, the joint action of input I2 and external stimulus displaces strand R in a fast reaction rate. Here, the external stimulus serves as a cofactor and mediates the reaction via the interaction with the mismatched domain. The stimulus-induced complex draws the toehold domain and binding domain together, thus accelerates the reaction. The design realizes the cooperative regulation of strand displacement reaction by DNA input and environmental stimulus.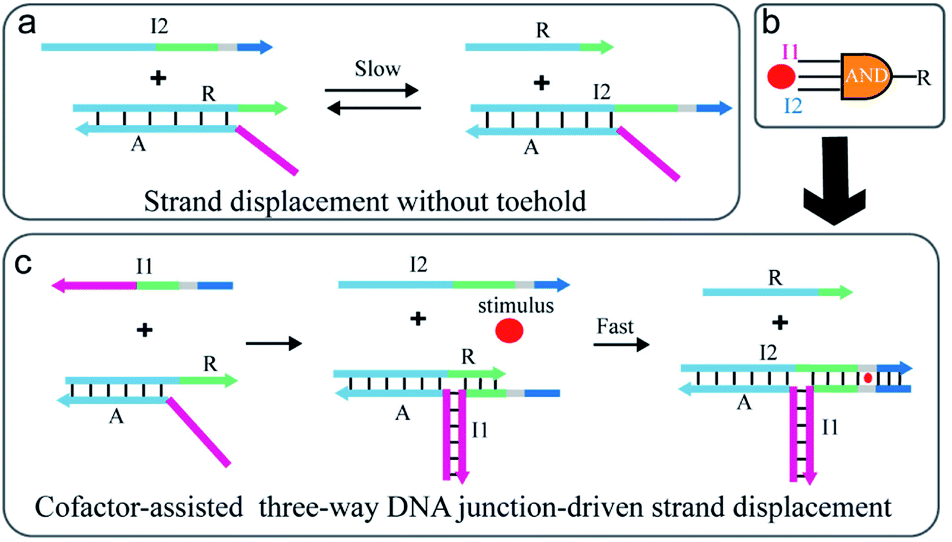 | ||
| Fig. 1 Schematic illustration of (a) the strand displacement reaction between A/R and I2 in the absence of the toehold provided by I1. (b) 3-Input AND logic gate using DNA and environmental stimulus as inputs. (c) Cofactor-assisted TWJ-driven strand displacement reaction. The sequence domains are colour-coded to indicate identical or complementary sequences except the specific domain (grey) that binds to the stimulus. | ||
Considering that the molecular logic gates are fundamental elements in constructing programmable molecular circuits, the proposed displacement reaction induced by various factors is suited for performing multi-input Boolean logic operations. Thus, this strategy is utilized to develop three-input AND logic gates where the complex A/R acts as the logic unit, I1, I2, and the stimulus as the inputs, and strand R as the output (Fig. 1b). Furthermore, combining with signal amplification methods,43–45 the proposed strategy also could be used to construct multi stimuli-fueled DNA nanodevices for biosensing and medical diagnosis.
ATP-assisted TWJ-driven strand displacement
To demonstrate the rationality of the scheme, a clinically relevant input ATP was chosen as the cofactor to drive the TWJ strand displacement. As illustrated in Fig. 2a, the hybridization of strand AI1 with AM and AA formed a three-way structure, which could supply a toehold for the displacement reaction. To make the structure further respond to ATP, the aptamer that could bind to ATP molecules with high specificity and affinity was introduced. Specifically, the ATP-binding aptamer sequence (orange) was split into two fragments and they were inserted between the 4-nt toehold region (dark blue) and branch migration region (light blue and green) of AI1 and AI3 respectively. Strands AI1 and AI3 constituted the complete aptamer and formed G-quadruplex nanostructure upon binding to ATP. Meanwhile, the G-quadruplex drew the toehold domain and binding domain together and thus accelerated the displacement reaction. Otherwise, without the aid of ATP, the displacement reaction would be slowed down due to the mismatch of the two fragments of the aptamer. To monitor and quantify the reaction, the strand AM was modified with a fluorophore (FAM, green dot) at 5′ end and a quencher (BHQ-1, black dot) at 3′ end. In the initial state, the fluorescence was high due to the long-distance induced by the hybridized structure AM/AA. Otherwise, the fluorescence was low when the strand AM was displaced and became single-stranded.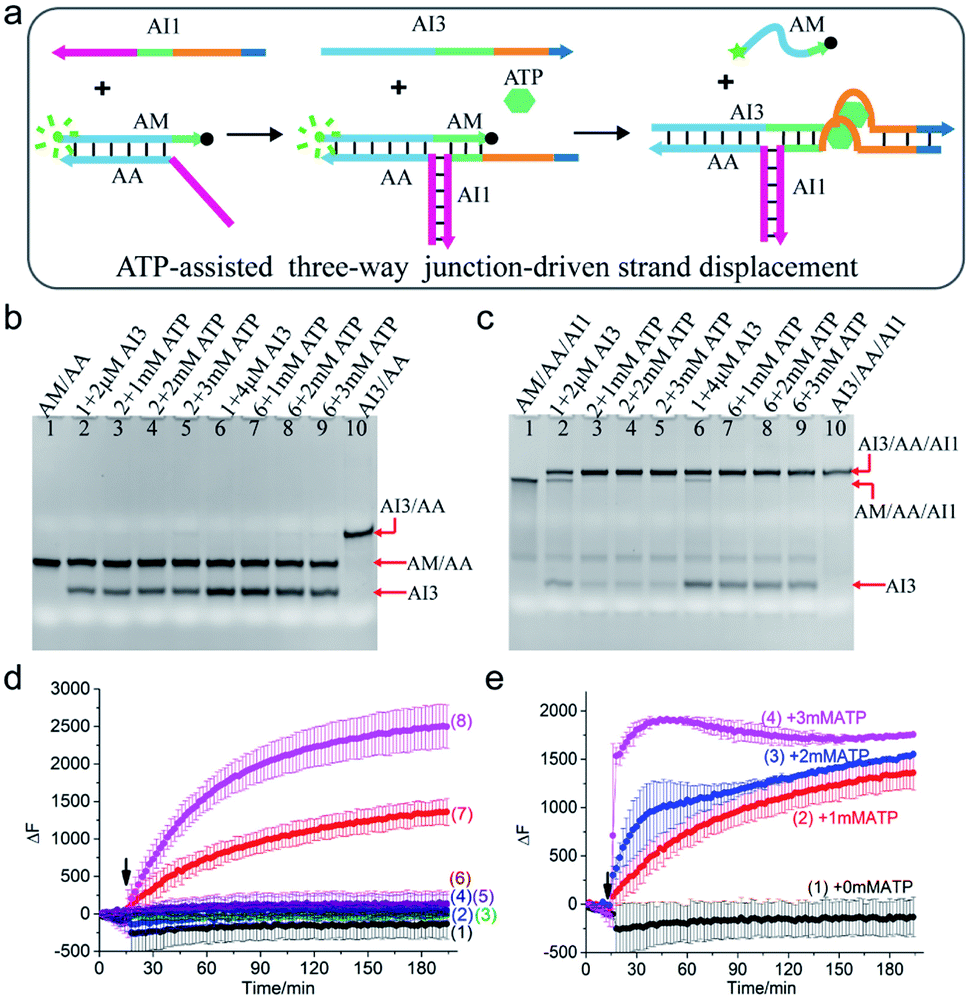 | ||
| Fig. 2 (a) Schematic illustration of ATP-assisted TWJ-driven strand displacement. (b and c) Native 15% polyacrylamide gel analysis of the reaction. For gel analysis, the samples were incubated at 25 °C for half an hour before electrophoresis. (b) Strand exchange reaction in the absence of toehold provided by AI1; (c) ATP-assisted TWJ-driven strand displacement reaction. DNA strands added in every lane are indicated above the image. The concentration of the DNA complex (AM/AA/AI1) or (AM/AA) in each lane is 2 μM. (d) Fluorescence intensity analysis of the reaction. Curve 1: AM/AA/AI1 + 50 nM AI3; curve 2: AM/AA/AI1 + 100 nM AI3; curve 3: AM/AA + 50 nM AI3; curve 4: AM/AA + 50 nM AI3 + 1 mM ATP; curve 5: AM/AA + 100 nM AI3; curve 6: AM/AA + 100 nM AI3 + 1 mM ATP; curve 7: AM/AA/AI1 + 50 nM AI3 + 1 mM ATP; curve 8: AM/AA/AI1 + 100 nM AI3 + 1 mM ATP. (e) Fluorescence intensity analysis of the reaction at different concentrations of ATP with 50 nM AI3. Curve 1: 0 mM ATP; curve 2: 1 mM ATP; curve 3: 2 mM ATP; curve 4: 3 mM ATP. For fluorescence assay, [AM/AA/AI1] = 50 nM, [AM/AA] = 50 nM. The arrow represents the addition of ATP and AI3. DNA sequences of single-stranded species were shown in ESI Table S1.† | ||
The ATP-assisted displacement reaction was confirmed by both native polyacrylamide gel electrophoresis (PAGE) experiment and time-dependent fluorescence assay. At first, the formation of the TWJ structure was identified by PAGE in Fig. S1.† In addition, to decrease the leak, the sequence of input AI3 was optimized (Fig. S2†). To analyse the displacement reaction under different conditions, several control experiments were carried out. For the reaction in the absence of the toehold provided by AI1, the band of complex AM/AA remained unchanged after adding 2 μM or 4 μM AI3 (Fig. 2b lane 2, lane 6). The results indicated that only AI3 was insufficient to displace AM from complex AM/AA. In contrast, upon addition of AI3 to the three-way structure formed by AI1 and AM/AA, apart from the band of three-way structure AM/AA/AI1 itself, a new slower band corresponding to AI3/AA/AI1 was also observed (Fig. 2c lane 2 and lane 6). With the aid of AI1, the mismatched region still triggered the displacement reaction. It is also found that the reaction with 4 μM AI3 (lane 6) consumed more AM/AA/AI1 than that of 2 μM (lane 2). In other words, increasing the concentration of input AI3 could promote the strand displacement reaction. To further speed up the reaction, different concentrations of ATP (1 mM, 2 mM, 3 mM) were added to the solution, the band of AM/AA/AI1 almost totally disappeared and a strong band of AI3/AA/AI1 was observed (Fig. 2c lane 3–5 and lane 7–9), demonstrating that the reaction was indeed affected by the combined action of AI1, AI3, and ATP. The results confirmed the mechanism of the ATP-assisted TWJ-driven strand displacement reaction.
To explore the reaction process, the time-dependent fluorescence assay was also performed. When only AI3 (50 nM or 100 nM) was added to AM/AA/AI1 or AM/AA respectively, no obvious fluorescence enhancement was observed (Fig. 2d curve 1, curve 2, curve 3, and curve 5). Nevertheless, upon the addition of ATP (1 mM) and AI3 (50 nM or 100 nM) at the same time, the fluorescence intensity of the complex AM/AA/AI1 changed significantly (Fig. 2d curve 7, curve 8). Meanwhile, with AI3 concentration increasing from 50 nM to 100 nM, the fluorescence signal increased faster, indicating that the concentration of AI3 influenced the reaction rate. As expected, for AM/AA with either 50 nM or 100 nM AI3 (curve 4, curve 6), almost no distinct change of fluorescence signal was observed after the addition of ATP. Moreover, in the presence of 50 nM AI3, with the concentration of ATP increasing, the reaction rate of the TWJ structure increased accordingly (Fig. 2e). These results demonstrated that the DNA inputs AI1, AI3, and ATP stimulus could cooperatively regulate the strand displacement reaction.
Furthermore, at higher concentrations (2 μM), even without ATP, a large amount of AM/AA/AI1 was consumed (Fig. 2c lane 2, lane 6). Whereas, at a relatively low concentration (50 nM), almost no AM was displaced which led to no increase of the fluorescence signal (Fig. 2d curve 1, curve 2). The results indicated that the concentration had a great effect on the reaction.
The above strategy relies on the specific binding of the G-quadruplex to ATP. Thus, it can be extended to other stimuli such as metal ions (K+, Na+, Sr2+) which could specifically interact with G-rich sequences.
Hg2+-Assisted TWJ-driven strand displacement
Mercury is a highly toxic metal ion widely distributed in the environment. To demonstrate the universality of the scheme, Hg2+ was chosen as a cofactor to regulate the TWJ strand displacement. As shown in Fig. 3a, the hybridization of strand HI1 with HM and HA formed a three-way structure, which could provide a toehold for the displacement reaction. To achieve Hg2+-assisted TWJ strand displacement, two T–T mismatched base pairs were inserted into the toehold domain of HI1 and HI3. In the presence of Hg2+, the formation of T–Hg2+–T base pairs strengthened the binding between HI1 and input HI3, thus promoted the following branch migration process. Otherwise, without the assistance of Hg2+, the displacement would be slowed down due to the two mismatched base pairs in the 6-nt toehold domain. To characterize the reaction, strand HM was modified with a fluorophore (FAM, green dot) at 5′ end and HA a quencher (BHQ-1, black dot) at 3′ end. If the displacement reaction did not occur, HM/HA brought the two DNA strands close and the fluorescence was low. However, when strand HM was released, the distance between the two DNA strands was enlarged, and the fluorescence would increase.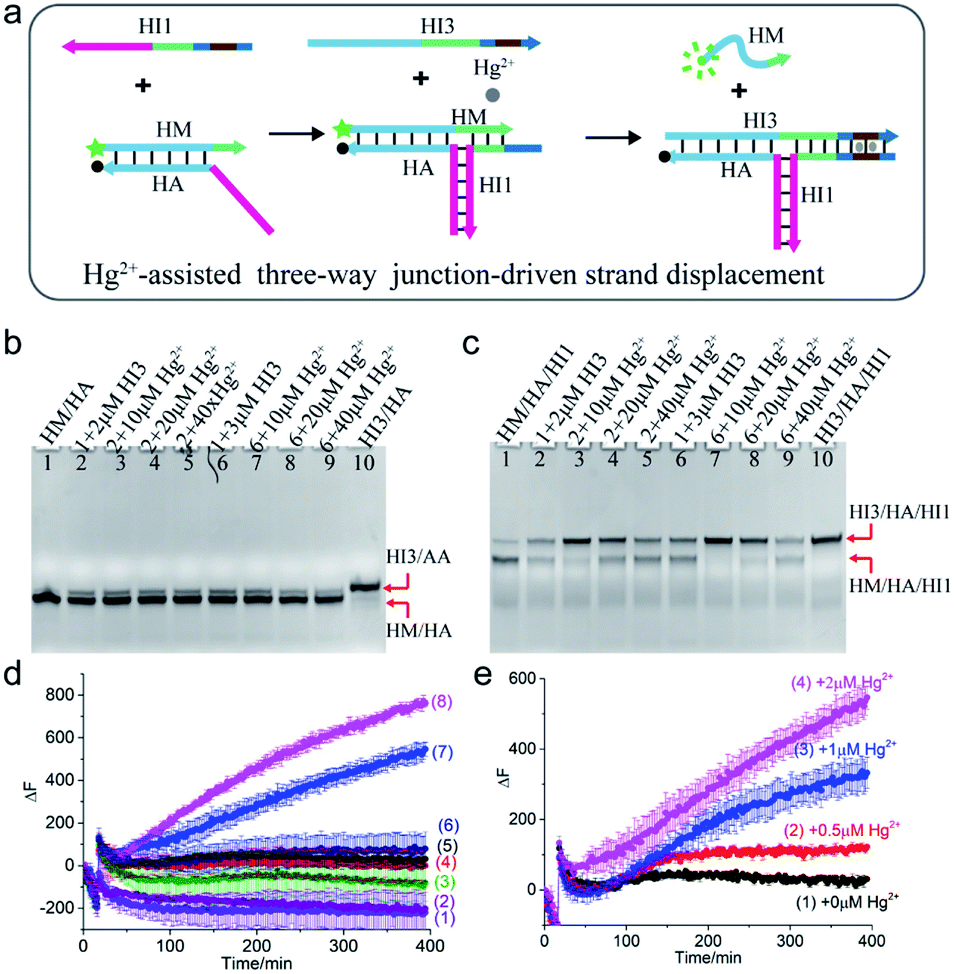 | ||
| Fig. 3 (a) Schematic illustration of Hg2+-assisted TWJ-driven strand displacement. (b and c) Native 15% polyacrylamide gel analysis of the reaction. For gel analysis, the samples were incubated at 25 °C for 4 hours before electrophoresis. (b) Strand exchange reaction in the absence of toehold provided by HI1; (c) Hg2+-assisted TWJ-driven strand displacement reaction. DNA strands added in every lane are indicated above the image. The concentration of DNA complex HM/HA/HI1 or HM/HA in each lane is 2 μM. (d) Fluorescence intensity analysis of the reaction. Curve 1: HM/HA + 100 nM HI3 + 2 μM Hg2+; curve 2: HM/HA + 150 nM HI3 + 2 μM Hg2+; curve 3: HM/HA + 100 nM HI3; curve 4: HM/HA + 150 nM HI3; curve 5: HM/HA/HI1 + 100 nM HI3; curve 6: HM/HA/HI1 + 150 nM HI3; curve 7: HM/HA/HI1 + 100 nM HI3 + 2 μM Hg2+; curve 8: HM/HA/HI1 + 150 nM HI3 + 2 μM Hg2+. (e) Fluorescence intensity analysis of the reaction with 100 nM AI3 at different concentrations of Hg2+. Curve 1: 0 μM; curve 2: 0.5 μM; curve 3: 1 μM; curve 4: 2 μM. For fluorescence assay, [HM/HA/HI1] = 100 nM, [HM/HA] = 100 nM. The arrow represents the addition of Hg2+ and HI3. DNA sequences of single-stranded species were shown in ESI Table S1.† | ||
The Hg2+-assisted displacement reaction was verified by both gel electrophoresis and time-course fluorescence assay. Firstly, the assembling of the TWJ structure was confirmed by PAGE in Fig. S3.† To compare the displacement reaction under different conditions, several control experiments were carried out including the presence or absence of the toehold and Hg2+. For the reaction without HI1 providing the toehold, when HI3 was added into HM/HA and reacted for 4 hours, a small amount of new product HI3/HA was observed (Fig. 3b lane 2 and lane 6). In contrast, after the addition of HI3 to the TWJ structure HM/HA/HI1, both bands corresponding to HM/HA/HI1 and HI3/HA/HI1 appeared (Fig. 3c lane 2 and lane 6). The above PAGE results indicated that under the condition without toehold or with mismatched toehold, the slow displacement reaction still occurred and mismatched toehold led to stronger reaction than without toehold. To further regulate the reaction, Hg2+ was involved. As shown in Fig. 3b lane 3–5 and lane 7–9, in the absence of the toehold region by H11, even with Hg2+, the band of HM/HA almost remained unchanged compared to lane 2. In the presence of both 10 μM Hg2+ and toehold region provided by 2 μM HI1, HM was hugely displaced from the TWJ structure HM/HA/HI1 (Fig. 3c lane 3). Moreover, with the same concentration of 10 μM Hg2+, a higher concentration of HI3 (3 μM) promoted the reaction (Fig. 3c lane 7). A similar situation was observed with other concentrations of mercury ions. However, upon the addition of 40 μM Hg2+ (Fig. 3c lane 5 and lane 9), there was still a large amount of HM/HA/HI1 remained. This indicated that moderate concentrations of Hg2+ promoted the reaction, and excessive Hg2+ inhibited the reaction.
In addition, the reaction time was extended to 20 hours and the electrophoresis images were obtained. For the TWJ structure containing mismatched toehold (Fig. S4a†), it could be observed that when 2 μM or 3 μM HI3 was added, no matter whether 10 μM or 20 μM Hg2+ was added, HM/HA/HI1 was almost exhausted and the displacement product HI3/HA/HI1 was generated; whereas, for samples without mercury ions, a large amount of HA/HM/HI1 still remained. As shown in Fig. S4b,† for the HM/HA structure without toehold, even after 20 hour reaction, the band remained unchanged.
Furthermore, the time-dependent fluorescence assay was also carried out. As can be seen in Fig. 3d, upon addition of 100 nM or 150 nM HI3 and 2 μM Hg2+, only the fluorescence intensity of HM/HA/HI1 had a dramatic increase (curve 7 for 100 nM HI3 and curve 8 for 150 nM HI3). Moreover, a higher concentration of HI3 led to a faster increase of fluorescence signal, demonstrating that the concentration of HI3 affected the reaction rate. Meanwhile, upon the addition of the same concentration of HI3 (100 nM), with the concentration of Hg2+ increasing from 0.5 μM to 2 μM, the fluorescence signal increased correspondingly (Fig. 3e), confirming the regulation effect of Hg2+. The fluorescence results with 150 nM input and various concentrations of Hg2+ could be found in Fig. S5.† It is worth noting that, at a lower concentration (100 nM) of DNA strands, only when inputs HI1, HI3, and Hg2+ were all present, the strand HM could be displaced. Whereas, at a higher concentration (2 μM) of DNA strands, even without HI1 or Hg2+, part of the strand HM was displaced.
The experimental results verified the mechanism of Hg2+-assisted three-way DNA junction-driven strand displacement reaction. In addition, just like T–Hg2+–T base pairs, the C–Ag+–C base pairs could also be incorporated in the three-junction structure to regulate the reaction.
pH-Assisted TWJ-driven strand displacement
Nature often utilizes finely pH-regulated biomolecules to tune several biological activities. Here, pH was employed as a cofactor to modulate the TWJ-driven strand displacement. As shown in Fig. 4a, the C-rich sequence was incorporated into the TWJ structure. Specifically, the purple domain of PI1 and PI3 corresponded to the C-rich sequence and the dark blue domain indicated the 12 nt toehold sequence. Under the neutral or basic condition, the C-rich sequence functioned as a mismatched segment between the toehold and branch migrating domains, and impeded the displacement reaction. At acidic conditions, the C-rich sequence on the strands PI1 and PI3 assembled into an i-motif structure, thus promoted the displacement reaction.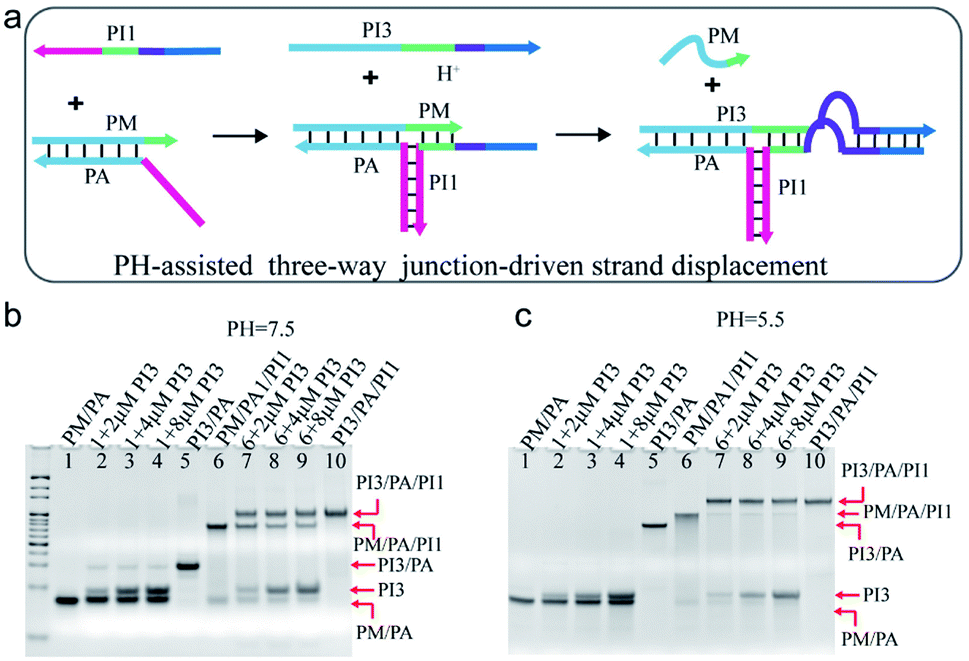 | ||
| Fig. 4 (a) Schematic illustration of pH-assisted TWJ-driven strand displacement. (b and c) Native 15% polyacrylamide gel comparison of TWJ-driven strand displacement and the reaction without the toehold provided by PI1 at (b) pH = 7.5 and (c) pH = 5.5. DNA strands added in every lane are indicated above the image. The concentration of the DNA complex in each lane is 2 μM. For gel analysis, the samples were incubated at 25 °C for 20 hours before electrophoresis. DNA sequences of single-stranded species were shown in ESI Table S1.† | ||
The pH-assisted displacement reaction was confirmed by the native PAGE experiment. At first, the formation of the TWJ structure was identified by PAGE in Fig. S6.† To analyse the displacement reaction under different conditions, several control experiments were performed, including (i) PM/PA + PI3 and (ii) PM/PA/PI1+PI3. Fig. 4b and c showed the PAGE results at neutral and acidic conditions after 20 h reaction respectively. The band of complex PM/PA almost remained unchanged upon addition of 2 μM, 4 μM, and 8 μM PI3 at pH = 7.5 (Fig. 4b lane 2–4) and pH = 5.5 (Fig. 4c lane 2–4). The results indicated that PI3 alone was insufficient to displace PM from complex PM/PA. Whereas, after PI3 was added to the TWJ structure PM/PA/PI1, a new slower band corresponding to PI3/PA/PI1 was observed at pH = 7.5 (Fig. 4b lane 7–9). This indicated that with the help of the TWJ structure, even with the mismatched sequence under neutral conditions, the displacement reaction still partly occurred. It is also found that with the concentration of input PI3 increasing, the remained PM/PA/PI1 decreased. However, at pH = 5.5, the complex PM/PA/PI1 was almost totally consumed (Fig. 4c lane 7–9), demonstrating that the i-motif structure at acidic condition promoted the displacement reaction.
The reaction time was further shortened to half an hour (Fig. S7†). At pH = 7.5, the gel showed similar results as 20 h reaction time (Fig. S7† lane 7–9). At pH = 5.5, after 30 min reaction, PI3 of concentration 2 μM was not able to totally displace strand PM from the TWJ structure PM/PA/PI1, but PI3 of higher concentration (4 μM or 8 μM) succeeded (Fig. S7† lane 2–4), indicating that higher concentration of PI3 could speed up the pH-assisted TWJ-driven strand displacement reaction.
Above all, the experimental results confirmed that the synergy between DNA input and pH regulated the strand displacement reaction. On the other hand, the leakage was observed at a relative higher sample concentration in the electrophoresis experiment. Furthermore, the proposed method compared the difference in the presence or the absence of the target analytes and detected the samples in a relative higher concentration without the assistance of other amplification method. Thus, to achieve a high sensitivity in the biosensing applications, the proposed method should be coupled with other amplification methods.
Conclusions
In summary, a cofactor-assisted TWJ strategy to regulate strand displacement reaction was reported, in which specific domains such as aptamer sequences were incorporated into the structure. In this way, the structure could respond to not only nucleic acid inputs but also environmental stimuli. As a demonstration of the strategy, ATP, Hg2+, and pH together with nucleic acid inputs were used as regulators to tune the displacement reaction. The experimental results verified that the regulation of the strand displacement was well performed. With the advantage of flexible tunability, the proposed strategy is suitable to construct dynamic nanodevices and multi-input circuits. Moreover, the detailed kinetics analysis of the cofactor-assisted strand displacement reaction and the introduction of more cofactors such as proteins into the system will increase the tunability of the strategy. Combined with signal amplification methods, the sensitivity of the nanodevices established by the proposed method could be improved. Thus, the strategy also provides a way to regulate strand displacement reaction in a modular and flexible manner and have potentials in multi-biosensing or biocomputing.Author contributions
Y. F. J. and Y. X. H. designed and performed the experiment. Y. F. J. analysed the data. Y. F. J. and Y. X. H. wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Notes and references
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS PubMed.
- J. H. Reif, Science, 2011, 332, 1156–1157 CrossRef CAS PubMed.
- G. Chatterjee, N. Dalchau, R. A. Muscat, A. Phillips and G. Seelig, Nat. Nanotechnol., 2017, 12, 920–927 CrossRef CAS PubMed.
- K. M. Cherry and L. Qian, Nature, 2018, 559, 370–376 CrossRef CAS PubMed.
- Y. Tang, Z. Wang, X. Yang, J. Chen, L. Liu, W. Zhao, X. C. Le and F. Li, Chem. Sci., 2015, 6, 5729–5733 RSC.
- X. Yang, Y. Tang, S. D. Mason, J. Chen and F. Li, ACS Nano, 2016, 10, 2324–2330 CrossRef CAS PubMed.
- P. Song, J. Shen, D. Ye, B. Dong, F. Wang, H. Pei, J. Wang, J. Shi, L. Wang, W. Xue, Y. Huang, G. Huang, X. Zuo and C. Fan, Nat. Commun., 2020, 11, 838 CrossRef CAS PubMed.
- H. Zhang, F. Li, B. Dever, X.-F. Li and X. C. Le, Chem. Rev., 2013, 113, 2812–2841 CrossRef CAS PubMed.
- H. Ramezani and H. Dietz, Nat. Rev. Genet., 2020, 21, 5–26 CrossRef CAS PubMed.
- S. M. Douglas, B. Ido and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- A. J. Thubagere, W. Li, R. F. Johnson, Z. Chen, S. Doroudi, Y. L. Lee, G. Izatt, S. Wittman, N. Srinivas and D. Woods, Science, 2017, 357, eaan6558 CrossRef PubMed.
- S. P. Li, Q. Jiang, S. L. Liu, Y. L. Zhang, Y. H. Tian, C. Song, J. Wang, Y. G. Zou, G. J. Anderson, J. Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. B. Zhou, G. J. Nie, H. Yan, B. Q. Ding and Y. L. Zhao, Nat. Biotechnol., 2018, 36, 258–264 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed.
- F. Li, Y. W. Lin and X. C. Le, Anal. Chem., 2013, 85, 10835–10841 CrossRef CAS PubMed.
- A. J. Genot, D. Y. Zhang, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 2177–2182 CrossRef CAS PubMed.
- N. E. C. Haley, T. E. Ouldridge, I. M. Ruiz, A. Geraldini, A. A. Louis, J. Bath and A. J. Turberfield, Nat. Commun., 2020, 11, 2562 CrossRef CAS PubMed.
- J. Cabello-Garcia, W. Bae, G.-B. V. Stan and T. E. Ouldridge, ACS Nano, 2021, 15, 3272–3283 CrossRef CAS PubMed.
- J. Zhu, F. Bošković, B.-N. T. Nguyen, J. R. Nitschke and U. F. Keyser, Nano Lett., 2021, 21, 1368–1374 CrossRef CAS PubMed.
- J. Zhu, L. Zhang, S. Dong and E. Wang, ACS Nano, 2013, 7, 10211–10217 CrossRef CAS PubMed.
- Z. Libing, G. Shaojun, Z. Jinbo, Z. Zhixue, L. Tao, L. Jing, D. Shaojun and W. Erkang, Anal. Chem., 2015, 87, 11295–11300 CrossRef PubMed.
- J. Zhu, L. Zhang, Z. Zhou, S. Dong and E. Wang, Anal. Chem., 2014, 86, 312–316 CrossRef CAS PubMed.
- X. Yang, Y. Tang, S. M. Traynor and F. Li, J. Am. Chem. Soc., 2016, 138, 14076–14082 CrossRef CAS PubMed.
- A. J. Genot, J. Bath and A. J. Turberfield, Angew. Chem., Int. Ed., 2013, 52, 1189–1192 CrossRef CAS PubMed.
- X. Chen, J. Am. Chem. Soc., 2012, 134, 263–271 CrossRef CAS PubMed.
- L. J. Li, W. K. Zhang, X. F. Tang, Z. J. Li, Y. Z. Wu and X. J. Xiao, Chem. Commun., 2020, 56, 8794–8797 RSC.
- W. Ding, W. Deng, H. Zhu and H. Liang, Chem. Commun., 2013, 49, 9953–9955 RSC.
- W. Tang, H. Wang, D. Wang, Y. Zhao, N. Li and F. Liu, J. Am. Chem. Soc., 2013, 135, 13628–13631 CrossRef CAS PubMed.
- B. M. G. Janssen, V. R. Martijn, V. B. Lotte and M. Maarten, Angew. Chem., Int. Ed., 2015, 54, 2530–2533 CrossRef CAS PubMed.
- G. D. McLachlan, S. M. Cahill, M. E. Girvin and S. C. Almo, Biochemistry, 2007, 46, 6931–6943 CrossRef CAS PubMed.
- Y. Xing, Z. Yang and D. Liu, Angew. Chem., Int. Ed., 2011, 50, 11934–11936 CrossRef CAS PubMed.
- A. Alessia, Z. Bin, P. Alessandro, I. Andrea, C. Matteo, F. Chunhai and R. Francesco, J. Am. Chem. Soc., 2014, 136, 16469–16472 CrossRef PubMed.
- W. Engelen, L. H. H. Meijer, B. Somers, T. F. A. de Greef and M. Merkx, Nat. Commun., 2017, 8, 14473 CrossRef CAS PubMed.
- J. Zhu, L. Zhang, Z. Zhou, S. Dong and E. Wang, Chem. Commun., 2014, 50, 3321–3323 RSC.
- W. Tang, S. Hu, H. Wang, Y. Zhao, N. Li and F. Liu, Chem. Commun., 2014, 50, 14352–14355 RSC.
- L.-L. Wang, Q.-L. Zhang, Y. Wang, Y. Liu, J. Lin, F. Xie and L. Xu, Chem. Sci., 2021, 12, 8698–8705 RSC.
- F. Li, H. Zhang, C. Lai, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2012, 51, 9317–9320 CrossRef CAS PubMed.
- Z. Wang, Y. Hu and L. Pan, Angew. Chem., Int. Ed., 2020, 59, 14979–14985 CrossRef CAS PubMed.
- R. Montagud-Martínez, M. Heras-Hernández, L. Goiriz, J.-A. Daròs and G. Rodrigo, ACS Synth. Biol., 2021, 10, 950–956 CrossRef PubMed.
- Y. Du, P. Peng and T. Li, Chem. Commun., 2018, 54, 6132–6135 RSC.
- H. H. Wang, J. Zheng, Y. D. Sun and T. Li, Biosens. Bioelectron., 2018, 117, 729–735 CrossRef CAS PubMed.
- K. Shi, S. Xie, R. Tian, S. Wang and Z. Nie, Sci. Adv., 2021, 7, eabc7802 CrossRef CAS PubMed.
- H. Peng, A. M. Newbigging, M. S. Reid, J. S. Uppal, J. Xu, H. Zhang and X. C. Le, Anal. Chem., 2020, 92, 292–308 CrossRef CAS PubMed.
- X. Zhou, Q. Zhu and Y. Yang, Biosens. Bioelectron., 2020, 165, 112422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05242j |
| This journal is © The Royal Society of Chemistry 2021 |