
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of xanthohumol and xanthohumol-d3 from naringenin†
Joanna Andrusiakab,
Kinga Mylkiecd,
Małgorzata Wysockab,
Jacek Ścianowski a,
Andrzej Wolanab and
Marcin Budny*b
a,
Andrzej Wolanab and
Marcin Budny*b
aDepartment of Organic Chemistry, Faculty of Chemistry, Nicolaus Copernicus University, Gagarina 7, 87-100 Toruń, Poland
bSynthex Technologies Sp. z o.o., Gagarina 7/134B, 87-100 Toruń, Poland. E-mail: budny@synthex.com.pl
cDepartment of Biomedical and Polymer Chemistry, Faculty of Chemistry, Nicolaus Copernicus University, Gagarina 7, 87-100 Toruń, Poland
dNoctiluca S.A., Gagarina 7/41B, 87-100 Toruń, Poland
First published on 31st August 2021
Abstract
A six-step synthesis of xanthohumol (1a) and its d3-derivative (1b) from easily accessible naringenin is reported. The prenyl side chain was introduced by Mitsunobu reaction followed by the europium-catalyzed Claisen rearrangement and base-mediated opening of chromanone gave access to an α,β-conjugated ketone system. Compound 1b was used as an internal standard in stable isotope dilution assays of 1a in two Polish beers.
Xanthohumol (1a, Scheme 1A) is a naturally occurring prenylated chalcone produced by lupulin glands in female inflorescences of hop plants.1 In recent years, 1a has attracted significant attention due to its vast range of biological activities including cancer-preventive, antioxidative, anti-inflammatory, and antiviral.2–8 Such properties combined with low toxicity to the human body make 1a a prospective therapeutic agent, diet supplement, or ingredient of cosmetics.9 Although 1a was isolated from natural sources in 1913,10 the first synthesis of this compound was reported as late as in 2007.11 Several other syntheses have been reported since then, but only minor improvements have been achieved.12–14
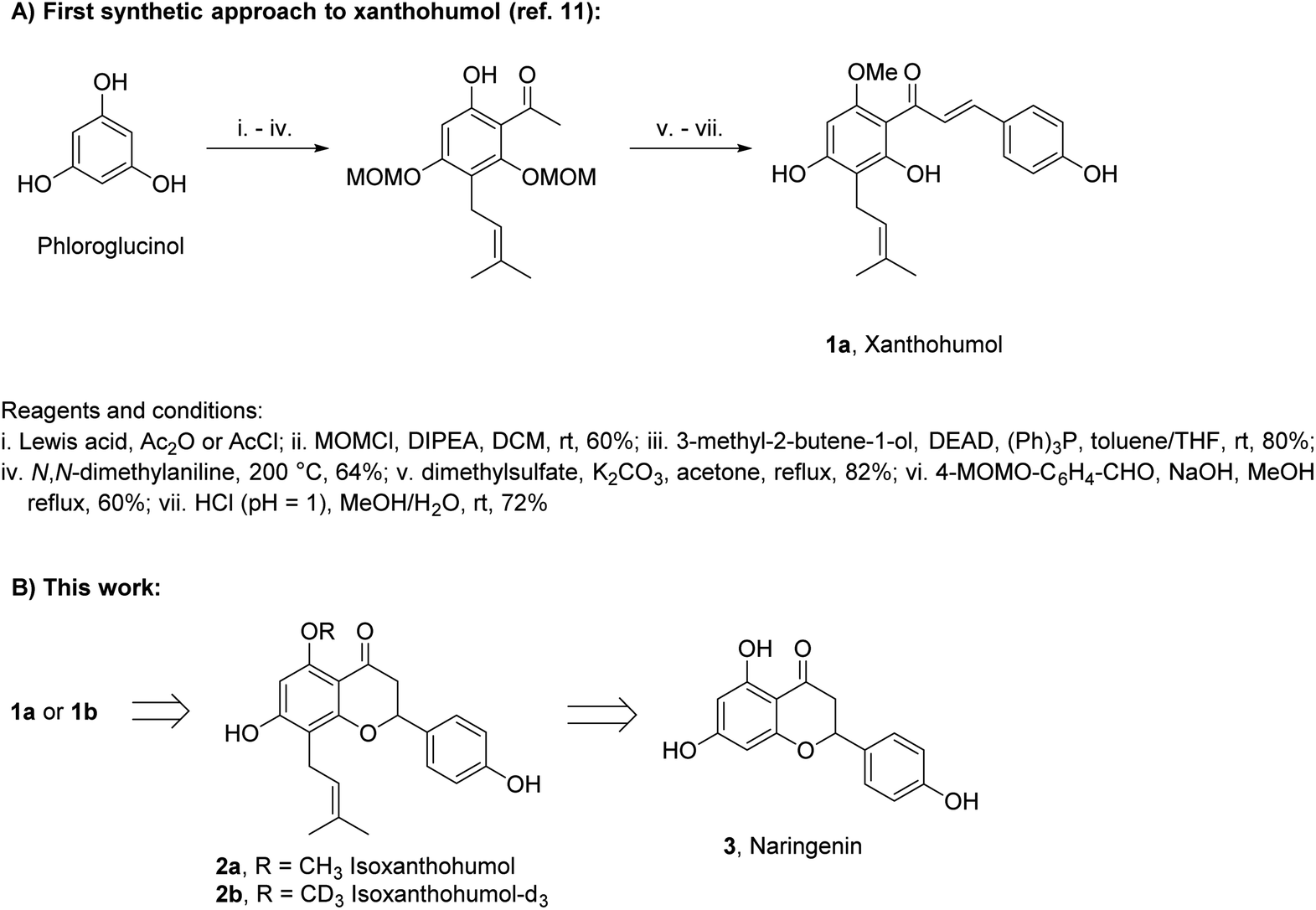 | ||
| Scheme 1 The background of the study. | ||
Bioactive compounds labeled with stable isotopes (deuterium, carbon-13) are widely applied in metabolomic studies for tracking metabolic pathways and as internal standards in stable isotope dilution assays.15,16 Deuterated compounds are also considered as attractive drug candidates due to the influence of the kinetic isotope effect on pharmacokinetics.17–19 Although approaches to 13C-enriched xanthohumol20,21 and hydrogen/deuterium exchange in 1a22 were reported, no scalable and cost-effective synthesis of the deuterium-labeled derivative of 1a (i.e. 1b) has been disclosed to date.
Two main challenges have to be faced in the synthesis of 1a: (i) construction of a pentasubstituted aromatic ring containing a prenyl side chain and (ii) selection of suitable protecting groups for phenols. In the case of (i), phloroglucinol is used as a precursor and an acyl-substituent is introduced by Friedel–Crafts acylation with a subsequent Claisen–Schmidt condensation. The prenyl side-chain is introduced by Mitsunobu alkylation, followed by Claisen rearrangement. In the case of (ii), acid-sensitive alkoxymethyl protecting groups, removable under conditions in which 1a does not cyclize to isoxanthohumol (2a), are used most often (Scheme 1A).
In this study, we have developed a synthetic approach for the formation of 1a and its deuterated analog 1b. We envisioned that both 1a and 1b can be directly obtained by the base-promoted chromanone ring-opening of 2a or 2b, which in turn can be obtained from easily accessible naringenin (3) (ca. 1 $/1 g) via two-step prenylation and Williamson etherification of the phenolic OH (Scheme 1B). The use of 3 as the starting material is beneficial as only one prenyl substituent has to be introduced.
The synthetic route leading to 1a and 1b is depicted in Scheme 2. Our synthesis commenced from naringenin (3), which was selectively converted to diester 4 (Ac2O, pyridine). O-Alkylation of 4 under Mitsunobu conditions (3-methyl-2-butene-1-ol, Ph3P, DIAD), followed by the catalytic Claisen rearrangement of 5 (Eu(fod)3, 1,2-dichloroethane, 80 °C) afforded prenyl-derivative 6. Notably, performing the latter reaction in 1,2-dichloroethane above its boiling point was superior in comparison to earlier reports.23–26 Alkylation of 6 (CH3I, Ag2O or CD3I, Ag2O) afforded 7a/7b in good yields. An alternative approach to 7b involving the alkylation of phenolic OH under Mitsunobu conditions (CD3OD, Ph3P, DIAD) required a large excess of reagents and afforded the product in moderate yield. Basic hydrolysis (KOH, MeOH) of esters afforded isoxanthohumols 2a/2b. Although the chromane ring was stable during the hydrolysis, it could be opened under more harsh conditions (DBU, DMF, 70 °C),27,28 leading to 1a/1b in good yields after a mild acidic workup.
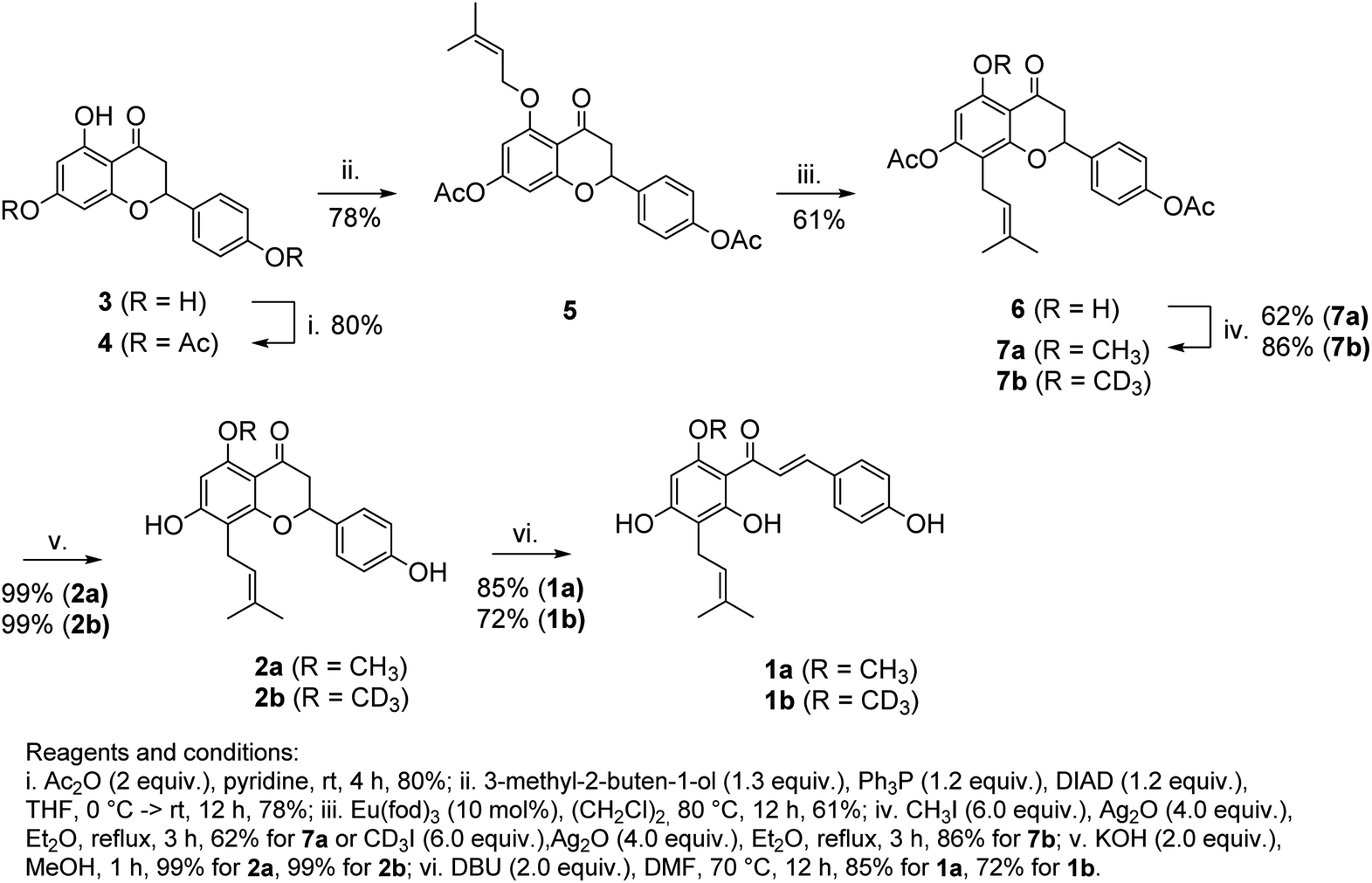 | ||
| Scheme 2 The synthetic route to 1a and 1b. | ||
With 1a and 1b in hand, we investigated their MS-fragmentation patterns in electrospray ionization in positive and negative ion modes. The MRM transitions were found by an automatic procedure and they are listed in Table 1 (see ESI† for details). In positive ion mode, the most intensive product ions in fragmentation of 1a were ions with m/z values of 178.9, 299.0, 113.0, and 150.9. Corresponding ions with m/z +3 values can be found in the fragmentation of 1b. On the other hand, in the negative ion mode, the same product ions are observed both in the case of 1a and 1b, indicating that the CH3/CD3 groups were lost during fragmentations.
| Ionization mode | 1a | 1b | ||
|---|---|---|---|---|
| Precursor ion | Product ion | Precursor ion | Product ion | |
| ESI(+) | 355.0 | 178.9 | 358.0 | 182.0 |
| 299.0 | 302.0 | |||
| 113.0 | 115.9 | |||
| 150.9 | 107.9 | |||
| 93.0 | 154.0 | |||
| ESI(−) | 353.0 | 119.1 | 356.0 | 119.1 |
| 233.0 | 236.0 | |||
| 295.1 | 295.2 | |||
| 218.2 | 175.0 | |||
| 175.0 | 218.1 | |||
| 189.2 | 168.2 | |||
One of the criteria for an effective internal standard is the coelution of the labeled and non-labeled compounds during the HPLC analysis. This is particularly important in case of deuterium-labeled compounds as with the increase in the number of deuterons in the molecule, retention times may be extended. The retention times of 1a and 1b under different HPLC conditions are listed in Table 2. Notably, coelution of 1a and 1b is observed in case of the XB-C18 stationary phase (entry 1). Minor differences were observed when separation was attempted on C18-PFP (entry 2) and polar-C18 (entry 3) stationary phases.
| Entry | Conditions | Retention time [min] | |
|---|---|---|---|
| 1a | 1b | ||
| 1 | Column: XB-C18, 100 × 3.0 mm, 2.6 μm, 100 Å; flow: 0.55 mL min−1; oven: 35 °C; gradient MeOH/0.1% HCO2H(aq): from 5% MeOH to 95% MeOH | 20.030 | 20.034 |
| 2 | Column: Ace 5 C18-PFP, 250 × 4.6 mm; flow: 1.0 mL min−1, oven: 35 °C; isocrat. MeOH/0.1% HCO2H(aq): 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 :20 |
19.615 | 19.700 |
| 3 | Column: polar-C18, 100 × 3.0 mm, 2.6 μm, 100 Å; flow: 0.55 mL min−1; oven: 35 °C; isocrat.: MeOH/0.1% HCO2H(aq): 65:35 |
6.590 | 6.520 |
As an example of applications, compound 1b was used as an internal standard in a stable isotope dilution assay of xanthohumol (1a) in two Polish beers (determined concentrations: 0.4069 mg L−1 and 0.5488 mg L−1, respectively). The developed MRM method allowed for the direct analysis of 1a and any preconcentration of the analyte was not needed.
In conclusion, we have developed a six-step synthesis of xanthohumol (1a) and its deuterated analog 1b from naringenin (3) in total 19.8% yield for 1a and 23.3% for 1b. In a key step, isoxanthohumols 2a/2b were converted to the target compounds under basic conditions. The overall synthetic route was scalable and was used in the synthesis of 1a on a 5 g scale. The MRM transitions of 1b and its coelution with 1a makes 1b a suitable internal standard for the stable isotope dilution assay.
Experimental section
Tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) were dried over activated molecular sieves 4 Å. Other solvents and reagents were of analytical grade and were used as received without further purification. NMR spectra were recorded on Bruker Avance III 300 MHz, 400 MHz, and 700 MHz spectrometers. Chemical shifts are reported as δ values in parts per million relative to the residual solvent signal (CDCl3: δ = 7.24 ppm for 1H and 77.23 ppm for 13C; CD3OD: δ = 3.31 ppm for 1H and 49.15 ppm for 13C). Coupling constants are in hertz (Hz). The following abbreviations are used for spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. HPLC-ESI MS analyses were performed on a triple quadrupole Shimadzu LCMS 8030. Infrared spectra were recorded on a PerkinElmer UATR two instrument and are reported in cm−1. Melting points were determined in open glass capillaries and are uncorrected. Silica Gel 60, Merck 230–400, was used for preparative column chromatography. Sigma-Aldrich TLC plates (silica gel on Al foil with fluorescent indicator 254 nm) were used for analytical TLC. UV lamp (λ = 254 nm) and solution of phosphomolybdic acid in ethanol were used for the visualization of TLC plates.4-(7-Acetoxy-5-hydroxy-4-oxochroman-2-yl)phenyl acetate (4)
To a suspension of naringenin (3) (1.0 equiv., 73.5 mmol, 20.0 g) in pyridine (60 mL), acetic anhydride (2.0 equiv., 147.0 mmol, 15.0 g) was added portionwise. The resulting solution was stirred until the complete consumption of the starting material. The reaction was quenched with conc. HCl (60 mL) and water (200 mL), extracted with EtOAc (3 × 100 mL), and washed with water (3 × 100 mL) and brine (100 mL). The extract was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was recrystallized from MeOH to afford 4 (20.94 g, 80%) as a white solid.Mp. 143–144 °C; IR (neat, cm−1): 2964, 1742, 1650, 1370, 1276, 1210, 1076, 1014, 840, 769; 1H NMR (700 MHz, CDCl3), δ (ppm): 11.81 (s, 1H), 7.46–7.43 (m, 2H), 7.16–7.13 (m, 2H), 6.30 (d, J = 2.1 Hz, 1H), 6.29 (d, J = 2.1 Hz, 1H), 5.44 (dd, J1 = 13.4 Hz, J2 = 2.9 Hz, 1H), 3.08 (dd, J1 = 17.2 Hz, J2 = 13.4 Hz, 1H), 2.85 (dd, J1 = 17.2 Hz, J2 = 2.9 Hz, 1H), 2.30 (s, 3H), 2.27 (s, 3H). 13C NMR (75 MHz, CDCl3), δ (ppm): 196.9, 169.4, 168.4, 163.6, 162.4, 158.7, 151.3, 135.8, 127.5, 122.3, 106.4, 103.6, 101.9, 79.0, 43.8, 21.4, 21.3; anal. calcd for C19H16O7: C, 64.04; H, 4.53. Found: C, 64.17; H, 4.60.
4-(7-Acetoxy-5-((3-methylbut-2-en-1-yl)oxy)-4-oxochroman-2-yl)phenyl acetate (5)
To a solution of 4 (1 equiv., 30.3 mmol, 10.792 g) in anhydrous THF (270 mL), 3-methylbut-2-en-1-ol (1.3 equiv., 39.4 mmol, 3.39 g) and triphenylphosphine (1.2 equiv., 36.4 mmol, 9.55 g) were added. The resulting solution was cooled to 0 °C, and then DIAD (1.2 equiv., 36.4 mmol, 7.36 g) was added portionwise. The resulting mixture was stirred at 0 °C for 30 min and then at room temperature (rt) overnight. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in MTBE (250 mL), and the resulting solution was stirred at 0 °C for 30 min. The precipitated triphenylphosphine oxide was filtered off and the solution was concentrated under a reduced pressure. The residue was purified by flash chromatography (petroleum ether-ethyl acetate = 70:30 → 60:40) affording 5 (10.03 g, 78%) as a pale yellow solid.
Mp. 113–116 °C; IR (neat, cm−1): 2979, 1764, 1680, 1596, 1373, 1197, 1108, 1076, 1031, 904, 843; 1H NMR (700 MHz, CDCl3), δ (ppm): 7.46–7.43 (m, 2H), 7.13–7.11 (m, 2H), 6.40 (d, J = 2.1 Hz, 1H), 6.29 (d, J = 2.1 Hz, 1H), 5.53–5.49 (m, 1H), 5.41 (dd, J1 = 13.2 Hz, J2 = 2.7 Hz, 1H), 4.62–4.56 (m, 2H), 2.99 (dd, J1 = 16.4 Hz, J2 = 13.2 Hz, 1H), 2.80 (dd, J1 = 16.4 Hz, J2 = 2.7 Hz, 1H), 2.29 (s, 3H), 2.28 (s, 3H), 1.77 (br s, 3H), 1.72 (br s, 3H). 13C NMR (175 MHz, CDCl3), δ (ppm): 189.2, 169.6, 168.6, 163.9, 161.4, 156.6, 151.0, 138.5, 136.3, 127.5, 122.2, 119.1, 109.8, 103.4, 100.1, 78.9, 66.3, 46.0, 26.0, 21.4, 21.3, 18.6; anal. calcd for C24H24O7: C, 67.91; H, 5.70. Found: C, 68.01; H, 5.68.
4-(7-Acetoxy-5-hydroxy-8-(3-methylbut-2-en-1-yl)-4-oxochroman-2-yl)phenyl acetate (6)
In a 50 mL pressure vial, 5 (1 equiv., 6.96 mmol, 2.95 g), Eu(fod)3 (0.1 equiv., 0.696 mmol, 722 mg) and 1,2-dichloroethane (6 mL) were placed. The resulting mixture was stirred at 80 °C (temp. of the oil bath) overnight. Then, the mixture was cooled to rt and directly purified by flash chromatography (petroleum ether-ethyl acetate = 90:10 → 70:30) to afford 6 (1.80 g, 61%) as a white solid.
Mp. 143–144 °C; IR (neat, cm−1): 2975, 1763, 1637, 1428, 1372, 1189, 1066, 1012, 896; 1H NMR (700 MHz, CDCl3), δ (ppm): 11.70 (s, 1H), 7.46–7.43 (m, 2H), 7.16–7.13 (m, 2H), 6.29 (s, 1H), 5.43 (dd, J1 = 13.3 Hz, J2 = 2.9 Hz, 1H), 5.06–5.01 (m, 1H), 3.18–3.10 (m, 2H), 3.06 (dd, J1 = 17.1 Hz, J2 = 13.3 Hz, 1H), 2.87 (dd, J1 = 17.1 Hz, J2 = 2.9 Hz, 1H), 2.30 (s, 3H), 2.28 (s, 3H), 1.64–1.62 (m, 3H), 1.58–1.56 (m, 3H). 13C NMR (175 MHz, CDCl3), δ (ppm): 197.3, 169.4, 168.4, 161.1, 160.0, 157.0, 151.2, 136.1, 132.1, 127.4, 122.2, 121.8, 113.8, 106.8, 104.3, 78.9, 43.8, 25.8, 22.9, 21.3, 21.1, 18.0; anal. calcd for C24H24O7: C, 67.91; H, 5.70; N, 14.42. Found: C, 67.94; H, 5.82.
4-(7-Acetoxy-5-methoxy-8-(3-methylbut-2-en-1-yl)-4-oxochroman-2-yl)phenyl acetate (7a)
To a suspension of 6 (1 equiv., 9.46 mmol, 4.012 g) and freshly prepared Ag2O (4 equiv., 37.85 mmol, 8.770 g) in Et2O (95 mL), MeI (6 equiv., 56.772 mmol, 8.058 g, 3.66 mL) was added. The resulting mixture was refluxed for 3 h. Then, the reaction mixture was cooled to rt, silver salts were filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (petroleum ether-ethyl acetate = 60:40) affording 7a (2.59 g, 62%) as a white solid.
Mp. 140–141 °C; IR (neat, cm−1): 2962, 1753, 1649, 1592, 1369, 1203, 1098, 1055, 902, 834; 1H NMR (700 MHz, CDCl3), δ (ppm): 7.46–7.42 (m, 2H), 7.14–7.10 (m, 2H), 6.28 (s, 1H), 5.41 (dd, J1 = 13.3 Hz, J2 = 2.8 Hz, 1H), 5.07–5.02 (m, 1H), 3.86 (s, 3H), 3.21–3.15 (m, 2H), 2.98 (dd, J1 = 16.4 Hz, J2 = 13.3 Hz, 1H), 2.83 (dd, J1 = 16.4 Hz, J2 = 2.8 Hz, 1H), 2.31 (s, 3H), 2.30 (s, 3H), 1.64–1.63 (m, 3H), 1.58–1.56 (m, 3H). 13C NMR (175 MHz, CDCl3), δ (ppm): 190.1, 169.6, 168.9, 161.9, 159.6, 154.8, 150.9, 136.5, 132.2, 127.4, 122.1, 121.7, 115.4, 109.9, 99.6, 78.8, 56.5, 45.8, 25.9, 23.2, 21.4, 21.1, 18.00; anal. calcd for C25H26O7: C, 68.48; H, 5.98. Found: C, 68.21; H, 6.11.
4-(7-Acetoxy-5-(methoxy-d3)-8-(3-methylbut-2-en-1-yl)-4-oxochroman-2-yl)phenyl acetate (7b)
The same procedure as for compound 7a was used. Starting from 6 (2.36 mmol, 1.0 g) 7b (0.89 g, 86%) as a pale yellow solid was obtained.Mp. 138–139 °C, IR (neat, cm−1): 1754, 1685, 1593, 1362, 1204, 1091, 902. 1H NMR (700 MHz, CDCl3), δ (ppm): 7.46–7.42 (m, 2H), 7.14–7.10 (m, 2H), 6.27 (s, 1H), 5.41 (dd, J1 = 13.1 Hz, J2 = 2.8 Hz, 1H), 5.07–5.03 (m, 1H), 3.21–3.14 (m, 2H), 2.98 (dd, J1 = 16.5 Hz, J2 = 13.2 Hz, 1H), 2.83 (dd, J1 = 16.5 Hz, J2 = 2.8 Hz, 1H), 2.30 (s, 3H), 2.29 (s, 3H), 1.64–1.62 (m, 3H), 1.58–1.56 (m, 3H). 13C NMR (175 MHz, CDCl3), δ (ppm): 189.9, 169.4, 168.8, 161.8, 159.7, 154.9, 151.0, 136.6, 132.2, 127.3, 122.1, 121.8, 115.4, 110.0, 99.6, 78.8, 78.8, 45.8, 25.8, 23.2, 21.3, 21.1, 18.0; anal. calcd for C25H23D3O7: C, 68.01; H, 6.62. Found: C, 68.07; H, 6.60.
7-Hydroxy-2-(4-hydroxyphenyl)-5-methoxy-8-(3-methylbut-2-en-1-yl)chroman-4-one (2a)
To a solution of 7a (1 equiv., 4.349 mmol, 1.905 g) in MeOH (30 mL), KOH (2 equiv., 8.699 mmol, 488 mg) was added. The reaction mixture was stirred until the complete consumption of the starting material. The reaction mixture was neutralized with 2 M HCl, extracted with EtOAc (3 × 60 mL), washed with water (2 × 30 mL) and brine (30 mL), dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (petroleum ether-ethyl acetate = 50:50) to afford 2a (1.524 g, 99%) as a pale yellow solid.
Mp. 157–161 °C; IR (neat, cm−1): 3150, 1646, 1590, 1452, 1410, 1349, 1270, 1088, 827; 1H NMR (700 MHz, CD3OD), δ (ppm): 7.33–7.28 (m, 2H), 6.83–6.79 (m, 2H), 6.11 (s, 1H), 5.28 (dd, J1 = 12.8 Hz, J2 = 3.0 Hz, 1H), 5.15–5.12 (m, 1H), 3.79 (s, 3H), 3.24–3.17 (m, 2H), 2.97 (dd, J1 = 16.7 Hz, J2 = 12.8 Hz, 1H), 2.66 (dd, J1 = 16.7 Hz, J2 = 3.0 Hz, 1H), 1.62 (s, 3H), 1.56 (s, 3H). 13C NMR (700 MHz, CD3OD), δ (ppm): 193.0, 164.4, 164.0, 159.0, 131.8, 131.7, 129.0, 124.1, 116.4, 110.2, 106.1, 93.8, 80.2, 56.1, 46.4, 26.0, 22.9, 18.0; anal. calcd for C21H22O5: C, 71.17; H, 6.26. Found: C, 71.38; H, 6.42.
7-Hydroxy-2-(4-hydroxyphenyl)-5-(methoxy-d3)-8-(3-methylbut-2-en-1-yl)chroman-4-one (2b)
The same procedure as for compound 2a was used. Starting from 7b (4.33 mmol, 1.799 g) 2b (1.530 g, 99%) as a pale yellow solid was obtained.Mp. 150–151 °C; IR (neat, cm−1): 3164, 1591, 1509, 1422, 1359, 1259, 1095, 823; 1H NMR (700 MHz, CD3OD), δ (ppm): 7.33–7.28 (m, 2H), 6.83–6.78 (m, 2H), 6.10 (s, 1H), 5.27 (dd, J1 = 12.8 Hz, J2 = 2.9 Hz, 1H), 5.18–5.11 (m, 1H), 3.24–3.17 (m, 2H), 2.97 (dd, J1 = 16.5 Hz, J2 = 12.8 Hz, 1H), 2.65 (dd, J1 = 16.5 Hz, J2 = 2.9 Hz, 1H), 1.61 (s, 3H), 1.56 (s, 3H). 13C NMR (100 MHz, CD3OD), δ (ppm): 193.0, 164.3, 164.0, 162.0, 158.9, 131.8, 131.7, 129.0, 124.0, 116.4, 110.1, 106.1, 93.6, 80.1, 46.4, 26.1, 22.9, 18.0; anal. calcd for C21H19D3O5: C, 70.57; H, 7.05. Found: C, 70.46; H, 6.96.
(E)-1-(2,4-Dihydroxy-6-methoxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (1a)
To a solution of 2a (1 equiv., 4.349 mmol, 1.544 g) in DMF (20 mL), DBU (2 equiv., 8.699 mmol, 1.324 g) was added dropwise and the reaction mixture was stirred at 70 °C for 12 h. Then, the reaction mixture was neutralized with 2 M HCl, extracted with EtOAc (4 × 60 mL), washed with water (3 × 30 mL) and brine (30 mL). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (petroleum ether-ethyl acetate = 60:40) affording 1a (1.31 g, 85%) as orange solid.
Mp. 151–152 °C; IR (neat, cm−1): 3184, 2916, 1596, 1511, 1437, 1340, 1100, 824, 804; 1H NMR (700 MHz, CD3OD), δ (ppm): 7.80 (d, J = 15.6 Hz, 1H), 7.67 (d, J = 15.6 Hz, 1H), 7.51–7.49 (m, 2H), 6.85–6.81 (m, 2H), 6.02 (s, 1H), 5.22–5.18 (m, 1H), 3.09 (s, 3H), 3.23 (d, J = 7.1 Hz, 2H), 1.76 (s, 3H), 1.65 (s, 3H). 13C NMR (75 MHz, CD3OD), δ (ppm): 194.2, 166.3, 163.6, 161.2, 143.4, 131.5, 131.4, 128.6, 126.0, 124.4, 117.0, 109.6, 106.7, 91.8, 56.3, 26.1, 22.4, 18.0; anal. calcd for C21H22O5: C, 71.17; H, 6.26. Found: C, 71.03; H, 6.20.
(E)-1-(2,4-Dihydroxy-6-(methoxy-d3)-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (1b)
The same procedure as for compound 1b was used.Starting from 2b (4.43 mmol, 1.581 g) 1b (1.141 g, 72%) as a pale yellow solid was obtained.
Mp. 149–151 °C, IR (neat, cm−1): 3302, 2915, 1521, 1439, 1337, 1213, 1157, 1098, 830; 1H NMR (700 MHz, CD3OD), δ (ppm): 7.79 (d, J = 15.5 Hz, 1H), 7.66 (d, J = 15.5 Hz, 1H), 7.51–7.46 (m, 2H), 6.85–6.79 (m, 2H), 6.01 (s, 1H), 5.22–5.18 (m, 1H), 3.23 (d, J = 7.0 Hz, 2H), 1.76 (s, 3H), 1.65 (s, 3H). 13C NMR (100 MHz, CD3OD), δ (ppm): 194.3, 166.3, 163.8, 162.6, 161.1, 143.4, 131.5, 131.4, 128.7, 126.1, 124.4, 117.0, 109.6, 106.7, 91.9, 26.1, 22.4, 18.0; anal. calcd for C21H19D3O5: C, 70.57; H, 7.05. Found: C, 70.55; H, 6.97.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support from Synthex Technologies Sp. z o.o. and Noctiluca S.A. is kindly acknowledged. Financial support from the Polish Ministry of Science and Higher Education (“Industrial Doctorates 1st Edition”) for Joanna Andrusiak is kindly acknowledged.References
- J. F. Stevens and J. E. Page, Xanthohumol and Related Prenylflavonoids from Hops and Beer: To Your Good Health!, Phytochemistry, 2004, 65(10), 1317–1330, DOI:10.1016/j.phytochem.2004.04.025.
- C. Gerhauser, A. Alt, E. Heiss, A. Gamal-Eldeen, K. Klimo, J. Knauft, I. Neumann, H.-R. Scherf, N. Frank, H. Bartsch and H. Becker, Cancer Chemopreventive Activity of Xanthohumol, a Natural Product Derived from Hop 1 Support for This Work Has Been Provided by Verein Zur Förderung Der Krebsforschung in Deutschland e.V. and by Wissenschaftsförderung Der Deutschen Brauwirtschaft e.V, Mol. Cancer Ther., 2002, 1(11), 959–969 CAS.
- M. Liu, P. E. Hansen, G. Wang, L. Qiu, J. Dong, H. Yin, Z. Qian, M. Yang and J. Miao, Pharmacological Profile of Xanthohumol, a Prenylated Flavonoid from Hops (Humulus Lupulus), Molecules, 2015, 20(1), 754–779, DOI:10.3390/molecules20010754.
- M. Stompor, K. Dancewicz, B. Gabryś and M. Anioł, Insect Antifeedant Potential of Xanthohumol, Isoxanthohumol, and Their Derivatives, J. Agric. Food Chem., 2015, 63(30), 6749–6756, DOI:10.1021/acs.jafc.5b02025.
- Z. A. Nowakowska, Review of Anti-Infective and Anti-Inflammatory Chalcones, Eur. J. Med. Chem., 2007, 42(2), 125–137, DOI:10.1016/j.ejmech.2006.09.019.
- C. Gerhäuser, Beer Constituents as Potential Cancer Chemopreventive Agents, Eur. J. Cancer, 2005, 41(13), 1941–1954, DOI:10.1016/j.ejca.2005.04.012.
- B. Orlikova, D. Tasdemir, F. Golais, M. Dicato and M. Diederich, Dietary Chalcones with Chemopreventive and Chemotherapeutic Potential, Genes Nutr., 2011, 6(2), 125–147, DOI:10.1007/s12263-011-0210-5.
- Y. Zhou, J. Zheng, Y. Li, D.-P. Xu, S. Li, Y.-M. Chen and H.-B. Li, Natural Polyphenols for Prevention and Treatment of Cancer, Nutrients, 2016, 8(8), 515, DOI:10.3390/nu8080515.
- D. I. Batovska and I. T. Todorova, Trends in Utilization of the Pharmacological Potential of Chalcones, Curr. Clin. Pharmacol., 2010, 1–29, DOI:10.2174/157488410790410579.
- F. B. Power, F. Tutin and H. Rogerson, CXXXV.—The Constituents of Hops, J. Chem. Soc., Trans., 1913, 103, 1267–1292, 10.1039/CT9130301267.
- R. S. Khupse and P. W. Erhardt, Total Synthesis of Xanthohumol, J. Nat. Prod., 2007, 70(9), 1507–1509, DOI:10.1021/np070158y.
- S. Vogel, S. Ohmayer, G. Brunner and J. Heilmann, Natural and Non-Natural Prenylated Chalcones: Synthesis, Cytotoxicity and Anti-Oxidative Activity, Bioorg. Med. Chem., 2008, 16(8), 4286–4293, DOI:10.1016/j.bmc.2008.02.079.
- J. Yao, B. Zhang, C. Ge, S. Peng and J. Fang, Xanthohumol, a Polyphenol Chalcone Present in Hops, Activating Nrf2 Enzymes To Confer Protection against Oxidative Damage in PC12 Cells, J. Agric. Food Chem., 2015, 63(5), 1521–1531, DOI:10.1021/jf505075n.
- B. Zhang, D. Duan, C. Ge, J. Yao, Y. Liu, X. Li and J. Fang, Synthesis of Xanthohumol Analogues and Discovery of Potent Thioredoxin Reductase Inhibitor as Potential Anticancer Agent, J. Med. Chem., 2015, 58(4), 1795–1805, DOI:10.1021/jm5016507.
- E. Ciccimaro and I. A. Blair, Stable-Isotope Dilution LC–MS for Quantitative Biomarker Analysis, Bioanalysis, 2010, 2(2), 311–341, DOI:10.4155/bio.09.185.
- A. Chokkathukalam, D.-H. Kim, M. P. Barrett, R. Breitling and D. J. Creek, Stable Isotope-Labeling Studies in Metabolomics: New Insights into Structure and Dynamics of Metabolic Networks, Bioanalysis, 2014, 6(4), 511–524, DOI:10.4155/bio.13.348.
- C. Schmidt, First Deuterated Drug Approved, Nat. Biotechnol., 2017, 35(6), 493–494, DOI:10.1038/nbt0617-493.
- J. F. Liu, S. L. Harbeson, C. L. Brummel, R. Tung, R. Silverman and D. Doller, Chapter Fourteen – A Decade of Deuteration in Medicinal Chemistry, in Platform Technologies in Drug Discovery and Validation, ed. R. A. Goodnow, Academic Press, 2017, vol. 50, pp. 519–542, DOI:10.1016/bs.armc.2017.08.010.
- S. Cargnin, M. Serafini and T. Pirali, A Primer of Deuterium in Drug Design, Future Med. Chem., 2019, 11(16), 2039–2042, DOI:10.4155/fmc-2019-0183.
- D. C. Ellinwood, M. F. El-Mansy, L. S. Plagmann, J. F. Stevens, C. S. Maier, A. F. Gombart and P. R. Blakemore, Total Synthesis of [13C]2-, [13C]3-, and [13C]5-Isotopomers of Xanthohumol, the Principal Prenylflavonoid from Hops, J. Labelled Compd. Radiopharm., 2017, 60(14), 639–648, DOI:10.1002/jlcr.3571.
- S. Berwanger, J. Zapp and H. Becker, 13C-Labelling of Xanthohumol in Hop Cones (Humulus Lupulus), Planta Med., 2005, 71(6), 530–534 CrossRef CAS PubMed.
- L. Buckett, S. Schinko, C. Urmann, H. Riepl and M. Rychlik, Stable Isotope Dilution Analysis of the Major Prenylated Flavonoids Found in Beer, Hop Tea, and Hops, Front. Nutr., 2020, 319 Search PubMed.
- S. Gester, P. Metz, O. Zierau and G. Vollmer, An Efficient Synthesis of the Potent Phytoestrogens 8-Prenylnaringenin and 6-(1,1-Dimethylallyl)Naringenin by Europium(III)-Catalyzed Claisen Rearrangement, Tetrahedron, 2001, 57(6), 1015–1018, DOI:10.1016/S0040-4020(00)01078-4.
- S. Tischer and P. Metz, Selective C-6 Prenylation of Flavonoids via Europium(III)-Catalyzed Claisen Rearrangement and Cross-Metathesis, Adv. Synth. Catal., 2007, 349(1-2), 147–151, DOI:10.1002/adsc.200600454.
- A. Keßberg and P. Metz, Utilizing an O-Quinone Methide in Asymmetric Transfer Hydrogenation: Enantioselective Synthesis of Brosimine A, Brosimine B, and Brosimacutin L, Angew. Chem., Int. Ed., 2016, 55(3), 1160–1163, DOI:10.1002/anie.201507269.
- O. Schaefer, R. Bohlmann, W.-D. Schleuning, K. Schulze-Forster and M. Hümpel, Development of a Radioimmunoassay for the Quantitative Determination of 8-Prenylnaringenin in Biological Matrices, J. Agric. Food Chem., 2005, 53(8), 2881–2889, DOI:10.1021/jf047897u.
- H. Zhang, Z. Wang, I. Ghiviriga, G. G. Pillai, F. Jabeen, J. A. Arami, W. Zhou, P. J. Steel, C. Dennis Hall and A. R. Katritzky, Synthesis, Characterization and Energetic Properties of Novel 1-Methyl-1,2,4-Triazolium N-Aryl/N-Pyridinyl Ylids, Tetrahedron Lett., 2017, 58(12), 1079–1085, DOI:10.1016/j.tetlet.2016.12.016.
- N. Jun, G. Hong and K. Jun, Synthesis and Evaluation of 2′,4′,6′-Trihydroxychalcones as a New Class of Tyrosinase Inhibitors, Bioorg. Med. Chem., 2007, 15(6), 2396–2402, DOI:10.1016/j.bmc.2007.01.017.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05443k |
| This journal is © The Royal Society of Chemistry 2021 |