
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-functional porous organic polymers for highly-efficient solid-phase extraction of β-agonists and β-blockers in milk†
Ci Wu *ad,
Xingshuang Ningc,
Xi Chenc,
Junfeng Mad,
Qun Zhaob,
Li Zhaob,
Guozhi Zhub and
Song Shi*b
*ad,
Xingshuang Ningc,
Xi Chenc,
Junfeng Mad,
Qun Zhaob,
Li Zhaob,
Guozhi Zhub and
Song Shi*b
aLiaoning Academy of Inspection and Quarantine, Dalian 116000, China. E-mail: wuci1111@126.com
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: shisong@dicp.ac.cn
cTechnology Centre of Dalian Customs District, Dalian 116600, China
dDepartment of Oncology, Lombardi Comprehensive Cancer Center, Georgetown University, Technology Centre of Dalian Customs District, Washington DC 20057, USA
First published on 31st August 2021
Abstract
A simple, accurate, and highly sensitive analytical method was developed in this study for the determination of ten β-agonists and five β-blockers in milk. In this method, new adsorbent phosphonic acid-functionalized porous organic polymers were synthesized through a direct knitting method. The synthesis procedure of the materials and the extraction conditions (such as the composition of loading buffer and eluent) were optimized. Benefitting from the high surface area (545–804 m2 g−1), multiple functional framework and good porosity, the phosphonic acid-functionalized porous organic polymers showed a high adsorption rate and high adsorption capacity for β-agonists (224 mg g−1 and 171 mg g−1 for clenbuterol and ractopamine, respectively). The analytes were quantified by ultra-high-performance liquid chromatography coupled to high-resolution tandem mass spectrometry. It showed a good linearity (with R2 ranging from 0.9950 to 0.9991 in the linear range of 3–5 orders of magnitude), with low limits of quantification ranging from 0.05 to 0.25 ng g−1. The limits of detection of the method for the analytes were measured to be in the range of 0.02 to 0.1 ng g−1. The recoveries of target analytes from real samples on the material were in the range of 62.4–119.4% with relative standard deviations of 0.6–12.1% (n = 4). Moreover, good reproducibility of the method was obtained with the interday RSD being lower than 11.7% (n = 5) and intraday RSD lower than 12.2% (n = 4). The proposed method was accurate, reliable and convenient for the simultaneous analysis of multiple β-agonists and β-blockers. Finally, the method was successfully applied for the analysis of such compounds in milk samples.
Introduction
β-Agonists, which possess a common β-phenyl-β-ethanol amine group but vary in different substituents on the amine nitrogen and phenyl ring, are normally used in symptomatic treatment of asthma and chronic bronchitis and also in prevention of exercise induced asthma.1 But an overdose of β-agonists can induce heart palpitations, muscle tremors, and even malignant tumors. Many countries including the European Union (EU) and China have banned using β-agonists as growth promoting agents in farm animals.2 However, they are still sometimes illegally used as feed supplements to increase feed efficiency and obtain greater muscle to fat ratio in animal husbandry because they can increase production of leaner meat.1 β-Blockers, which also have an ethanol amine side chain bonded to the aromatic ring via a CH2–O-bond in the structure, are among the most prescribed drugs and frequently recommended in patients with hypertension, angina, myocardial infarction, arrhythmias, and heart failure.3 Furthermore, β-blockers may be abused by athletes to lower anxiety in some competitions. Thus, β-blocker residues may be present in environmental samples due to high consumption. Those kinds of misapplication or illegal use could lead to the presence of β-agonist and β-blocker residues in food of animal origin (such as milk, one of the most important components of the human diet).4 Extensive consumption of food containing β-agonists or β-blockers could be very harmful for humans. Therefore, developing a sensitive and specific analytical method is of critical importance for monitoring the abuse of β-agonists and β-blockers to ensure food safety.Due to the complexity of the composition of matrices and the trace amounts of analytes of interest, an effective extraction/purification strategy prior to final analysis is a prerequisite for determination of β-agonists and β-blockers. In recent years, various pretreatment techniques for the extraction of such residues from biological samples have been developed, including the QuEChERS (Quick, Easy, Cheap, Effective, Rugged, and Safe) technique,5 solid phase extraction (SPE),6–8 immunoaffinity chromatography (IAC)9 and supercritical fluid extraction (SFE).10 Among these strategies, SPE has been the most frequently used method owing to several advantages: low reagent consumption and no requirement of expensive apparatus,11,12 high sensitivity and good recovery with minimal sample transfer,13–16 enhanced simplicity of operation with robustness,17 and great safety with less exposure to toxic agents.18,19 The performance of the SPE technique depends on both the adsorption capacity and adsorption/desorption kinetics, and the SPE adsorbent directly affects the extraction efficiency and the analysis sensitivity. Thus, during the past several years, significant efforts have been made towards the development and characterization of advanced sorbent materials, such as molecularly imprinted polymers (MIPs),20,21 graphene-based nanocomposites,22–25 functionalized covalent organic frameworks (COFs)26–28 and magnetic nanoparticles,29–32 to enhance selectivity or specificity towards target analytes and improve adsorption capacity.33–35 Currently, SPE adsorbents developed for the extraction of β-agonists and β-blockers from complex samples are C18 based materials,21,36 polymeric cation-exchange resins,37 divinylbenzene38 and mixed-mode cation exchange (MCX)39,40 materials (based on reversed phase and ion exchange interaction). Nevertheless, it is still a challenge for the determination of β-agonists and β-blockers by using SPE adsorbents with high adsorption capacity and fast sorption/desorption.41
Porous organic polymers (POPs)42–45 are a class of newly developed porous materials synthesized from organic monomers. A number of such materials have been designed, including hyper-crosslinked polymers (HCPs), polymers of intrinsic microporosity (PIMs), covalent organic frameworks (COFs), conjugated microporous polymers (CMPs), covalent triazine frameworks (CTFs) and porous aromatic frameworks (PAFs). Recently, this kind of material has gained popularity as SPE absorbents with the advantages of good physicochemical stability and high diversity in structure and functionality. Li et al.46 synthesized hydrazine linked COFs for enrichment of Sudan dyes in food samples. Shortly afterwards a magnetic polyphenylene CMP and microporous organic polymers (MOPs) based on a Schiff base network (SNW-1) were reported by this group for the enrichment of hydroxylated polycyclic aromatic hydrocarbons (OH-PAHs)47 and volatile fatty acids,48 respectively. Qiu et al.49 used a kind of magnetic CTF with high surface area to remove organic dye from aqueous solutions. Zhang et al. prepared a CTF-based material for reversed-phase/hydrophilic interaction mixed-mode separation of organic pollutants50 and hydrophilic–lipophilic balance/cation-exchange extraction of benzimidazole fungicides.51 Dubey et al.52 reported CTFs as a promising sorbent for analysis of chemical weapons convention related compounds. Wang et al.53 developed carbazole-based heterocyclic conjugated POPs for SPE of triazine herbicides in vegetables. The application of POPs as adsorbents for extraction of other compounds is still under explored. Considering the phenyl-ethanolamines structure in β-agonists and β-blockers, POP materials hold great promise for high-efficient and selective extraction of them by taking advantage of the large surface area, hydrophobic nature and easy modification of the framework.
In this manuscript, phosphonic acid-functionalized porous organic polymers (PPOPs) were developed through a direct knitting method and exploited as novel adsorbents for the solid-phase extraction (SPE) of β-agonists and β-blockers in milk samples. With the high adsorption capacity, adsorption rate and good selectivity of the PPOPs, ten β-agonists and five β-blockers in real milk samples were confidently identified by using the prepared materials for the SPE sample work-up prior to analysis by liquid chromatography coupled with high resolution mass spectrometry (HRMS). To our knowledge, this is the first example of employing porous organic polymers for the analysis of veterinary drug residues in human diet milk.
Materials and methods
Materials and apparatus
Phenylphosphonic acid (PPA, >98.0%) was purchased from TCI (Shanghai, China). 1,2-Dichloroethane (≥99%) and trichloroacetic acid (TCA, ≥99%) were purchased from Kemiou Chemical Reagent Co., Ltd (Tianjin, China). Dimethoxymethane (DMM, 98%) was purchased from Alfa Aesar Chemical Reagent Co. Ltd (Tianjin, China). FeCl3 (≥97.0%) and formic acid (FA, ∼98%) were obtained from J&K Scientific Ltd (Beijing, China). Ammonium hydroxide (28–30 wt%, AR) was purchased from Shanghai Hushi Laboratorial Equipment Co. Ltd. Toluene (Kemiou, Tianjin, China) was refluxed (110 °C) over sodium and distilled (136 °C). Methanol and acetonitrile were purchased from Merck (Darmstadt, Germany). Ractopamine, salbutamol, terbutaline, cimaterol, clenbuterol, tulobuterol, propranolol, clorprenaline, atenolol, betaxolol, nadolol, sotalol, cimbuterol, and mabuterol (>97.0%) were purchased from Dr Ehrenstorfer (Augsburg, Germany). Deionized water was used in the experiments (Millipore, Milford, MA).For the nitrogen adsorption–desorption measurements (QuadraSorb SI4, U.S.A.), the materials were pretreated at 100 °C for 12 h under vacuum. Morphology of the material was characterized by a JSM-6330F (JEOL, Tokyo, Japan) scanning electron microscopy (SEM). Transmission electron microscope images (TEM) were collected on a JEM 2000EX. The nuclear magnetic resonance spectra of 13C cross polarization/magic-angle spinning nuclear magnetic resonance (13C CP/MAS NMR) were collected on a Bruker DRX-400 spectrometer equipped with a magic angle spin probe in a 4 mm ZrO2 rotor. The experimental parameters for 13C CP MAS NMR experiments: 6 kHz spin rate, 2 s pulse delay, 6 ms contact time, 1500–3000 scans. Thermal gravity analysis (TGA) measurements were carried out on a NETZSCH STA 409 PC instrument. The P element content of the material was measured using Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES/PE Optima 8000, Thermo-Fisher, San Jose, CA). Milk samples were decomposed with a full automatic microwave (WX-8000KJ160, Preekem, Shanghai, China), with solid-phase extraction performed on a device Supelco-24 (USA).
Synthesis of phosphonic acid-functionalized porous organic polymers
The phosphonic acid-functionalized porous organic polymers (PPOP) was synthesized via the Friedel–Crafts alkylation reaction, a reaction which had been adopted previously.54 The mixture of monomers (toluene and phenylphosphonic acid with various ratios), cross-linker (DMM, 0.04 mol, 3.04 g) and catalyst (FeCl3, 0.04 mol, 6.5 g) were dissolved in 1,2-dichloroethane (DCE, 20 mL) in a round bottom flask. Then the flask was heated at 45 °C for 5 h to form a primary framework, followed by heating at 80 °C for 19 h to complete the condensation reaction to produce a meso- and microporous polymer. After washing with 15 mL methanol three times, the solid product was Soxhlet extracted in methanol for 24 h followed by drying in a vacuum oven at 60 °C for 24 h. PPOPs with different phosphate contents were synthesized with various molar ratios of the monomer mixture, producing dark brown powders.Sample preparation procedure
Five grams of milk samples were weighed and placed into a 50 mL centrifuge tube. Nine mL of water with 3% TCA solution and 1 mL of ACN were added into the sample. Then, the sample was thoroughly mixed and extracted by vortex for 5 min. The extracts were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C and the supernatant was collected for further clean-up.
000 rpm for 10 min at 4 °C and the supernatant was collected for further clean-up.
The PPOP adsorbent (60 mg) was added into a Teflon tube, serving as a SPE column. SPE extraction was performed using gravity, without external vacuum or pressure applied. The column was initially pre-activated by adding 2 mL of ACN onto the top of the SPE column and then equilibrated by adding 2 mL of 0.1% FA. 5 mL of extracted sample in 10% ACN and 3% TCA buffer was loaded onto the pre-conditioned SPE column. After washing with 0.1% FA (2 mL), analytes were eluted from the SPE column by acetonitrile with 5% aqueous ammonia (5 mL). The eluate was collected and evaporated to dryness. After being reconstituted with 0.5 mL of water/ACN (5:95, v/v), the resulting solution was filtered through a 0.2 μm nylon filter and analyzed by UPLC-MS/MS.
UPLC-HRMS analysis
Sample analyses were performed on a UPLC-HRMS equipped with a UPLC pump (Dionex UltiMate 3000, U.S.A.), controlled by Xcalibur software version 4.0. A reversed-phase Accucore RP-MS column (Thermo Fisher Scientific, 100 × 2.1 mm, 2.6 μm particle size) was employed for the separation of analytes. The column temperature was set at 40 °C. The flow rate was 0.3 mL min−1. The mobile phases were H2O with 0.1% FA as phase A and methanol as phase B. The UPLC gradient was set as follows: 5% B (0 min)–5% B (2 min)–30% B (7 min)–90% B (11 min)–90% B (13 min)–5% B (16 min). The temperature of the sample manager was set as 5 °C. The sample injection volume was 5 μL.Mass spectrometry analysis was performed with a Q-Exactive plus (Thermo Fisher Scientific, Rockford, IL, U.S.A.). A heated electrospray ionization source was used for the ionization of analytes. The spray voltage was set as 3.20 kV in positive mode, and the included charge state was set at 1. The capillary temperature was set as 320 °C. Sheath gas flow rate and aux gas flow rate were set at 40 and 10 (in arbitrary units), respectively. The aux gas heater temperature was 350 °C. The S-lens RF level was 50.0. All MS and MS/MS spectra were acquired by Orbitrap. Chromatographic peak width was set as 10 s. The scan range was at m/z 100–1000. In full scan MS mode, the resolution was set as 70000. The AGC (automatic gain control) target and maximum IT (injection time) were set as 1 × 106 ions capacity and 50 ms, respectively. The resolution of MS/MS mode was 17500, and the AGC target was set as 2 × 105. The isolation window was set as 2.0 m/z, loop count was 1 and MSX count was 1. The normalized collision energy (NCE) was 30%.
Adsorption of analytes by PPOPs
The adsorption experiment was performed with ractopamine and clenbuterol, two commonly used model analysts previously.25,55 To determine the adsorption isotherms at 25 °C, 2 mg of adsorbents were added to a series of ractopamine and clenbuterol solutions (1 mL) with various initial concentrations (0.01–1 mg mL−1), with 2 mL centrifuge tubes containing the mixture solution shaken on a shaker at 25 °C for 2 h. Then, the adsorbents were centrifuged and separated from the suspension, with the remaining analyte concentration determined by UPLC-MS/MS. Adsorption capacity (Qe, mg m−2) of the prepared materials is calculated as follows (eqn (1)):| Qe = 1000 × (C0 − Ce) × V/(m × SBET), | (1) |
The amounts of analytes adsorbed at different contact times were calculated from (eqn (2)):
| Qt = 1000 × (C0 − Ct) × V/(m × SBET), | (2) |
Method validation
Matrix-matched standards were used to obtain calibration curves for the 15 target compounds (including ten β-agonists: ractopamine, salbutamol, terbutaline, cimaterol, clenbuterol, tulobuterol, clorprenaline, mabuterol, cimbuterol and brombuterol; and five β-blockers: atenolol, betaxolol, nadolol, sotalol, and propranolol). Under the positive ionization mode, calibration curves were obtained by a linear regression analysis on the standard solution areas versus analyte concentrations with a series of external standard concentrations.Mixed working solutions were prepared by spiking different amounts of standards into blank milk samples. Eighteen concentration levels of 0.02, 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10, 25, 50, 100, 250, 500, 1000, 2500, 5000, and 10000 ng g−1 were used to investigate linear ranges and limits of detection (LODs). Three concentration levels (0.5, 1.0 and 5 ng g−1), obtained by spiking standards into blank milk samples before extraction, were prepared for measurement of recovery and reproducibility. The relative recovery was evaluated by comparing the response of 15 compounds in spiked solutions before and after extraction at above low, medium, and high concentration levels.
The accuracy and precision of the whole analytical procedure were evaluated by spiking into whole milk samples at 0.5, 1, and 5 ng g−1 (four replicates at each level). The analytes were extracted as described in the extraction and purification procedure.
Results and discussion
Preparation and characterization of PPOPs
In this study, PPOPs were synthesized based on the direct knitting method. Specifically, dimethoxymethane (DMM) was used as an external cross-linker to react with two aromatic monomers (i.e., toluene and phenylphosphonic acid), as shown in Fig. 1. Materials with various amounts of phosphonic acid groups were obtained by adjusting the ratio of toluene and phenylphosphonic acid (PPA). Additionally, to remove residual FeCl3 and unreacted monomers, the products have to be washed with a Soxhlet extractor using methanol. It was found that the amount of Fe was lower than 0.068% (wt%), demonstrating the successful removal of residual FeCl3 in this work. Typical synthesis conditions and properties (including P content and porosity) of these materials were summarized in Table 1. The results showed that the P content, representing the phosphonic acid group amount in the materials, was increased with the increase of the PPA/toluene molar ratio in the reaction mixture.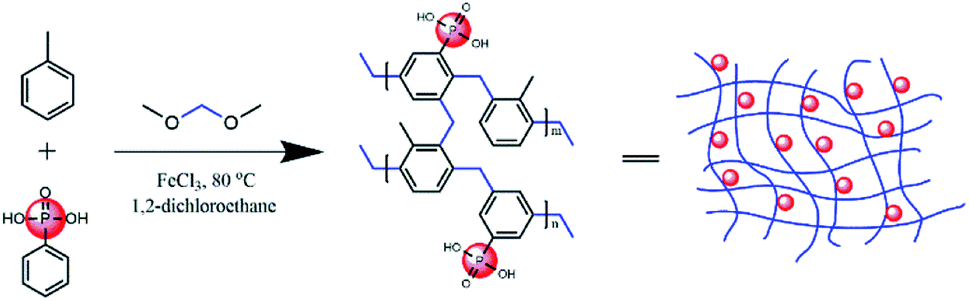 | ||
| Fig. 1 Schematic of the preparation of PPOPs. | ||
| Materials | PPA/toluene (mol mol−1) | P content (mmol g−1) | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|---|---|
| POP10 | 1/9 | 0.233 | 784.900 | 0.609 | 3.102 |
| POP20 | 1/4 | 0.252 | 789.414 | 0.607 | 3.077 |
| POP40 | 2/3 | 1.479 | 804.127 | 0.705 | 3.508 |
| POP60 | 3/2 | 2.116 | 728.048 | 0.582 | 3.197 |
| POP80 | 4/1 | 2.695 | 545.085 | 0.473 | 3.472 |
Microstructures and porous characteristics of these PPOPs were characterized by SEM images (Fig. S1†). It was revealed that the surface of PPOPs was homogeneous and porous with micrometer pores. The porous structure of PPOPs was also supported by TEM images (Fig. S2†). A translucent feature could be observed in the electron beam, probably due to the very high porosity. This porosity of the absorbent is anticipated to render good permeability which would contribute to the rapid mass transfer during SPE.
The pore properties of the as-prepared PPOPs were further investigated by using N2 adsorption–desorption isotherms (Fig. 2a), with pore diameter distributions shown in Fig. 2b and porosity parameters listed in Table 1. According to IUPAC classification, the isotherm shapes of all samples exhibit type-IV isotherms.57,58 All PPOPs displayed a sharp gas uptake at low relative pressure (P/P0 < 0.002), demonstrating the existence of extensive micropores. Pore size distribution analyses using the Barrett–Joyner–Halenda (BJH) method revealed a broad range of pore diameter distribution from 1 to 70 nm, reflecting the presence of both micropores and mesopores. The BET specific surface areas (BET) of the as-synthesized composites varied from 545 to 804 m2 g−1. Total pore volumes and average pore diameter of all polymers were around 0.6 cm3 g−1 and 3 nm, respectively.
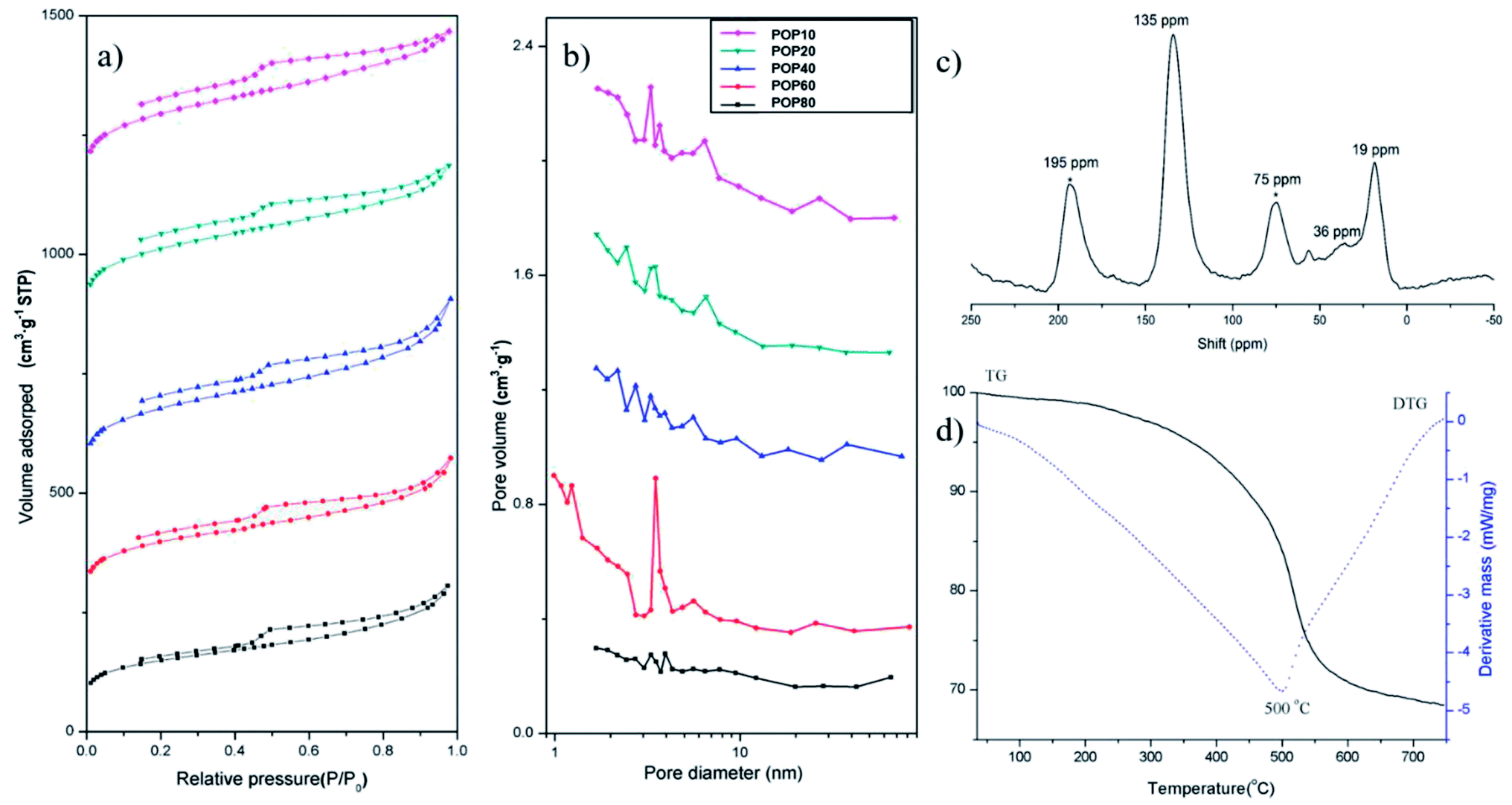 | ||
| Fig. 2 (a) N2 sorption–desorption isotherms and (b) pore diameter distributions of PPOPs; (c) solid-state 13C NMR spectrum of POP20 and (d) TGA curve of POP20. | ||
The material POP20 was selected as the representative material for further characterization, since it was approved to have the best extraction performance for target analytes in our work. Specifically, POP20 was characterized with solid-state 13C polarization/magic-angle spinning nuclear magnetic resonance (13C CP/MAS NMR) NMR spectra and thermal gravimetric analysis (TGA). The 13C NMR spectrum (Fig. 2c) clearly showed the resonance peaks near 135 ppm due to substituted aromatic carbon, and the resonance peak near 36 ppm due to carbon in the methylene linker.59 The network based on toluene showed a resonance peak at 19 ppm which can be assigned to methyl carbon connected to the benzene ring. The thermal stability of the material was evaluated by TGA under a nitrogen atmosphere at a heating rate of 10 °C min−1 (Fig. 2d). It could be seen that there was a little mass loss before 100 °C due to the removal of adsorbed water, and further heating showed no obvious weight loss up to 320 °C until thermal decomposition at 500 °C. The TGA result suggested that the PPOP adsorbent (POP20) had high thermal stability.
Optimization of extraction conditions
Milk samples contain a large amount of proteins and other polar compounds, the chemical properties of which are similar to β-agonists and β-blockers to some extent. Since these compounds may also be adsorbed and desorbed in mixed-mode SPE materials, they should be removed before SPE. Herein, samples were pretreated with an aqueous acidic solution. Proteins were precipitated out based on adjusting pH using trichloroacetic acid (TCA).40 The content of TCA in the extraction solution was further investigated in this study. The semitransparent extraction solution was observed with TCA content lower than 1% (w/v), which might be caused by insufficient protein precipitation. As shown in Fig. 4a, the highest extraction efficiency was achieved with 3% TCA (w/v). The samples were then processed by SPE to enhance the detection performance.Parameters that might affect the performance of SPE were investigated to obtain the best detection sensitivity and selectivity for β-agonists and β-blockers. Firstly, eight typical agonists were used to evaluate the extraction efficiency of PPOPs with various contents of phosphate groups. As shown in Fig. 3, with the increase of phosphate groups content in the PPOP materials, the capture efficiency of targets was increased at first and decreased subsequently, with the highest efficiency achieved with a PPA molar ratio of 20% (named POP20). This trend could be ascribed to the interaction between PPOPs and β-agonists or β-blockers. Besides cation exchange interaction from –PO43− groups, hydrophobic interactions could also exist, considering the presence of a large number of phenyl groups. This was proved by the effect of ACN content in the adsorption solvent on the extraction efficiency, as shown in Fig. 4b. It could be seen that the extraction efficiency of targets is decreased with the increase of the ACN concentration in the loading buffer. These results indicated that the retention mechanism was a combination of ion exchange and hydrophobic interaction, thus it is crucial to achieve a balance between the –PO43− groups and phenyl groups amounts in the material. Moreover, the effect of salt addition on the extraction efficiency was also investigated by adding different concentrations of NaCl. As shown in Fig. 4c, the extraction efficiency of targeted β-agonists decreased with the increase of NaCl concentration in the loading buffer. With that, POP20 was used as the optimized adsorbent and the loading buffer with 10% ACN (v/v) was selected to get the optimum extraction efficiency of analytes for further experiments.
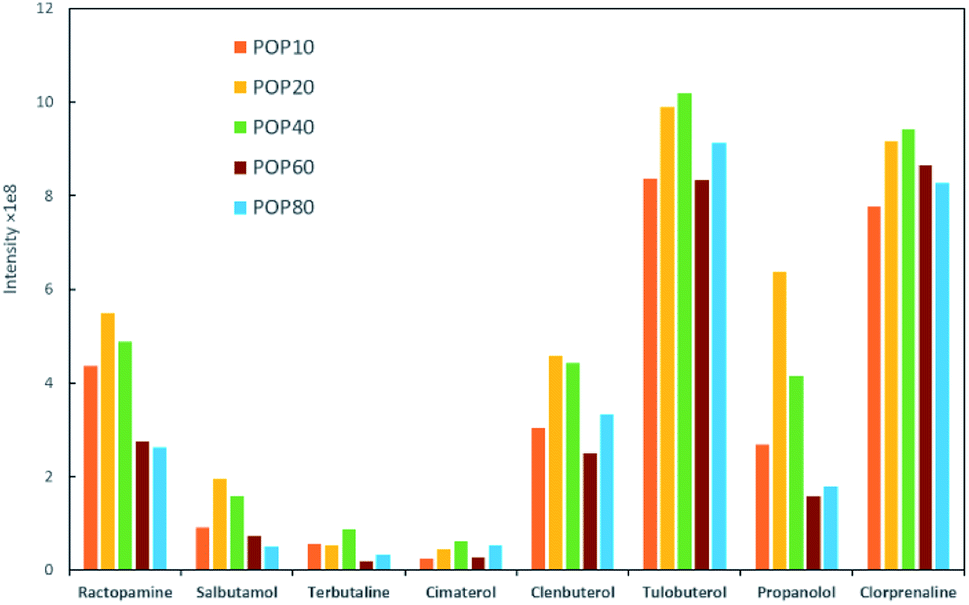 | ||
| Fig. 3 Effect of different PPOPs of on the extraction performance of β-agonists and β-blockers under identical conditions. | ||
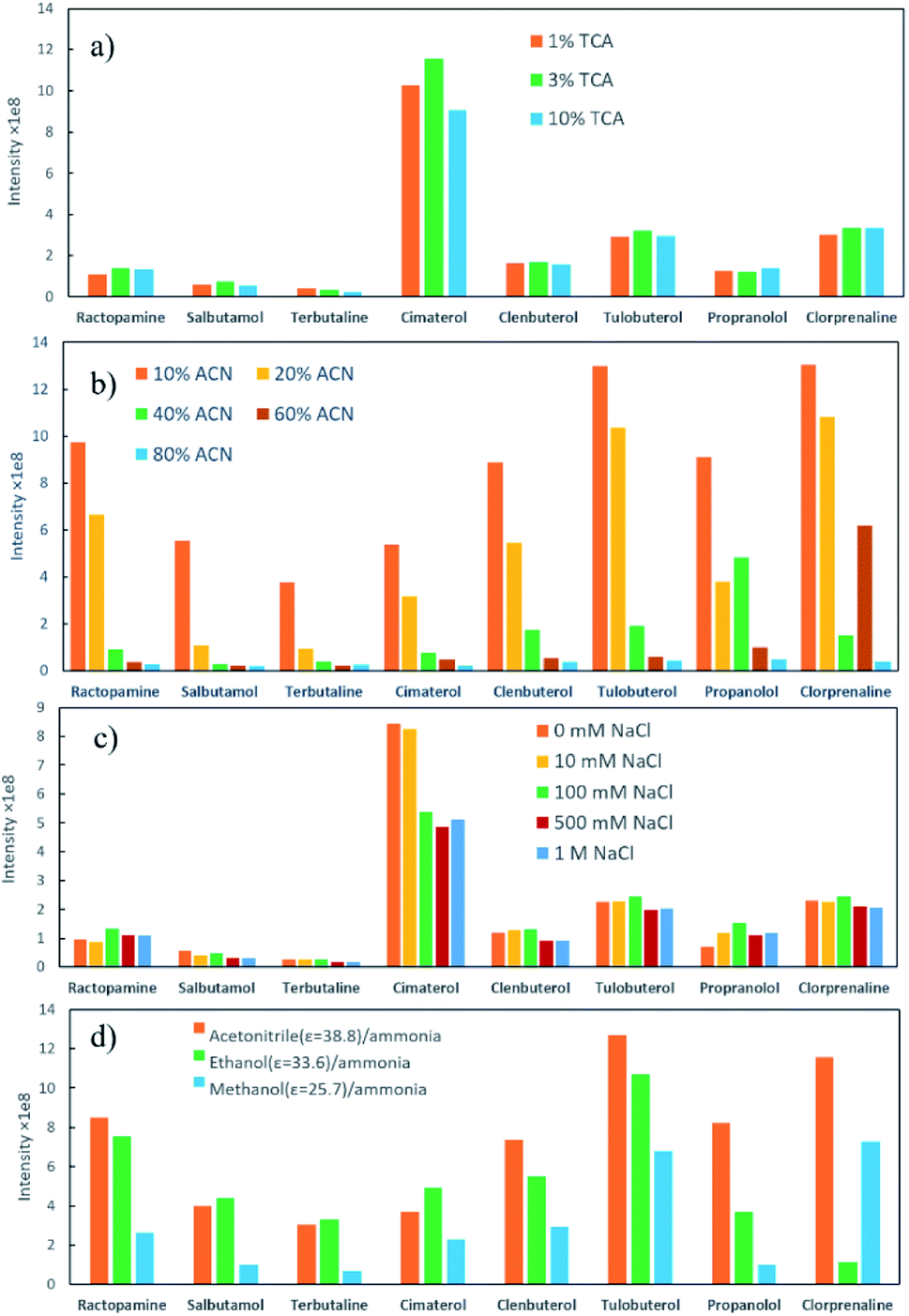 | ||
| Fig. 4 (a) Effect of TCA concentration in loading buffer on the extraction efficiency of analytes; (b) effect of ACN content in loading buffer on the extraction efficiency; (c) effect of salt concentration in loading buffer on the extraction efficiency; (d) effect of eluent type on the extraction efficiency (ε is the dielectric constant). | ||
Additionally, the elution efficiency of three kinds of organic solvents (including methanol/ammonia, ethanol/ammonia and acetonitrile/ammonia) was evaluated (Fig. 4d). Among them, acetonitrile/ammonia as desorption solvent provided the highest extraction efficiency towards most compounds. One reason might be that the dielectric constant (ε) of ACN is higher than the other two solvents, resulting in better solubility for β-agonists and β-blockers.
Adsorption isotherms
Adsorption properties were demonstrated by using POP20 as the adsorbent and clenbuterol and ractopamine as model analytes. Fig. 5 shows the adsorption isotherms of clenbuterol and ractopamine. It can be seen that the adsorption capacities of clenbuterol and ractopamine increase with the increase of the agonist concentration. Moreover, the shape of the isotherms reveals L behavior according to the Giles classification,60 confirming a high affinity between the PPOP and target molecules. The initial sharp rise indicates that a large amount of analyte is adsorbed at a lower concentration as more active sites are available. As the concentration increases, it becomes difficult for β-agonist molecules to find vacant sites, with saturated adsorption achieved. By fitting the equilibrium adsorption data with the Langmuir adsorption model, the adsorption capacity of the agonist onto the composite was calculated from (eqn (3)):56| Ce/qe = 1/(b × qm) + Ce/qm; | (3) |
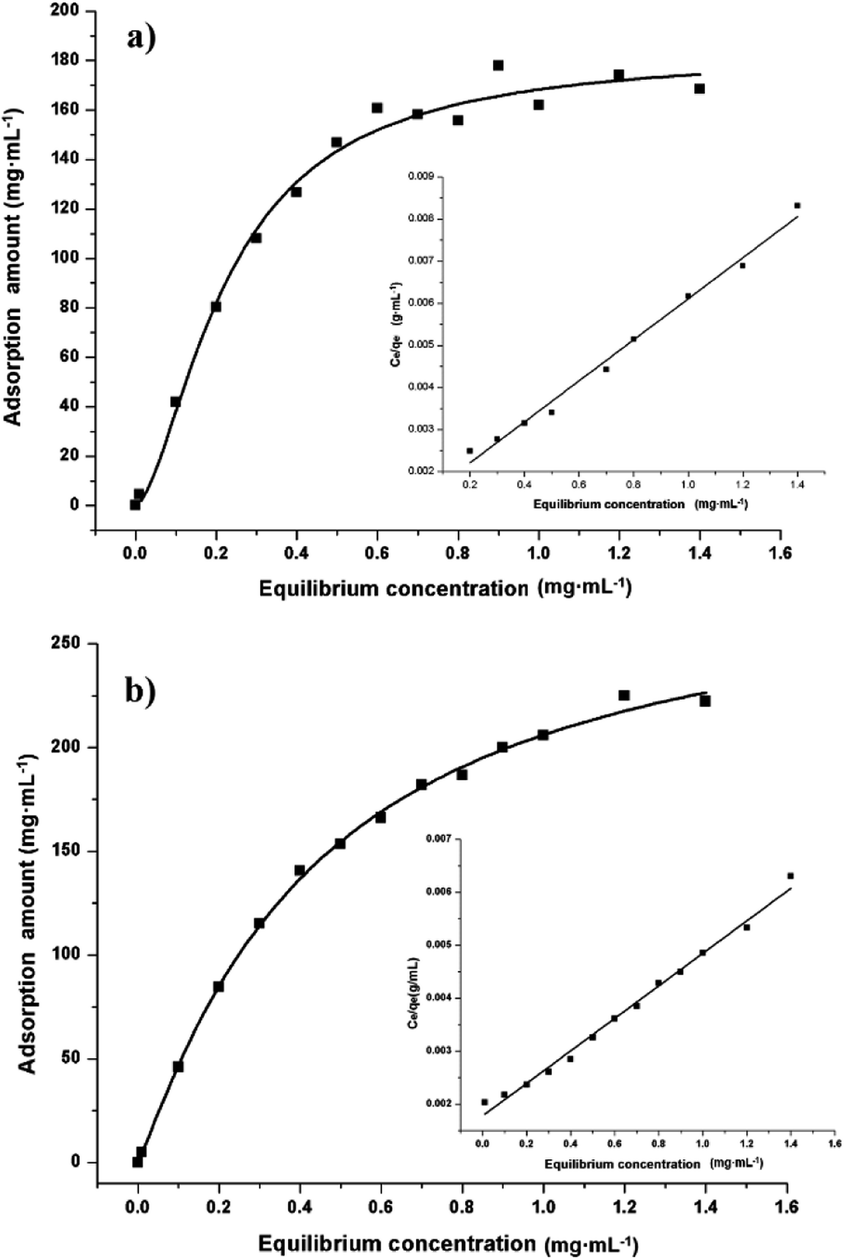 | ||
| Fig. 5 Adsorption isotherms of (a) clenbuterol and (b) ractopamine onto POP20, and the linear regression by fitting the equilibrium adsorption data with the Langmuir adsorption model. | ||
Adsorption kinetics
To have a better understanding of adsorption kinetics, the effect of contact time on the adsorption of β-agonist was investigated with an initial concentration of 1.0 mg mL−1. As shown in Fig. 6, the adsorption capacities increase rapidly in a short contact time and reach equilibrium at 10 min, indicating both a high adsorption capacity and high adsorption efficiency of the adsorbent for the extraction of β-agonists and β-blockers. This could be attributed to a broad pore diameter distribution from micropores to mesopores of the material. Since the adsorption kinetics of our materials is quite similar to that of hierarchically micro- and mesoporous materials, we calculated the kinetics parameters by the pseudo-second order kinetic equation (eqn (4)):61| t/qt = 1/(k2 × qe2) + t/qe; | (4) |
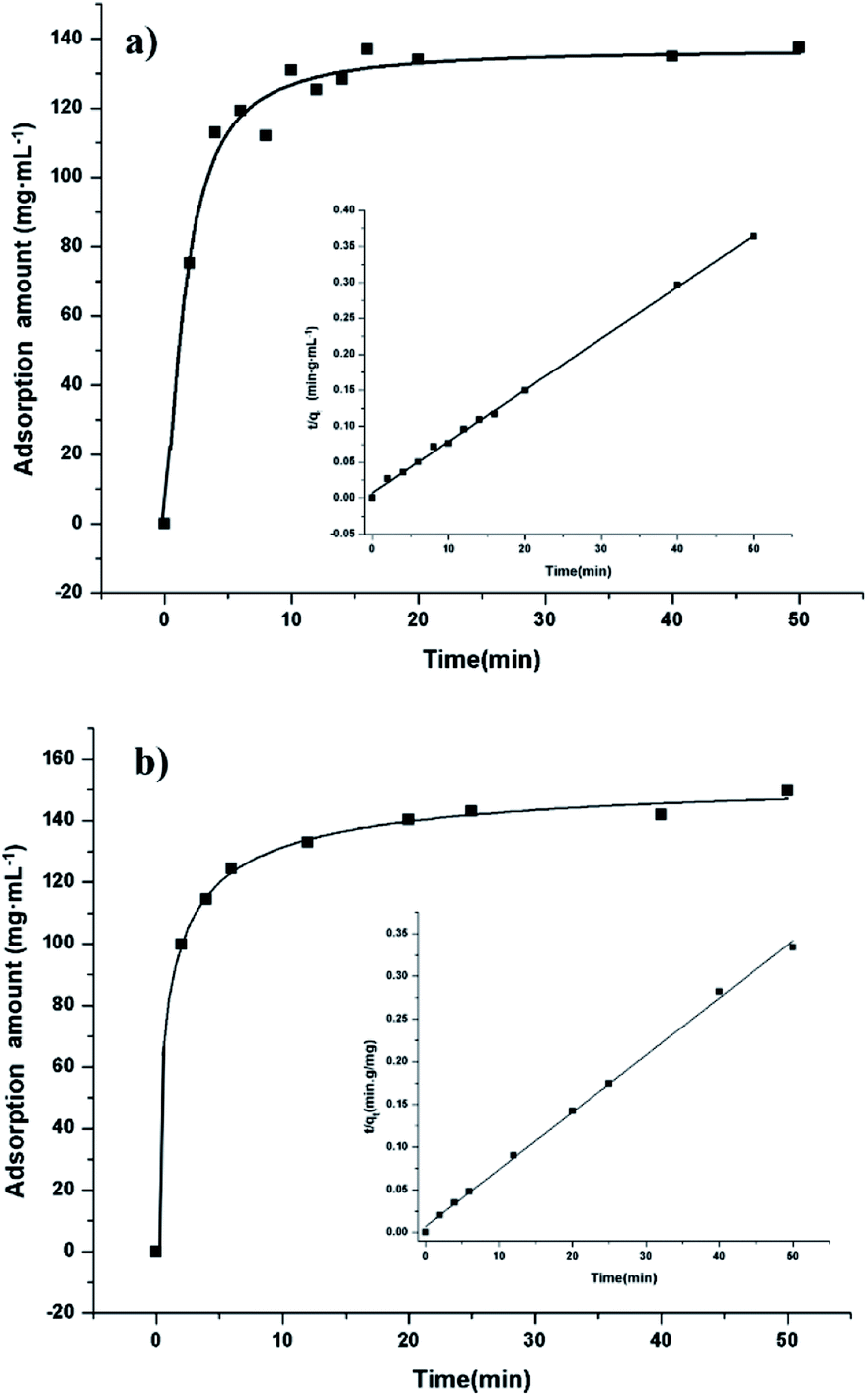 | ||
| Fig. 6 Adsorption curves of analytes onto POP20 versus contact time in aqueous solution, and the insets of the figure show the pseudo-second-order kinetic plot for the adsorption (analytes concentration: 1000 mg L−1): (a) clenbuterol and (b) ractopamine. | ||
Method validation
The refined sample pre-treatment followed by UHPLC-HRMS was then applied for the analysis of ten β-agonists and five β-blockers (see the Materials and methods section). For investigation of linearity and LOQs, solutions spiked with 18 concentration levels were analyzed, with the results displayed in Table S1.† It showed a good linearity with regression coefficients ranging from 0.9950 to 0.9991 and with the linear range of 3–5 orders of magnitude. The LOQs ranged from 0.05 to 0.25 ng g−1. The limits of detection of the method for the analytes were measured with 0.02 to 0.1 ng g−1. Relative recovery and reproducibility of the extraction method were measured by spiking standards into blank milk samples before and after the extraction process at three concentration levels (0.5, 1.0 and 5.0 ng g−1) with four replicates. The recoveries of target analytes from real samples on the material were in the range of 62.4–119.4% with relative standard deviations of 0.6–12.1% (n = 4). Detailed information for all analysts is displayed in Table 2. Moreover, the precision was further determined by calculating the RSD for repeated measurements by determining the inter-day and intraday RSD values. As shown in Table S2,† the inter-day and intraday RSD were lower than 11.7% (n = 5) and 12.2% (n = 4), respectively, which were in good agreement with 2002/657/EC requirements, demonstrating the excellent method precision.| Compound | Spiked (0.5 ng g−1) | Spiked (1.0 ng g−1) | Spiked (5.0 ng g−1) | ||||
|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| β-Agonists | Ractopamine | 106.8 | 5.1 | 101.4 | 1.3 | 97.4 | 3.1 |
| Salbutamol | 117.1 | 2.1 | 119.2 | 1.1 | 110.9 | 8.2 | |
| Terbutaline | 97.1 | 4.5 | 102.2 | 5.2 | 89.4 | 12.1 | |
| Cimaterol | 96.3 | 9.8 | 84.6 | 5.7 | 89.4 | 5.3 | |
| Clenbuterol | 107.3 | 3.5 | 112.5 | 1.2 | 110.5 | 2.3 | |
| Tulobuterol | 83.2 | 8.1 | 80.3 | 3.6 | 116.6 | 3.5 | |
| Clorprenaline | 85.1 | 11.1 | 98.6 | 7.3 | 83.4 | 2.3 | |
| Cimbuterol | 103.6 | 4.2 | 85.4 | 8.3 | 80.8 | 0.6 | |
| Brombuterol | 80.3 | 2.7 | 84.3 | 1.9 | 87.9 | 7.1 | |
| Mabuterol | 112.4 | 3.0 | 112.9 | 1.6 | 111.8 | 2.8 | |
| β-Blockers | Nadolol | 89.1 | 3.3 | 82.0 | 2.2 | 83.1 | 4.1 |
| Atenolol | 62.4 | 2.6 | 69.2 | 2.7 | 67.7 | 0.6 | |
| Sotalol | 112.9 | 8.3 | 109.8 | 4.0 | 101.0 | 5.3 | |
| Betaxolol | 110.6 | 5.7 | 119.4 | 1.0 | 110.6 | 2.7 | |
| Propranolol | 88.8 | 5.4 | 73.9 | 3.2 | 68.5 | 7.2 | |
Table S3† shows the performance comparison between the proposed strategy based on PPOP and other analytical methods reported for the determination of β-agonists. Our method showed advantages in terms of detection throughput and LOQs. For example, the LOQs of this method are equal to or more favorable than that of some traditional methods. These results demonstrate that the proposed method can fulfil the need for trace analysis of β-agonists and β-blockers in complex matrix samples, with reliable processing and precise quantification.
Application to real samples
The method was applied for analysis of 10 real raw milk samples collected from different manufacturers (Dalian) in China. All milk samples were processed according to the method described, and no detectable β-agonist or β-blocker residues were identified, probably due to stringent regulations of the government.Conclusions
In conclusion, a type of phosphonic acid-functionalized porous organic polymer (PPOP) with high surface area was synthesized through a direct knitting method. The material was exploited as a novel adsorbent for the solid-phase extraction (SPE) of β-agonists and β-blockers in milk, with the eluents analyzed by UPLC-HRMS. This new analytical approach exhibited high adsorption capacity, good linearity (R2 = 0.9950–0.9991) in the linear range of 3–5 orders of magnitude and high sensitivity (LOQ: 0.05–0.25 ng g−1). The recoveries of target analytes from real samples on the material were in the range of 62.4–119.4% with relative standard deviations of 0.6–12.1% (n = 4). Moreover, good reproducibility of the method was obtained with the inter-day RSD being lower than 11.7% (n = 5) and intraday RSD lower than 12.2% (n = 4). Finally, the method was successfully applied to the detection of ten β-agonists and five β-blockers and the screening of them in real samples. Taken together, these results indicate that our method can be a valuable tool in regulatory laboratories for the control of illegal usage of such drugs in milk products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from National Natural Science Foundation (21705065). Dr Song Shi thanks for the National Natural Science Foundation of China of 22072147.References
- D. Courtheyn, B. Le Bizec, G. Brambilla, H. F. De Brabander, E. Cobbaert, M. Van de Wiele, J. Vercammen and K. De Wasch, Anal. Chim. Acta, 2002, 473, 71–82 CrossRef CAS.
- H. A. Kuiper, M. Y. Noordam, M. M. H. v. Dooren-Flipsen, R. Schilt and A. H. Roos, J. Anim. Sci., 1998, 76, 13 CrossRef.
- S. Yıldırım, C. Erkmen and B. Uslu, Crit. Rev. Anal. Chem., 2020, 1–39 CrossRef.
- J. F. Martínez-Navarro, Lancet, 1990, 336, 1311 CrossRef.
- Y.-P. Lin, Y.-L. Lee, C.-Y. Hung, W.-J. Huang and S.-C. Lin, J. Food Drug Anal., 2017, 25, 275–284 CrossRef CAS PubMed.
- T. Li, J. Cao, Z. Li, X. Wang and P. He, Food Chem., 2016, 192, 188–196 CrossRef CAS PubMed.
- M. Behbahani, S. Bagheri, M. M. Amini, H. Sadeghi Abandansari, H. Reza Moazami and A. Bagheri, J. Sep. Sci., 2014, 37, 1610–1616 CrossRef CAS.
- M. Behbahani, A. Veisi, F. Omidi, M. Y. Badi, A. Noghrehabadi, A. Esrafili and H. R. Sobhi, New J. Chem., 2018, 42, 4289–4296 RSC.
- L. Mei, B. Cao, H. Yang, Y. Xie, S. Xu and A. Deng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 945, 178–184 CrossRef PubMed.
- M. J. O'Keeffe, M. O'Keeffe and J. D. Glennon, Analyst, 1999, 124, 1355–1360 RSC.
- H. R. Fouladian and M. Behbahani, Food Anal. Methods, 2015, 8, 982–993 CrossRef.
- M. Behbahani, S. Bagheri, F. Omidi and M. M. Amini, Microchim. Acta, 2018, 185, 505–512 CrossRef PubMed.
- M. Behbahani, M. Najafi, M. M. Amini, O. Sadeghi, A. Bagheri and M. Salarian, Microchim. Acta, 2013, 180, 911–920 CrossRef CAS.
- M. Behbahani, A. Veisi, F. Omidi, A. Noghrehabadi, A. Esrafili and M. H. Ebrahimi, Appl. Organomet. Chem., 2018, 32, 4134–4143 CrossRef.
- S. Parvizi, M. Behbahani and A. Esrafili, New J. Chem., 2018, 42, 10357–10365 RSC.
- M. Behbahani, A. Aliakbari, M. M. Amini, A. S. Behbahani and F. Omidi, RSC Adv., 2015, 5, 68500–68509 RSC.
- M. G. Kakavandi, M. Behbahani, F. Omidi and G. Hesam, Food Anal. Methods, 2017, 10, 2454–2466 CrossRef.
- A. A. Salem, I. Wasfi, S. S. Al-Nassib, M. Allawy Mohsin and N. Al-Katheeri, J. Chromatogr. Sci., 2017, 55, 846–856 CAS.
- F. Karamolegou, M. Dasenaki, V. Belessi, V. Georgakilas and N. Thomaidis, Food Anal. Methods, 2018, 11, 2925–2942 CrossRef.
- P. Wang, X. Liu, X. Su and R. Zhu, Food Chem., 2015, 184, 72–79 CrossRef CAS PubMed.
- H. Liu, X. Lin, T. Lin, Y. Zhang, Y. Luo and Q. Li, J. Sep. Sci., 2016, 39, 3594–3601 CrossRef CAS PubMed.
- Q. Liu, J. Shi, J. Sun, T. Wang, L. Zeng and G. Jiang, Angew. Chem., Int. Ed., 2011, 50, 5913–5917 CrossRef CAS PubMed.
- C. Zhang, T. Tao, W. Yuan, L. Zhang, X. Zhang, J. Yao, Y. Zhang and H. Lu, Anal. Chem., 2017, 89, 4566–4572 CrossRef CAS PubMed.
- W. Zhang, G. Ruan, X. Li, X. Jiang, Y. Huang, F. Du and J. Li, Anal. Chim. Acta, 2019, 1071, 17–24 CrossRef CAS PubMed.
- Y. Yuan, H. Nie, J. Yin, Y. Han, Y. Lv and H. Yan, Food Chem., 2020, 313, 126155–126163 CrossRef PubMed.
- Q. Lu, Y. Ma, H. Li, X. Guan, Y. Yusran, M. Xue, Q. Fang, Y. Yan, S. Qiu and V. Valtchev, Angew. Chem., Int. Ed., 2018, 57, 6042–6048 CrossRef CAS PubMed.
- R.-Q. Wang, X.-B. Wei and Y.-Q. Feng, Chem.–Eur. J., 2018, 24, 10979–10983 CrossRef CAS.
- Y.-F. Ma, F. Yuan, Y. Yu, Y.-L. Zhou and X.-X. Zhang, Anal. Chem., 2020, 92, 1424–1430 CrossRef CAS PubMed.
- W. Jing, Y. Zhou, J. Wang, M. Ni, W. Bi and D. D. Y. Chen, Anal. Chem., 2019, 91, 11240–11246 CrossRef CAS PubMed.
- Y. Zhou, J. Zhu, J. Yang, Y. Lv, Y. Zhu, W. Bi, X. Yang and D. D. Y. Chen, Anal. Chim. Acta, 2019, 1066, 49–57 CrossRef CAS PubMed.
- A. Liu, W. Kou, H. Zhang, J. Xu, L. Zhu, S. Kuang, K. Huang, H. Chen and Q. Jia, Anal. Chem., 2020, 92, 4137–4145 CrossRef CAS PubMed.
- I. H. Šrámková, B. Horstkotte, J. Erben, J. Chvojka, F. Švec, P. Solich and D. Šatínský, Anal. Chem., 2020, 92, 3964–3971 CrossRef PubMed.
- H. Piri-Moghadam, F. Ahmadi, G. A. Gómez-Ríos, E. Boyacı, N. Reyes-Garcés, A. Aghakhani, B. Bojko and J. Pawliszyn, Angew. Chem., Int. Ed., 2016, 55, 7510–7514 CrossRef CAS PubMed.
- A. Andrade-Eiroa, M. Canle, V. Leroy-Cancellieri and V. Cerdà, TrAC, Trends Anal. Chem., 2016, 80, 641–654 CrossRef CAS.
- L. Xia, J. Yang, R. Su, W. Zhou, Y. Zhang, Y. Zhong, S. Huang, Y. Chen and G. Li, Anal. Chem., 2020, 92, 34–48 CrossRef CAS PubMed.
- M. Fiori, C. Cartoni, B. Bocca and G. Brambilla, J. Chromatogr. Sci., 2002, 40, 92–96 CAS.
- C. Juan, C. Igualada, F. Moragues, N. León and J. Mañes, J. Chromatogr. A, 2010, 1217, 6061–6068 CrossRef CAS PubMed.
- L. Giannetti, G. Ferretti, V. Gallo, F. Necci, A. Giorgi, F. Marini, E. Gennuso and B. Neri, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1036, 76–83 CrossRef.
- P. Guo, J. Wan, C. Zhan, C. Zhu, W. Jiang, Y. Ke, S. Ding and D. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1096, 122–134 CrossRef CAS PubMed.
- L. Xiong, Y.-Q. Gao, W.-H. Li, T.-F. Guo and X.-L. Yang, Food Sci. Biotechnol., 2015, 24, 1629–1635 CrossRef CAS.
- Y. Yuan, S. Liang, H. Yan, Z. Ma and Y. Liu, J. Chromatogr. A, 2015, 1408, 49–55 CrossRef CAS PubMed.
- Z. Chang, D.-S. Zhang, Q. Chen and X.-H. Bu, Phys. Chem. Chem. Phys., 2013, 15, 5430–5442 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- C. Zhang, G. Li and Z. Zhang, J. Chromatogr. A, 2015, 1419, 1–9 CrossRef CAS PubMed.
- L. Zhou, Y. Hu and G. Li, Anal. Chem., 2016, 88, 6930–6938 CrossRef CAS PubMed.
- J. Pan, S. Jia, G. Li and Y. Hu, Anal. Chem., 2015, 87, 3373–3381 CrossRef CAS PubMed.
- W. Zhang, F. Liang, C. Li, L.-G. Qiu, Y.-P. Yuan, F.-M. Peng, X. Jiang, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2011, 186, 984–990 CrossRef CAS PubMed.
- H. Zuo, Y. Guo, W. Zhao, K. Hu, X. Wang, L. He and S. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 46149–46156 CrossRef CAS PubMed.
- W. Zhao, X. Wang, J. Guo, Y. Guo, C. Lan, F. Xie, S. Zong, L. He and S. Zhang, J. Chromatogr. A, 2020, 1618, 460847–460856 CrossRef CAS PubMed.
- K. Sinha Roy, R. Goud D, A. Mazumder, B. Chandra, A. K. Purohit, M. Palit and D. K. Dubey, ACS Appl. Mater. Interfaces, 2019, 11, 16027–16039 CrossRef CAS PubMed.
- G. Li, X. Meng, J. Wang, Q. Wang, J. Zhou, C. Wang, Q. Wu and Z. Wang, Food Chem., 2020, 309, 125618–125625 CrossRef CAS PubMed.
- R. Dawson, T. Ratvijitvech, M. Corker, A. Laybourn, Y. Z. Khimyak, A. I. Cooper and D. J. Adams, Polym. Chem., 2012, 3, 2034–2038 RSC.
- J. Tang, J. Wang, S. Shi, S. Hu and L. Yuan, J. Anal. Methods Chem., 2018, 9053561, 1151–1160 Search PubMed.
- L. Ai, H. Huang, Z. Chen, X. Wei and J. Jiang, Chem. Eng. J., 2010, 156, 243–249 CrossRef CAS.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef CAS.
- X. Zhu, S. M. Mahurin, S.-H. An, C.-L. Do-Thanh, C. Tian, Y. Li, L. W. Gill, E. W. Hagaman, Z. Bian, J.-H. Zhou, J. Hu, H. Liu and S. Dai, Chem. Commun., 2014, 50, 7933–7936 RSC.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- C. H. Giles, T. H. MacEwan, S. N. Nakhwa and D. Smith, J. Chem. Soc., 1960, 111, 3973–3993 RSC.
- Y. S. Ho and G. McKay, Chem. Eng. J., 1998, 70, 115–124 CrossRef CAS.
- E. Haque, J. E. Lee, I. T. Jang, Y. K. Hwang, J.-S. Chang, J. Jegal and S. H. Jhung, J. Hazard. Mater., 2010, 181, 535–542 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04481h |
| This journal is © The Royal Society of Chemistry 2021 |