
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the abnormal capacity rising mechanism of MoS2 anode during long-term cycling for sodium-ion batteries†
Yucheng Zhua,
Haoyu Lia,
Yuanming Wua,
Liwen Yanga,
Yan Sunb,
Guang Chen c,
Yang Liud,
Zhenguo Wua,
Chuhong Zhang*e and
Xiaodong Guo*a
c,
Yang Liud,
Zhenguo Wua,
Chuhong Zhang*e and
Xiaodong Guo*a
aCollege of Chemical Engineering, Sichuan University, Chengdu 610065, Sichuan, China. E-mail: xiaodong2009@163.com
bSchool of Mechanical Engineering, Chengdu University, Chengdu 610106, Sichuan, China
cCollege of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Institutes of Biomedical Sciences, Shandong Normal University, Jinan 250014, Shandong, China
dSchool of Materials Science and Engineering, Henan Normal University, Xinxiang 453007, Henan, China
eState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute, Sichuan University, Chengdu 610065, Sichuan, China. E-mail: chuhong.zhang@scu.edu.cn
First published on 24th August 2021
Abstract
Transition metal sulfides are considered as one of the most potential anode materials in sodium-ion batteries due to their high capacity, low cost, and rich resources. Among plenty of options, molybdenum sulfide (MoS2) has been the focus of research due to the graphene-like layered structure and unique electrochemical properties. Importantly, an abnormal capacity increase phenomenon was observed in the MoS2 anode of sodium-ion batteries, but the mechanisms involved are still unclear. In this study, by analyzing the composition and structure of the material after a different number of cycles, we confirmed that the (002) plane shows a significant expansion of the interlayer spacing after the sodium ion insertion process and a phase transformation from the hexagonal phase MoS2 (2H-MoS2) to the trigonal phase MoS2 (1T-MoS2). Moreover, the ratio of 1T-MoS2 presented an increasing trend during cycling. The dual-phase co-existence leads to enhanced electrical conductivity, higher Na affinity, and higher Na+ mobility, thus increasing the capacity. Our work provides a new perspective on the anomalous electrochemical behavior of sulfide anodes during long-term cycling.
Introduction
The increase in clean energy has triggered a growing demand for energy storage systems, thus providing an opportunity for the dominance of rechargeable lithium ion batteries (LIBs) owing to their high energy density and long cycle life.1–4 However, the intrinsic problems (e.g., rare abundance and uneven geographical distribution of Li) of LIBs make the cost gradually evolve into an obstacle for grid-scale applications.5–8 Accordingly, the search for a cost-effective alternative with promising electrochemical performance becomes extremely pressing.9–11 Sodium ion batteries (SIBs) with analogical rocking-chair principles to LIBs are emerging as a viable choice for grid-level energy storage and conversion for renewable energy sources due to their low cost, wide global distribution, and excellent electrochemical performance.12–16 However, charge storage based on Na+/e− is quite challenging to enable both swift and frequent Na+ transfer because of the large ionic radius of Na+ (1.06 Å vs. 0.76 Å for Li+).17–19 Thus, one of the most urgent issues is exploiting suitable Na+-host materials, especially anodic materials, to reach prominent comprehensive electrochemical performances.Two-dimensional (2D) materials have presented great potential as anode materials in SIBs owing to their fascinating layered structure, which is capable of hosting Na+, delivers a large surface area for Na+ adsorption, and abundant defects.13,20–23 Compared with metal oxides, sulfides, as typical 2D materials, exhibited impressive ability in overcoming such problems (i.e., huge volume variation, severe structure collapse, limited high-rate capability) due to its weakened M–S bonds than M–O bonds.24 Among them, MoS2 has gained enormous attention because of its fantastic 2D S–Mo–S sandwich-like structure where a single Mo atom is covalently bonded by six S atoms.25–27 The laminar MoS2 with adjacent planes stacked through the van der Waals forces delivers large interlayer spacing (0.62 nm) for Na+ transport.27 Conventionally, MoS2 in the natural state presented a stable 2H phase with trigonal structure, which endow 2H-MoS2 with semi-insulating characteristics with a band gap of ∼1.9 eV, thus delivering a weak conductivity. In contrast, another 1T-MoS2 based on tetragonal symmetry with edge-sharing [MoS6] octahedra in the metallic phase delivers a much higher conductivity (∼107 times) than 2H-MoS2.28–31 These merits of MoS2 make it an appealing anode material and stimulated much investigation to explore the underlying sodium storage mechanism, especially the distinct rise in capacity during cycling. Some theories were proposed: (1) the decrease in MoS2 size along the cycling could boost the infiltration between MoS2 and electrolyte.32 (2) The electrolyte decomposition could lead to the reversible generation of organic polymeric/gel-like layers and the activation of the MoS2 electrode.33 (3) The repeated Na+ intercalation/deintercalation could result in gradual expanding and interlayer exfoliation.34 On the other hand, some investigations revealed that the formation of 1T-MoS2 in 2H-MoS2 improved the electrochemical performance via creating the internal electric field.33,35,36 However, so far, there is rarely any systematic study to explore the rise in capacity during cycling in terms of phase transition, which is available to enable a completely different crystalline structure, thus delivering totally distinct sodium storage behavior.
In this work, we synthesized molybdenum sulfide in the form of flowers by hydrothermal synthesis, and annealing at 350 °C allowed the molybdenum sulfide to achieve a stable structure. The structural and compositional changes of the material after long cycling were characterized by X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM), and it was found that Mo4+ peaks showed a significant shift toward lower binding energies after cycling, with a corresponding enhancement of the shift as the number of cycles increased. According to the results of the split-peak fitting and TEM patterns, we confirmed that the MoS2 of the metal phase was generated, and its content increased during the cycling process. Also, the layer spacing of some of the molybdenum sulfides widens after the first cycle and remains constant during the subsequent charge/discharge process. These results reveal the influence of structural and compositional changes of molybdenum disulfide on the storage electrodes of long-life sodium ion batteries.
Experimental section
Material preparation
Molybdenum sulfide was synthesized by a one-step simple solvothermal method using deionized water as the solvent, ammonium molybdate tetrahydrate as the molybdenum source, and thiourea as the sulfur source. Among them, 1.2 g of ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) and 2.2 g of thiourea (CH4N2S) were dissolved in 35 ml of deionized water and mixed well by stirring for half an hour at room temperature. The solution was then poured into a 100 ml Teflon-lined autoclave, which was maintained at 200 °C for 16 h. After washing with ethanol, it was dried under a vacuum at 80 °C overnight. Finally, it was heated for 4 h at 350 °C under Ar.Structural and morphological characterization
The crystalline structure was analyzed from powder X-ray diffraction (XRD, D/Max 2500 PC) with Cu Kα radiation. The XPS spectra were recorded applying an ESCALAB 250Xi (Thermo Fisher) with a monochromatic Al Kα source. Morphologies of the products were investigated by scanning electron microscope (SEM, Zeiss SUPRA 55) and transmission electron microscopy (TEM, JEOL JEM 2100).Electrochemical measurements
The working electrode was composed of the active material, acetylene black, and PVDF (Sigma-Aldrich) with a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 dissolved in N-methyl-2-pyrrolidinone (Sinopharm). After coating the slurries onto a copper foil current collector and drying at 80 °C overnight, the Cu foil with active material was cut into disk electrodes with a radius of 14 mm. The mass density of the active material in each electrode was 1.0–2.0 mg cm−2. The electrochemical measurements of these materials were carried out via CR2025 type coin cells assembled in an argon-filled glovebox (Dellis company). The electrolyte was composed of NaCF3SO3 (1 M) in diglyme and 1 M NaClO4 in propylene carbonate (PC) with 5 vol% fluoroethylene carbonate (FEC). Na metal was set as the counter and reference electrodes and glass microfiber as the separator (Whatman, GF/D). The cycling performances of the as-prepared samples were tested on a LAND-BT2013A measurement system with a voltage ranging from 0.4 to 3.0 V at 25 °C.
:2:1 dissolved in N-methyl-2-pyrrolidinone (Sinopharm). After coating the slurries onto a copper foil current collector and drying at 80 °C overnight, the Cu foil with active material was cut into disk electrodes with a radius of 14 mm. The mass density of the active material in each electrode was 1.0–2.0 mg cm−2. The electrochemical measurements of these materials were carried out via CR2025 type coin cells assembled in an argon-filled glovebox (Dellis company). The electrolyte was composed of NaCF3SO3 (1 M) in diglyme and 1 M NaClO4 in propylene carbonate (PC) with 5 vol% fluoroethylene carbonate (FEC). Na metal was set as the counter and reference electrodes and glass microfiber as the separator (Whatman, GF/D). The cycling performances of the as-prepared samples were tested on a LAND-BT2013A measurement system with a voltage ranging from 0.4 to 3.0 V at 25 °C.
Results and discussion
The crystal structures of as-prepared MoS2 were examined by X-ray diffraction (XRD). Fig. 1b displays four major diffraction peaks at 13.6, 33.2, 39.7, and 58.7° assigned to the (002), (100), (103), and (110) planes of 2H-MoS2 (JCPDS No. 37-1492). Based on the main diffraction peak at 13.6°, the interlayer distance of MoS2 is 0.61 nm via calculation by Bragg's Law. According to the previous reports, XPS can be used to identify the 1T phase in the sample because Mo 3d spectra of the 1T phase will shift towards lower binding energy. As shown in Fig. 1c, the two peaks of the annealed sample are located at 229.7 eV and 232.6 eV, there is no offset of the peak, and the results indicate that the annealed sample is pure 2H-MoS2. Inevitably, the two faint peaks show the presence of Mo6+ and S 2p, which are located at 235.7 eV and 226.7 eV, indicating that inevitable contact with air during the synthesis and preservation process caused partial oxidation of MoS2.37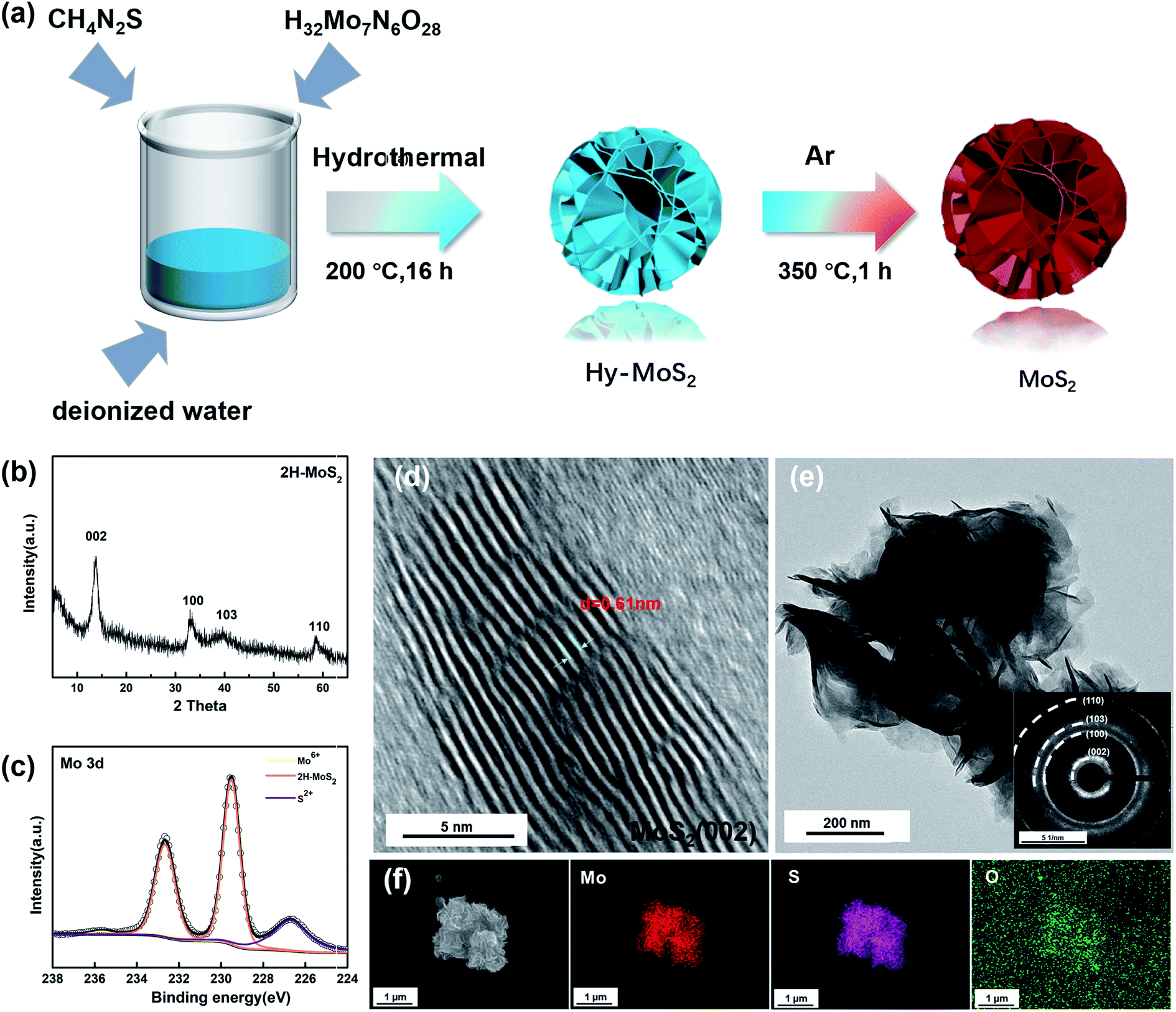 | ||
| Fig. 1 (a) Schematic illustration of flower-like MoS2 through a hydrothermal technique. (b) XRD patterns of 2H-MoS2. (c) High-resolution XPS of Mo 3d. (d) HRTEM images of the nanostructured MoS2. (e) SAED pattern of MoS2. (f) SEM images of MoS2 and mapping images (Mo, S, and O) of MoS2. | ||
The morphology and structure of the samples were observed by SEM, TEM, and high-resolution TEM. In Fig. 1d, we can observe that the layer spacing of the (002) plane is roughly 0.61 nm, which is consistent with the position of the (002) diffraction peak in XRD. The low magnification TEM (Fig. 1e) shows the different layers in the sample. The thinner edge areas are more susceptible to oxidation, and it can be judged that the particle size is about 500 nm. The (002), (100), (103), and (110) planes in the selected area electron diffraction pattern (Fig. 1e) also correspond to the four peaks in the XRD patterns. As shown in Fig. 1f, the sample has a flower-like structure, and S and Mo are uniformly distributed throughout the particles. Moreover, a clear distribution of O on the particles can also be observed, which also confirms that the samples were indeed partially oxidized.
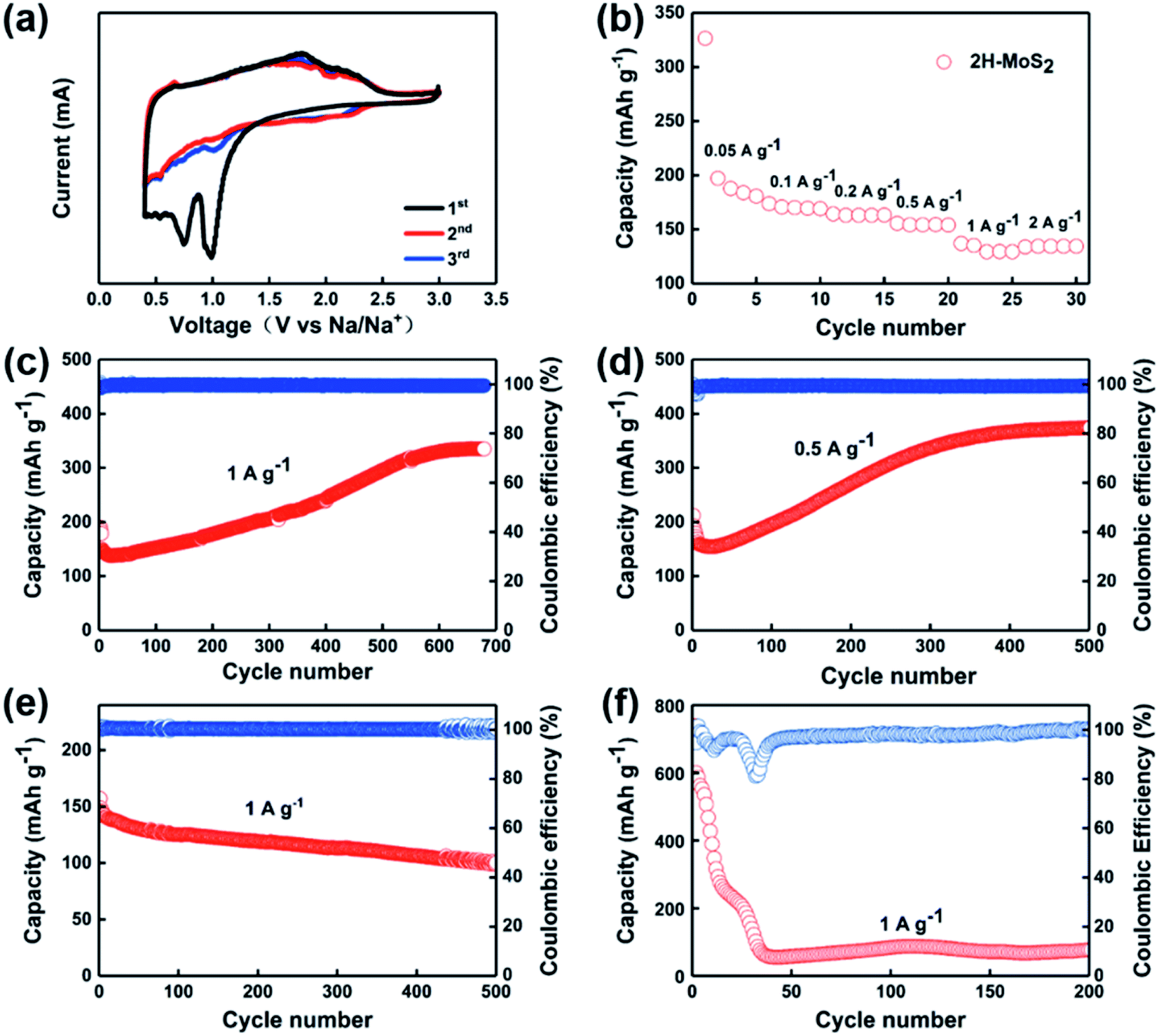 | ||
| Fig. 2 (a) Cyclic voltammetry curves of the as-prepared MoS2. (b) Rate capability of the as-prepared MoS2. (c) and (d) Cycling performances of as-prepared MoS2 nanosheets at 1 A g−1 and 0.5 A g−1. (e) Cycling performances of as-prepared MoS2 nanosheets at 1 A g−1 with 1 M NaClO4 in propylene carbonate (PC) with 5 vol% fluoroethylene carbonate. (f) Cycling performance of as-prepared MoS2 nanosheets at 1 A g−1 with a cut-off voltage in the range of 3–0.01 V. | ||
According to the previously reported studies, the cut-off voltage affects the charge/discharge performance of the battery because conversion reactions occur below 0.4 V, which leads to poor reversibility of the whole process.34,38 Intercalation and conversion reaction are shown in eqn (1) and (2), which are similar to that of LIBs.39,40 Therefore, during electrochemical tests, we have chosen the voltage range of 3.0–0.4 V for our experiments.
| MoS2 + xNa = NaxMoSx (above 0.4 V, x < 2) | (1) |
| NaxMoSx + (4 − x)Na = Mo + 2Na2S (below 0.4 V) | (2) |
In order to study the redox reactions occurring during the charge and discharge process, CV tests were performed on the samples. As shown in Fig. 2a, there are two cathodic peaks at 0.92 V and 0.76 V, respectively, during the first discharge, which corresponds to the process of Na+ inserting into MoS2 and accompanied by the occurrence of phase transition processes. The peaks at positions 1.9 V and 2.2 V appearing during charging indicate the process of Na+ extracting from NaxMoS2.31,38 The excellent overlap shown in the curves of both the second cycle and the third cycle confirms the ideal reversibility of the sample. Fig. 2b shows the rate performances of the electrodes. The reversible capacity of the MoS2-based anode cell can be obtained as 175, 170, 164, 154, 135, and 134 mA h g−1 at the current densities of 0.05, 0.1, 0.2, 0.5, 1, and 2 A g−1, respectively. Fig. 2c and d show the cycling stability of MoS2 as anode for SIBs at a current density of 1 A g−1 and 0.5 A g−1, respectively. Interestingly, after continuous reduction of capacity after the first 30 cycles, the capacity of the MoS2 anode showed an anomalous rise. The first discharge mass specific capacity of MoS2 at 1 A g−1 is 198 mA h g−1. After 30 cycles, the capacity drops to nearly 150 mA h g−1, which can be attributed to the generation of the SEI and the activation process of the batteries.41 However, the capacity of the MoS2 anode completely reversed the previous decreasing trend after the phenomena taking place at the 30th cycle. The capacity rose in a steady manner until it stabilized after 650 cycles, as it rose from 134 mA h g−1 to around 340 mA h g−1. In the subsequent cycles, the capacity stays stable and remains at 340 mA h g−1 with the generation of Na1.0MoS2.42 Similarly, the same situation of capacity falling and then rising occurs at a current of 0.5 A g−1. The discharge capacity is 215 mA h g−1 for the first cycle and then decreases to 150 mA h g−1 after 20 cycles. During the subsequent cycles, the capacity rises to 370 mA h g−1 after about 400 cycles and remains stable. On the contrary, there was no abnormal rise in capacity when 1 M NaClO4 in propylene carbonate (PC) with 5 vol% fluoroethylene carbonate was applied as the electrolyte (Fig. 2e). The ether electrolyte itself inhibits the shuttle effect of the generated polysulfide, and the use of ether electrolyte instead of ester electrolyte can reduce the organic components contained in the negative SEI film of the sodium ion battery so that the interface formed between the SEI film and the negative material is more dense and stable.43,44 Meanwhile, when the cut-off voltage was in the range of 0.01–3 V, the capacity dropped steadily from 739 mA h g−1 to 76 mA h g−1 due to an irreversible conversion reaction ranging from 0.4 V to 0.1 V (Fig. 2f). The reason for the phenomenon is mainly the formation of Mo and S monomers, which cannot completely regenerate MoS2 during cycling.
The anomaly of capacity rising is a relatively common phenomenon in MoS2 electrodes, and a number of explanations have been given based on different aspects. Along with the increase in capacity, a decrease in impedance is also observed. It is interesting to note that this phenomenon is usually limited to the use of ether electrolytes with controlled cut-off voltage intervals.
As shown in Fig. 3a, we chose four points to test during the first cycle of charging and discharging at a current of 0.02 A g−1. Combined with the position of the peak in the first-turn CV, we chose four points for characterization tests for discharging to 0.9 V, 0.4 V and charging to 1.5 V, 3.0 V. In the ex situ XRD results (Fig. 3b), we can see that the position of peak (002) has shifted about 9° left from 14°. The offset is universally considered to be caused by the expansion of the layer spacing of the (002) plane.45,46 In previous reports, the phenomenon and corresponding explanation that the layer spacing of MoS2 expands due to the insertion of Na+ were observed and concluded by researchers through some characterizations.47 Surprisingly, the expanded layer spacing after going through the Na+ insertion process was not reduced by Na+ extraction. The graph shows that the (002) peak position also remains stable at around 9° when charged to 1.5 V and 3.0 V. Combined with Fig. 3d and e, it can be confirmed that the layer spacing is not restored during the charging process after the expansion. Furthermore, the ex situ XRD diagram shows the presence of Na2MoO4 due to partial MoS2 oxidation as well. According to Michael et al., MoS2 is more likely to be present in the form of MoO42+ rather than MoO3 when oxidized.48 In the ex situ XPS, we can clearly see a peak of Mo at 235.5 eV, again proving that some MoS2 was oxidized during the cycle. Curiously, in the ex situ XPS for the samples discharging to 0.9 V and 0.5 V and charging to 1.5 V, we can see a clear peak at 228.0 eV, showing the existence of Mo, which is not consistent with the previous paper. We speculate that it is because some of the molybdenum oxide produced Mo during the reaction. It is confirmed that no chemical reaction occurred after assembling the battery without discharging and just leaving it for 12 h (Fig. S1†). After one cycle, the XPS of the sample charged to 3.0 V shows that the material consists of Mo4+ and Mo6+, but the Mo4+ peak is shifted toward the lower binding energy with respect to the original sample. Combined with what is shown in Fig. S2† and the previous reports, it is confirmed that there is 1T-MoS2 generation.49–51 Through heating or aging, unstable 1T-MoS2 will usually undergo phase transition back to 2H-MoS2.52,53 However, from the ex situ XPS analysis, it is obvious that the 1T-MoS2 produced by the phase transition does not return to the 2H-MoS2 phase but is basically stable in the sample. From the composition, the material after one cycle charge and discharge process contains Na2MoO4, 1T-MoS2, and 2H-MoS2. According to Fig. S3,† we can clearly see that the material composition after 200 cycles remains the same as that after the first charge and discharge process. The intercalation of Na+ tends to enhance the stability of 1T-MoS2, as shown in Fig. 3c. It can be clearly observed that there is still a co-existence of two phases, and the experimental results show that 1T-MoS2 still is stable and exists after Na+ is extracted. The explanation, which Xu proposed that MoO3 can also stabilize 1T-MoS2 without phase change, also applies to our experimental results.54 It is consistent with the calculation of Wang et al. that 1T-MoS2 is the dominant phase after Na+ extraction.55
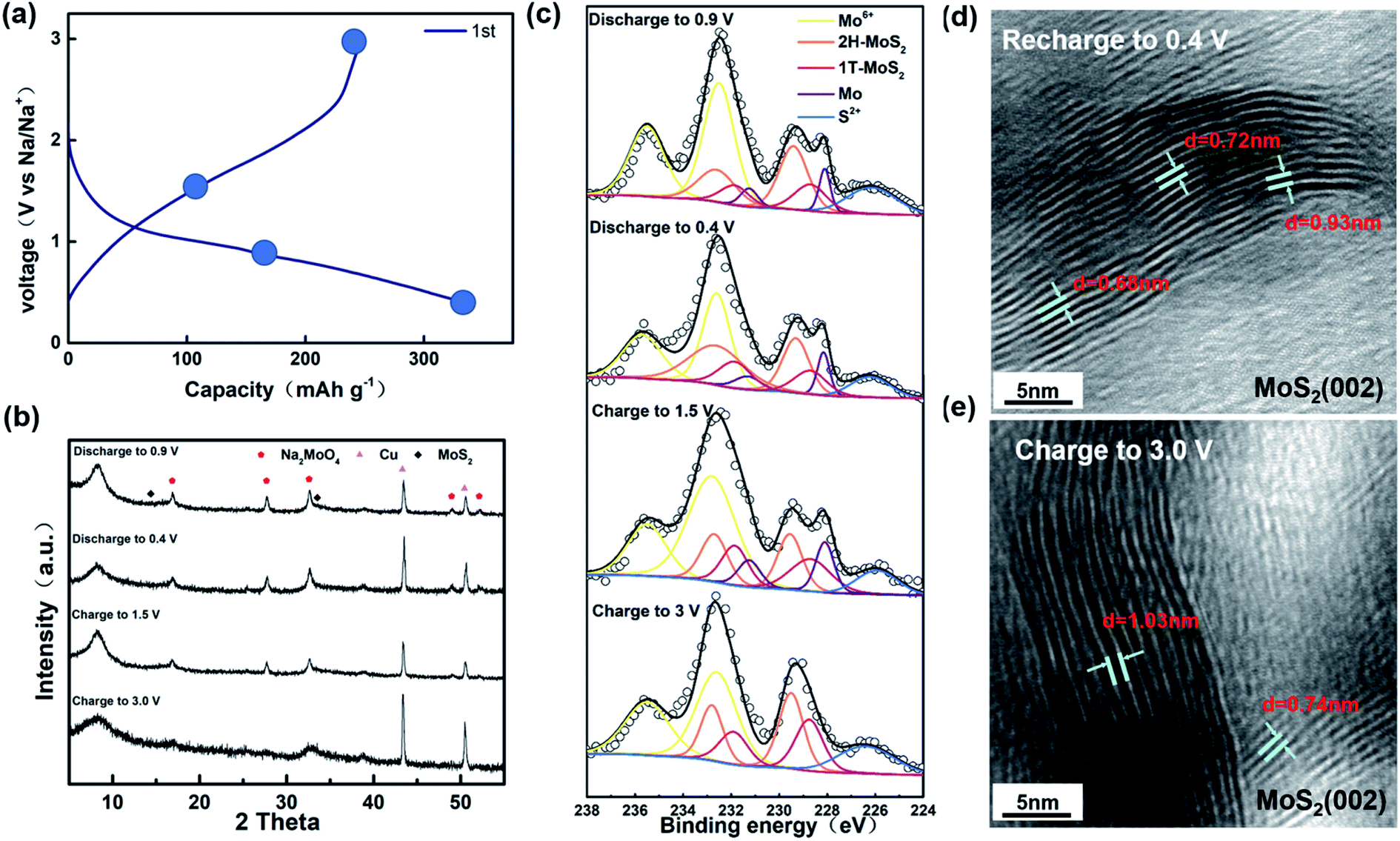 | ||
| Fig. 3 (a) Charge/discharge curves of the MoS2 electrode for the first cycle at a current density of 0.02 A g−1. (b) and (c) ex situ XRD and XPS pattern of different charge/discharge depths. (d) and (e) HRTEM of MoS2 after recharging to 0.4 V and charge to 3.0 V. | ||
In a subsequent study, we have tried to analyze the composition of phases in a different number of cycles. According to Fig. 2c and d, the capacity rising continues until the cycling reaches stability at around 600 cycles. The cycling process of the first lap has been analyzed, and the partial phase change during its recharge/discharge process has been confirmed. We selected cells after cycling 100th and 200th during the capacity rising to discover their composition and morphology. As shown in Fig. 4a and b, it is clearly observed that there is still a co-existence of the two phases. Furthermore, as shown in the diagram, there is always the presence of Mo6+, and combined with Fig. S3† we can determine its component as MoO42−. The Na2MoO4 in the cathode material provides good protection to the electrode and contributes to better cycling stability.54,56 According to Fig. 4c, the same two phases co-exist after 100 cycles, 200 cycles, and 650 cycles, in agreement with the results of ex situ XPS. It can be seen that the 1T (octahedral) and 2H (trigonal) phases co-exist in the same nanosheet, which can be classified as type IV with a 2D in-plane 1T/2H MoS2 heterogeneous structure.20,57 There are no significant defects in the boundaries between them. Meanwhile, the petal-like structure can still be observed in the morphology of the whole material (Fig. S4†).
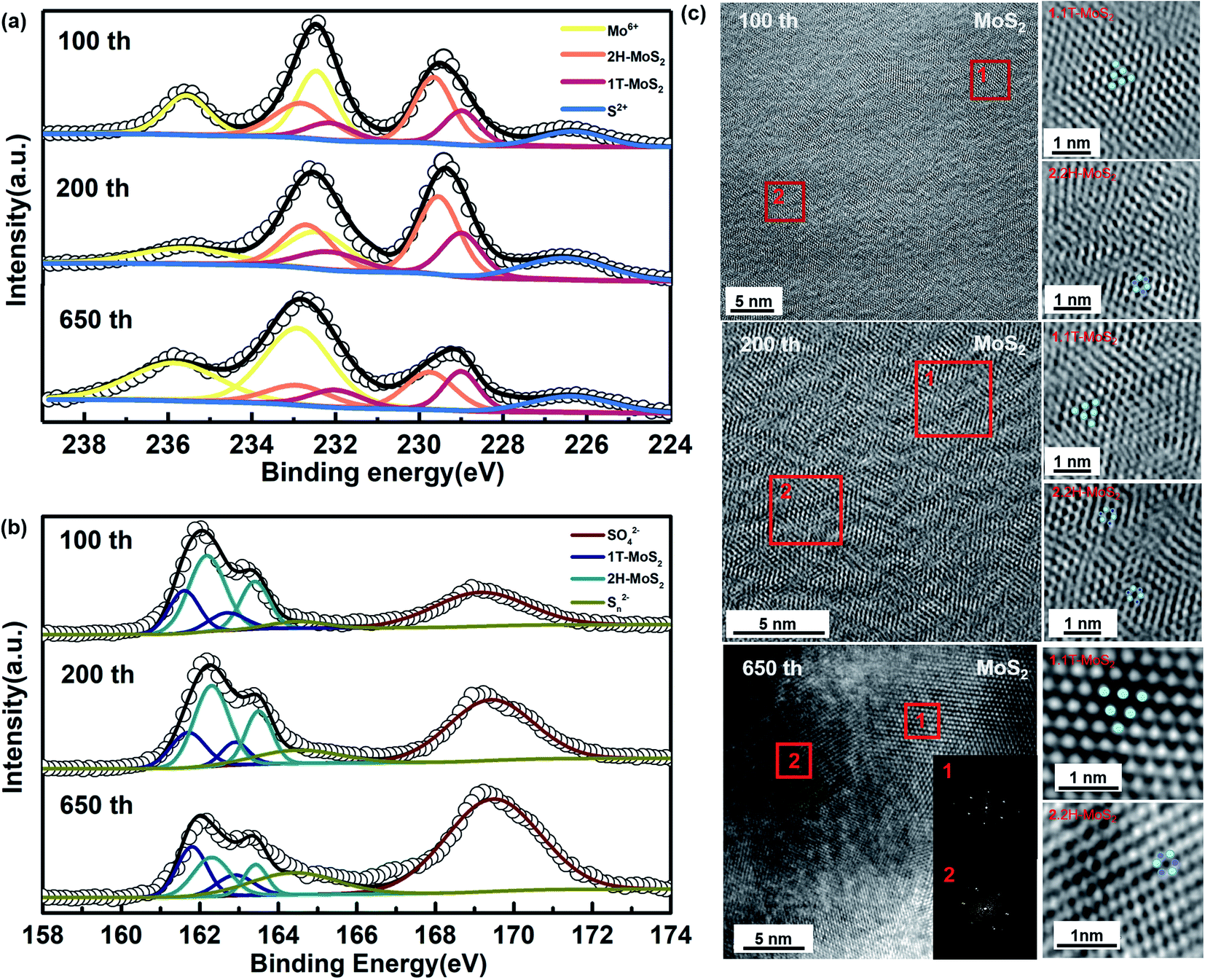 | ||
| Fig. 4 (a) and (b) Ex situ XPS of MoS2 anode after charge/discharge for different cycles, (c) HRTEM patterns and the corresponding SAED pattern of MoS2 anode after 650 cycles. | ||
According to Fig. S5,† there is a significant widening of the layer spacing on the (002) surface, and this widening appears and is maintained from the first cycle onwards. From this aspect, there is a relationship between the increase in capacity and the expansion of the layer spacing. Meanwhile, when analyzed in terms of composition, it can be found that the Mo4+ peaks show a significant shift toward lower binding energies, which means that the ratio of MoS2 in different phases has changed. From the 1st cycle to the 650th cycle, the 1T phase appears and continues to increase, showing the same trend as the rising capacity. It is noteworthy that S 2p3/2 and S 2p1/2 show the same peak shift in agreement with Mo4+ (Fig. 4b). Moreover, the generation of metallic phases as well as the increase in content lead to a continuous increase in capacity.
According to the results, the appearance and increase in the amount of 1T-MoS2 can be determined. The contribution of 1T-MoS2 to the capacity is usually explained in terms of these aspects. Benefiting from different coordination between Mo and S atoms, the 1T-MoS2 has a much higher electronic conductivity than the semiconducting 2H counterpart. Moreover, 1T-MoS2 and 2H-MoS2 could induce an internal electric field at the corresponding heterointerface because of the different Fermi energies. The phase change reduction is also an important reason for the capacity increase. During the discharge, a phase transition process from 2H-MoS2 to 1T-MoS2 occurs, but after the partial 1T phase stabilization, only the embedding process of sodium ions and no more phase transition occurs. According to the reports, the process is particularly similar to SnS, which has superior cycling performance relative to SnS2 during cycling because it undergoes only two phase transitions less than the three phase transformation of SnS2.58
Conclusions
In general, we synthesized pure 2H-MoS2 by hydrothermal method and applied it as the anode material for sodium-ion batteries. We controlled the charge/discharge cut-off voltage and used the DME electrolyte, and the battery capacity showed an abnormal increase. Based on the analysis of electrode sheets after cycling for a different number of revolutions, we found that 1T-MoS2 was generated after cycling and its content tended to increase with capacity during cycling. By analyzing the effect of 1T-MoS2, the capacity rising is due to the formation of a biphasic co-existence of MoS2 during the cycle and the progressive increase in the 1T-MoS2 content. At the same time, the layer spacing expanded from 0.62 nm to nearly 1.03 nm during the cycling process, which also contributed to the capacity of the cell. Finally, this study provides an explanation for the anomalous phenomenon of transition metal sulfides as anode for SIBs, which provides a new method and idea for future research on transition metal sulfides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21805198, 21878195, and 21805018), Distinguished Young Scholars of Sichuan University (2017SCU04A08), the Huohua Ku project of Sichuan University (2018SCUH0094), Research Foundation for the Postdoctoral Program of Sichuan University (No. 2017SCU12018 and 2018SCU12045), the Research Foundation for the Sichuan University, and Zigong City Joint research project (2018CDZG-16). The authors thank Dr Zhuo Zheng for help with Raman spectrum analysis. We would like to thank Yanping Huang from the Center of Engineering Experimental Teaching, School of Chemical Engineering, Sichuan University for the help with TEM images.Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- J. Zhu, P. Zhao, S. Yang, L. Chen, Q. Zhang and Q. Yan, Fuel, 2021, 292, 120263 CrossRef CAS.
- C. Geng, W. Hua, D. Wang, G. Ling, C. Zhang and Q. H. Yang, SusMat, 2021, 1, 51–65 CrossRef.
- E. Goikolea, V. Palomares, S. J. Wang, I. R. de Larramendi, X. Guo, G. X. Wang and T. Rojo, Adv. Energy Mater., 2020, 10, 2002055 CrossRef CAS.
- Y. Zhao, L. P. Wang, M. T. Sougrati, Z. X. Feng, Y. Leconte, A. Fisher, M. Srinivasan and Z. C. Xu, Adv. Energy Mater., 2017, 7, 1601424 CrossRef.
- H. L. Pan, Y. S. Hu and L. Q. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 RSC.
- M. Fan, X. Chang, Q. Meng, L. J. Wan and Y. G. Guo, SusMat, 2021, 1, 241–254 CrossRef.
- Y. F. Yuan, K. Amine, J. Lu and R. Shahbazian-Yassar, Nat. Commun., 2017, 8, 15806 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- Z. W. Yang, T. Chen, D. Q. Chen, X. Y. Shi, S. Yang, Y. J. Zhong, Y. X. Liu, G. K. Wang, B. H. Zhong, Y. Song, Z. G. Wu and X. D. Guo, Angew. Chem., Int. Ed., 2021, 60, 12539–12546 CrossRef CAS PubMed.
- Y. Von Lim, X. L. Li and H. Y. Yang, Adv. Funct. Mater., 2021, 31, 2006761 CrossRef.
- S. W. Kim, D. H. Seo, X. H. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- D. Q. Chen, W. Zhang, K. Y. Luo, Y. Song, Y. J. Zhong, Y. X. Liu, G. K. Wang, B. H. Zhong, Z. G. Wu and X. D. Guo, Energy Environ. Sci., 2021, 14, 2244–2262 RSC.
- H. Zhang, Y. Chen, C. Li and M. Armand, SusMat, 2021, 1, 24–37 CrossRef.
- Z. Yang, L. Qiu, M. Zhang, Y. Zhong, B. Zhong, Y. Song, G. Wang, Y. Liu, Z. Wu and X. Guo, Chem. Eng. J., 2021, 423, 130127 CrossRef CAS.
- D. Sun, D. Ye, P. Liu, Y. Tang, J. Guo, L. Wang and H. Wang, Adv. Energy Mater., 2018, 8, 1702383 CrossRef.
- Z. G. Wu, J. T. Li, Y. J. Zhong, J. Liu, K. Wang, X. D. Guo, L. Huang, B. H. Zhong and S. G. Sun, J. Alloys Compd., 2016, 688, 790–797 CrossRef CAS.
- X. H. Liu, G. L. Feng, Z. G. Wu, Z. G. Yang, S. Yang, X. D. Guo, S. H. Zhang, X. T. Xu, B. H. Zhong and Y. Yamauchi, Chem. Eng. J., 2020, 386, 123953 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- Y. H. Xiao, D. C. Su, X. Z. Wang, S. D. Wu, L. M. Zhou, Y. Shi, S. M. Fang, H. M. Cheng and F. Li, Adv. Energy Mater., 2018, 8, 1800930 CrossRef.
- T. J. Wu, M. J. Jing, Y. Liu and X. B. Ji, J. Mater. Chem. A, 2019, 7, 6439–6449 RSC.
- Z. Hu, Q. N. Liu, S. L. Chou and S. X. Dou, Adv. Mater., 2017, 29, 1700606 CrossRef.
- W. H. Lai, Y. X. Wang, J. Z. Wang, S. L. Chou and S. X. Dou, Manipulating 2D Few-Layer Metal Sulfides as Anode Towards Enhanced Sodium-Ion Batteries, Batter. Supercaps, 2020, 3(3), 236–253 CrossRef CAS.
- X. Xie, T. Makaryan, M. Zhao, K. L. Van Aken, Y. Gogotsi and G. Wang, Adv. Energy Mater., 2016, 6, 1502161 CrossRef.
- Q. Pan, Q. Zhang, F. Zheng, Y. Liu, Y. Li, X. Ou, X. Xiong, C. Yang and M. Liu, ACS Nano, 2018, 12, 12578–12586 CrossRef CAS PubMed.
- X. L. Hu, W. Zhang, X. X. Liu, Y. N. Mei and Y. Huang, Chem. Soc. Rev., 2015, 44, 2376–2404 RSC.
- L. Wang, Z. Xu, W. Wang and X. Bai, J. Am. Chem. Soc., 2014, 136, 6693–6697 CrossRef CAS PubMed.
- Y. Fang, J. Pan, J. He, R. Luo, D. Wang, X. Che, K. Bu, W. Zhao, P. Liu, G. Mu, H. Zhang, T. Lin and F. Huang, Angew. Chem., Int. Ed. Engl., 2018, 57, 1232–1235 CrossRef CAS PubMed.
- Y. Huang, Y. Sun, X. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan, N. Williams, J. Hu, J. Ge, N. Pu, X. Yan, X. Pan, L. Zhang, Y. Wei and J. Gu, Nat. Commun., 2019, 10, 982 CrossRef CAS PubMed.
- X. Zhao, S. Ning, W. Fu, S. J. Pennycook and K. P. Loh, Adv. Mater., 2018, 30, e1802397 CrossRef.
- D. Sun, D. Huang, H. Wang, G.-L. Xu, X. Zhang, R. Zhang, Y. Tang, D. Abd Ei-Hady, W. Alshitari, A. Saad Al-Bogami, K. Amine and M. Shao, Nano Energy, 2019, 61, 361–369 CrossRef CAS.
- J. Wu, J. Liu, J. Cui, S. Yao, M. Ihsan-Ul-Haq, N. Mubarak, E. Quattrocchi, F. Ciucci and J.-K. Kim, J. Mater. Chem. A, 2020, 8, 2114–2122 RSC.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed. Engl., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- C. Z. Zhang, F. Han, J. M. Ma, Z. Li, F. Q. Zhang, S. H. Xu, H. B. Liu, X. K. Li, J. S. Liu and A. H. Lu, J. Mater. Chem. A, 2019, 7, 11771–11781 RSC.
- L. B. Fang, Z. Y. Lan, W. H. Guan, P. Zhou, N. Bahlawane, W. P. Sun, Y. H. Lu, C. Liang, M. Yan and Y. Z. Jiang, Energy Storage Mater., 2019, 18, 107–113 CrossRef.
- J. J. Wang, C. Luo, T. Gao, A. Langrock, A. C. Mignerey and C. S. Wang, Small, 2015, 11, 473–481 CrossRef CAS PubMed.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- M. Minakshi, M. Barmi, D. R. G. Mitchell, A. J. Barlow and M. Fichtner, Mater. Today Energy, 2018, 10, 1–14 CrossRef.
- M. M. Sundaram, T. Watcharatharapong, S. Chakraborty, R. Ahuja, S. Duraisamy, P. T. Rao and N. Munichandraiah, Dalton Trans., 2015, 44, 20108–20120 RSC.
- M. Minakshi, D. R. G. Mitchell, A. R. Munnangi, A. J. Barlow and M. Fichtner, Nanoscale, 2018, 10, 13277–13288 RSC.
- Q. Li, Z. Yao, J. Wu, S. Mitra, S. Hao, T. S. Sahu, Y. Li, C. Wolverton and V. P. Dravid, Nano Energy, 2017, 38, 342–349 CrossRef CAS.
- J. Zhang, D. W. Wang, W. Lv, L. Qin, S. Z. Niu, S. W. Zhang, T. F. Cao, F. Y. Kang and Q. H. Yang, Adv. Energy Mater., 2018, 8, 1801361 CrossRef.
- K. K. Li, J. Zhang, D. M. Lin, D. W. Wang, B. H. Li, W. Lv, S. Sun, Y. B. He, F. Y. Kang, Q. H. Yang, L. M. Zhou and T. Y. Zhang, Nat. Commun., 2019, 10, 1248 CrossRef PubMed.
- T. Xiang, Q. Fang, H. Xie, C. Wu, C. Wang, Y. Zhou, D. Liu, S. Chen, A. Khalil, S. Tao, Q. Liu and L. Song, Nanoscale, 2017, 9, 6975–6983 RSC.
- J. Wang, L. Han, X. Li, L. Zeng and M. Wei, J. Colloid Interface Sci., 2019, 548, 20–24 CrossRef CAS PubMed.
- K. Zhang, P. Li, S. Y. Guo, J. Y. Jeong, B. Jin, X. M. Li, S. L. Zhang, H. B. Zeng and J. H. Park, J. Mater. Chem. A, 2018, 6, 22513–22518 RSC.
- L. Xu, A. R. Tetreault and M. A. Pope, Chem. Mater., 2019, 32, 148–156 CrossRef.
- K. Yao, Z. W. Xu, J. F. Huang, M. Ma, L. C. Fu, X. T. Shen, J. Li and M. S. Fu, Small, 2019, 15, 1805405 CrossRef.
- X. F. Wang, X. Shen, Z. X. Wang, R. C. Yu and L. Q. Chen, ACS Nano, 2014, 8, 11394–11400 CrossRef CAS.
- S. Hao, X. Shen, M. Tian, R. Yu, Z. Wang and L. Chen, Nano Energy, 2017, 41, 217–224 CrossRef CAS.
- S. J. Sandoval, D. Yang, R. F. Frindt and J. C. Irwin, Phys. Rev. B, 1991, 44, 3955–3962 CrossRef.
- D. Yang, S. J. Sandoval, W. M. R. Divigalpitiya, J. C. Irwin and R. F. Frindt, Phys. Rev. B, 1991, 43, 12053–12056 CrossRef CAS.
- X. Xue, J. Zhang, I. A. Saana, J. Sun, Q. Xu and S. Mu, Nanoscale, 2018, 10, 16531–16538 RSC.
- J. U. Choi, J. H. Jo, C.-H. Jo, M. K. Cho, Y. J. Park, Y. Jin, H. Yashiro and S.-T. Myung, J. Mater. Chem. A, 2019, 7, 13522–13530 RSC.
- P. Sharma, M. Minakshi, J. Whale, A. Jean-Fulcrand and G. Garnweitner, Nanomaterials, 2021, 11, 11030580 Search PubMed.
- Y. L. Zhang, Z. J. Mu, C. Yang, Z. K. Xu, S. Zhang, X. Y. Zhang, Y. J. Li, J. P. Lai, Z. H. Sun, Y. Yang, Y. G. Chao, C. J. Li, X. X. Ge, W. X. Yang and S. J. Guo, Adv. Funct. Mater., 2018, 28, 1707578 CrossRef.
- T. F. Zhou, W. K. Pang, C. F. Zhang, J. P. Yang, Z. X. Chen, H. K. Liu and Z. P. Guo, ACS Nano, 2014, 8, 8323–8333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05518f |
| This journal is © The Royal Society of Chemistry 2021 |