
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the impact of calcination parameters on the crystal structure, morphology, and optical properties of electrospun Fe2TiO5 nanofibers†
Zorka Ž. Vasiljević *a,
Milena P. Dojčinovića,
Jelena D. Vujančevićb,
Matjaž Spreitzerc,
Janez Kovačc,
Dragana Bartolića,
Smilja Markovićb,
Ivona Janković-Čaštvand,
Nenad B. Tadiće and
Maria Vesna Nikolića
*a,
Milena P. Dojčinovića,
Jelena D. Vujančevićb,
Matjaž Spreitzerc,
Janez Kovačc,
Dragana Bartolića,
Smilja Markovićb,
Ivona Janković-Čaštvand,
Nenad B. Tadiće and
Maria Vesna Nikolića
aInstitute for Multidisciplinary Research, University of Belgrade, Serbia. E-mail: zorkav@imsi.rs
bInstitute of Technical Sciences of the Serbian Academy of Sciences and Arts, Serbia
cJozef Stefan Institute, Slovenia
dFaculty of Technology and Metallurgy, Serbia
eFaculty of Physics, University of Belgrade, Serbia
First published on 1st October 2021
Abstract
Nanostructured Fe2TiO5 (pseudobrookite), a mixed metal oxide material holds significant promise for utilization in energy and environmental applications. However, its full application is still hindered due to the difficulty to synthesize monophasic Fe2TiO5 with high crystallinity and a large specific surface area. Herein, Fe2TiO5 nanofibers were synthesized via a versatile and low-cost electrospinning method, followed by a calcination process at different temperatures. We found a significant effect of the calcination process and its duration on the crystalline phase in the form of either pseudobrookite or pseudobrookite–hematite–rutile and the morphology of calcined nanofibers. The crystallite size increased whereas the specific surface area decreased with an increase in calcination temperature. At higher temperatures, the growth of Fe2TiO5 nanoparticles and simultaneous coalescence of small particles was noted. The highest specific surface area was obtained for the sample calcined at 500 °C for 6 h (SBET = 64.4 m2 g−1). This work opens new opportunities in the synthesis of Fe2TiO5 nanostructures using the electrospinning method and a subsequent optimized calcination process for energy-related applications.
Introduction
Nanostructured metal oxides (MO) play a very important role in different technological areas such as photocatalysis, catalysis, biotechnology, electronic devices, etc.1,2 Their small particle size and a large specific surface area to volume determine their physical, chemical, and optical properties.3,4 Nanostructured materials can be classified as zero (0D) – nanoparticles, quantum dots, and nanospheres (1D) – nanotubes, nanowires, and nanorods, (2D) – nanoplates, nanosheets, and nanodisks, and (3D) – nanoflowers, nanocones, and nanoballs.5 Thus, different methods (physical, chemical, or biological) have been used for the synthesis of nanostructures depending on the expected properties correlated with the application of the material.6–8TiO2 is one of the most investigated nanomaterials used in different applications such as wastewater treatment, photovoltaics, fillers in food packaging, etc. due to its superior optical, electronic, and catalytic properties. However, its application in industry is still limited due to its wide electronic band gap (3.1 eV) and fast recombination rate between electrons and holes. Recently, nanostructured Fe2TiO5 (pseudobrookite, or iron-titanate, with a band gap of 2.1 eV) has gained a lot of attention due to its good charge separation and transport properties, photochemical and electrochemical stability, nontoxicity, and low-cost production.9–11 Different methods have been used to obtain Fe2TiO5 in powder or film form, pure or as a nanocomposite to achieve efficient photoelectrochemical performance for solar energy conversion,9,11–16 visible light photocatalysis,17–20 or gas sensing ability.10,21,22 Courtin et al. reported on a multi-layer template-directed sol–gel technique for the synthesis of mesoporous nanostructured Fex–TiO2 films consisting of three phases: pseudobrookite, hematite, and anatase.11 A modified sol–gel method was used to obtain Fe2TiO5 nanoparticles that showed improved degradation efficiency of methylene blue in an alkaline medium under natural sunlight.19 Bassi et al. used a hydrothermal method to synthesize pure phase of pseudobrookite and investigated its potential use in photoelectrochemical water splitting.12 Gas sensing properties of nanocomposite Fe2TiO5/Fe2O3 prepared by impregnating method using carbonaceous polysaccharide microspheres as hard templates were reported by Yu et al.,21 while the simple solid-state synthesis method was used to obtain Fe2TiO5 that showed promising humidity sensing properties.10,21 Also, porous TiO2/Fe2TiO5 nanocomposites were fabricated via a solvothermal method followed by a sintering process and showed improved electrochemical capacity.23 Recently, Fe2TiO5 has been used as a co-catalyst to improve the photoelectrochemical water splitting performance of BiVO4.9 In spite of its excellent properties, its full application is still hindered due to the difficulty to synthesize monophasic pseudobrookite with high crystallinity and a large specific surface area.24 Formation of an interconnected web of nanofibers could overcome these problems.
Nanofibers are one-dimensional (1D) nanostructures, mostly prepared by the electrospinning technique. Electrospinning is a simple, low cost and versatile method to generate 1D continuous nanomaterial from different materials such as polymers, metal oxides, etc. under a high electric field at an industrial scale.25,26 Electrospun nanofibers and their mats have a large specific surface area, small and uniform diameter from less than 10 nm to 500 nm, interconnected nanopores, and improved mechanical and electronic properties. Moreover, the nanofiber surface can be modified and functionalized for the growth of secondary nanostructures.27 All these properties make nanofibers multifunctional, especially in the case of polymer–inorganic composite nanofibrous structures, since they have remarkable properties, which combine the properties of polymers such as flexibility and lightweight with the properties of inorganic structures as excellent electronic and optical properties, high mechanical and thermal stability, etc.28 They have found use in fields such as biomedicine and healthcare,29–31 energy storage,32,33 membranes,34 environmental protection.35–38
An electrospinning setup usually consists of a spinneret (plastic syringe with a metallic needle) containing a viscous solution, injection pump, fiber collector, and high voltage supply. The electrospinning process starts after the viscous solution emerges from the nozzle tip into an electric field developed between the collector and nozzle tip. Under the electrostatic forces, a liquid drop deforms into a Taylor cone. When the electrostatic force overcomes the surface tension of the solution, a liquid jet will be ejected from the bottom of the Taylor cone. During elongation of the jet in an electric field, the solvent evaporates and fibers are formed and deposited with a reduced diameter on the collector.39
Herein we report on the synthesis of Fe2TiO5, pseudobrookite (PSB) nanostructures via the simple electrospinning technique and followed by calcination (heat treatment) at different temperatures. Polyvinylpyrrolidone (PVP) was used as a mesoporous template for the synthesis of Fe2TiO5 nanofibers. We have focused on analyzing the influence of heat treatment temperature (500–750 °C) and its duration on the morphology, structural and optical properties of as-synthesized nanofibers. In this regard, this study will contribute to a better understanding of Fe2TiO5 nanofiber development and utilization in applications such as chemical sensors, photoelectrochemical water splitting, photocatalysis in which the specific surface area of nanofibers is of great importance.
Experimental
Materials
Iron(III) nonahydrate (Fe(NO3)3·9H2O, ACS reagent, ≥98%), titanium isopropoxide (Ti[OCH(CH3)2]4 purity 98%), N,N-dimethylformamide (DMF, puriss, ACS reagent, ≥99.8%), absolute ethanol (ACS reagent) and polyvinylpyrrolidone (PVP, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000) were purchased from Merck (Sigma Aldrich) and used as received.
300000) were purchased from Merck (Sigma Aldrich) and used as received.
Synthesis of Fe2TiO5 (PSB) nanofibers
PSB NFs were prepared by the electrospinning technique as follows. In a typical procedure, 0.808 g Fe(NO3)3·9H2O, 0.284 g Ti[OCH(CH3)2]4 and 0.937 g of PVP were dissolved in a mixture of DMF and ethanol. The solution was continuously magnetically stirred for 24 h at room temperature to provide a viscous precursor sol–gel solution. Next, the prepared solution was transferred into a 10 ml plastic syringe equipped with a stainless steel nozzle (0.8 mm inner diameter). A high voltage power supply (20 kV) was applied between the rotating drum collector (cathode) and needle tip (anode) with a distance of 15 cm. The solution flow rate of 1 ml h−1 was maintained using the syringe pump equipment. The PVP–PSB nanofibers were deposited on a baking paper collector during the electrospinning process. The as-spun fibers were then calcined in a chamber furnace (Elektron, ELP-06, Banja Koviljaca, Serbia), in air, at different temperatures to remove the polymer carrier and also to convert the precursors to pseudobrookite, without decomposition of fibers. In order to investigate the effect of calcination temperature and duration taking into account TG/DTA measurements, nanofibers mats were calcined at five different temperatures of 500, 550, 600, 650, 700, and 750 °C for 3 h, 550 °C with the holding time of 4 h and 500 °C with the holding time of 6 h.Materials characterization
The thermal stability of electrospun nanofibers was evaluated by TG/DTA analysis on a SETSYS Evolution 24000 Setaram Instrumentation device up to 1000 °C with a heating rate of 5° min−1 in air. The fiber morphology was investigated using transmission electron microscopy (TEM) and field emission scanning electron microscopy (FESEM) on JEM-2100 200 kV (Japan) and TESCAN MIRA3 XM (Czech Republic) devices, respectively. The mean fiber diameter was determined using Gatan Micrograph® software. Fourier transform infrared spectroscopy (FT-IR) spectra of as-spun and calcined fibers were recorded on a FT-IR Nicolet 6700 ATR device (Thermo Fisher, UK) in the range 400–4000 cm−1 in order to confirm the formation of pseudobrookite.The crystal structure and phase identification were investigated by X-ray diffraction (XRD) analysis using Ni-filtered Cu Kα radiation with the wavelength of 1.542 Å, in the range of 2θ = 10–90°, with the step of 0.05° and acquisition rate of 1 step per min (for phase identification) and on selected samples with the step of 0.02° and acquisition rate of 0.25 step per min (for structural analysis) on a Rigaku Ultima IV diffractometer (Japan). Structural refinement was performed using the Rietveld method and the GSAS II software package.40 X-ray photoelectron spectra XPS were recorded on a PHI-TFA XPS spectrometer (Physical Electronics Inc., USA) equipped with an Al-monochromatic X-ray source to determine the elemental composition and chemical state of the elements that exist in calcined nanofibers. The analyzed area was 0.4 mm in diameter and the analyzed depth was about 3–5 nm. The accuracy of binding energies was about ±0.7 eV. A low-energy electron gun was used for charge compensation and C 1s spectrum due to surface contamination was aligned to 284.8 eV. XPS spectra were fitted with Gauss–Lorentz functions and the Shirley function was used for background removal. UV-vis spectroscopy was used to determine the band gap from diffuse reflection spectra recorded on a Shimadzu UV-2600 device with an ISR2600 Plus Integrating sphere attachment (Japan) in the measuring range 200–1400 nm. The photoluminescence (PL) spectra of the analyzed samples were recorded using a FL3-221 spectrofluorimeter (Jobin Yvon Horiba, Paris, France), equipped with a 450 W Xe lamp and a photomultiplier tube, and FluorEssence 3.5 software (Horiba Scientific, Kyoto, Japan). The spectra were measured at room temperature in the front-face configuration of the measuring cavity. To reduce Rayleigh scattering which is a limiting factor to the measurement's sensitivity and accuracy, Rayleigh masking was applied. The fluorescence emission spectra were recorded in the range from 380 to 550 nm, with a 1 nm increment. The excitation wavelength was set to 360 nm. A spectral bandwidth of 3 nm was used for both the excitation and the emission slits.
Texture parameters of the prepared PSB nanostructures were determined based on N2 adsorption–desorption isotherms measured at 77 K using a Micromeritics ASAP 2020 instrument (USA). Before measurement samples were prepared by degassing at 150 °C for 10 h under reduced pressure. The specific surface area (SBET) of samples was calculated according to the Brunauer, Emmett, Teller (BET) method from the linear part of the nitrogen adsorption isotherm. The volume of the mesopores (Vmeso) was calculated according to the Barrett, Joyner, and Halenda method from the desorption branch of the isotherm. The volume of micropores (Vmicro) was calculated from the alpha-S plot.
Results and discussion
As-spun fibers
FESEM images of as-spun fibers are presented in Fig. 1a. Fibers were smooth, straight, beadless, and uniform forming a nonwoven fibrous mat, with an average diameter of ca. 205 nm. As mentioned above, TG/DTA analysis was performed on as-spun fibers (Fig. 1b) to evaluate thermal stability. The total mass loss was ≈87%. It started with volatilization of the residual solvents present in fibers (DMF, ethanol) and loss of moisture/desorption of water (H2O),41,42 corresponding to the initial minor mass loss of ≈7% and endothermic peak at ≈70 °C. Two distinct sharp exothermic peaks can be noted at ≈259.4 and 328.3 °C, with corresponding significant mass loss of a further ≈49.4 and 28%, respectively. This can be attributed to the process of degradation of PVP42 and decomposition/oxidation of the Fe(NO3)3·9H2O and Ti[OCH(CH3)2]4, involving dehydration on the polymer side chain and burning out of the organic composites.42,43 Formation of anatase, accompanied by the formation of pseudobrookite (Fe2TiO5) and the phase transformation of anatase to rutile also occurred in this temperature interval according to.44–47 There was no mass loss after ≈410 °C, though heat absorption continued slightly longer until ≈470 °C indicating a continuation of the phase change and solid-phase reaction.41 The crystal structure and phase composition of the as-synthesized fibers were investigated by XRD analysis. The measured XRD pattern of as spun is given in Fig. 2a. Only a broad peak centered at about 23° is observed indicating the amorphous (semi-crystalline) nature of the polymer PVP in the fibers.48,49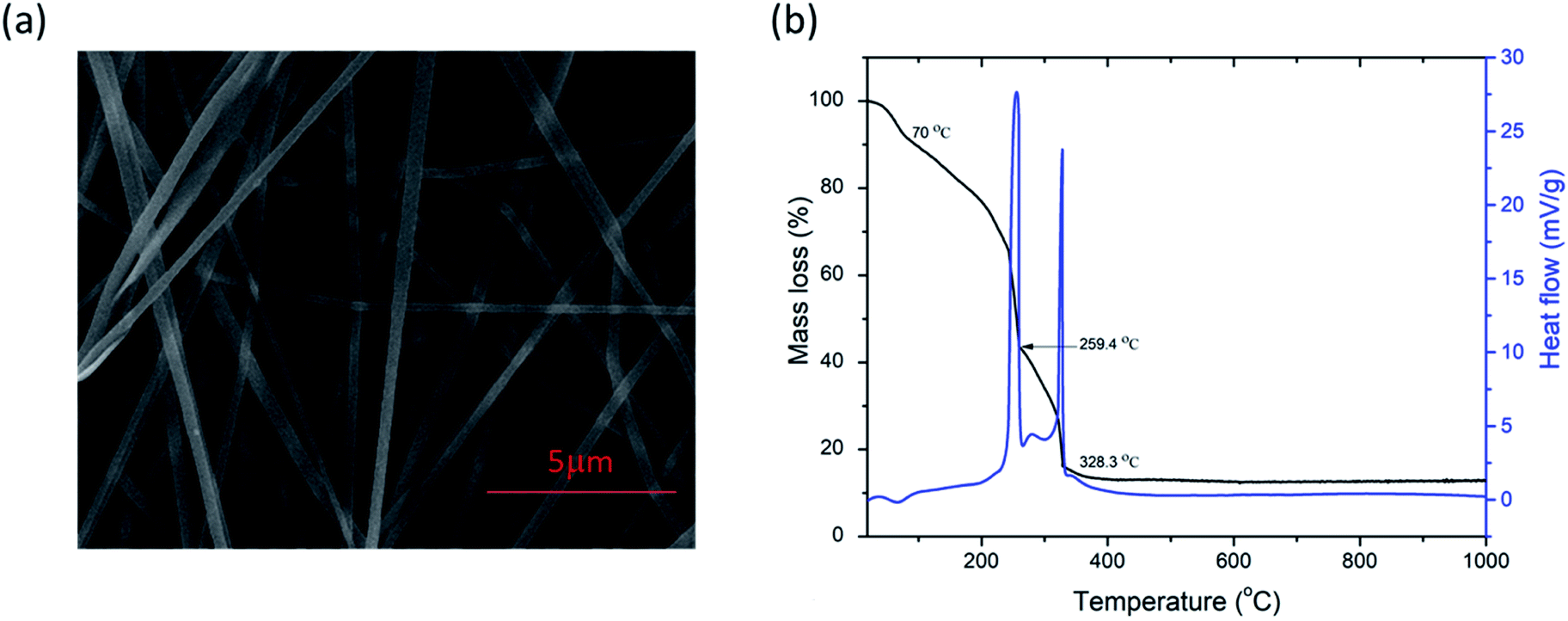 | ||
| Fig. 1 FESEM image (a) and TG/DTA curves (b) of as-spun PVP/PSB nanofibers. | ||
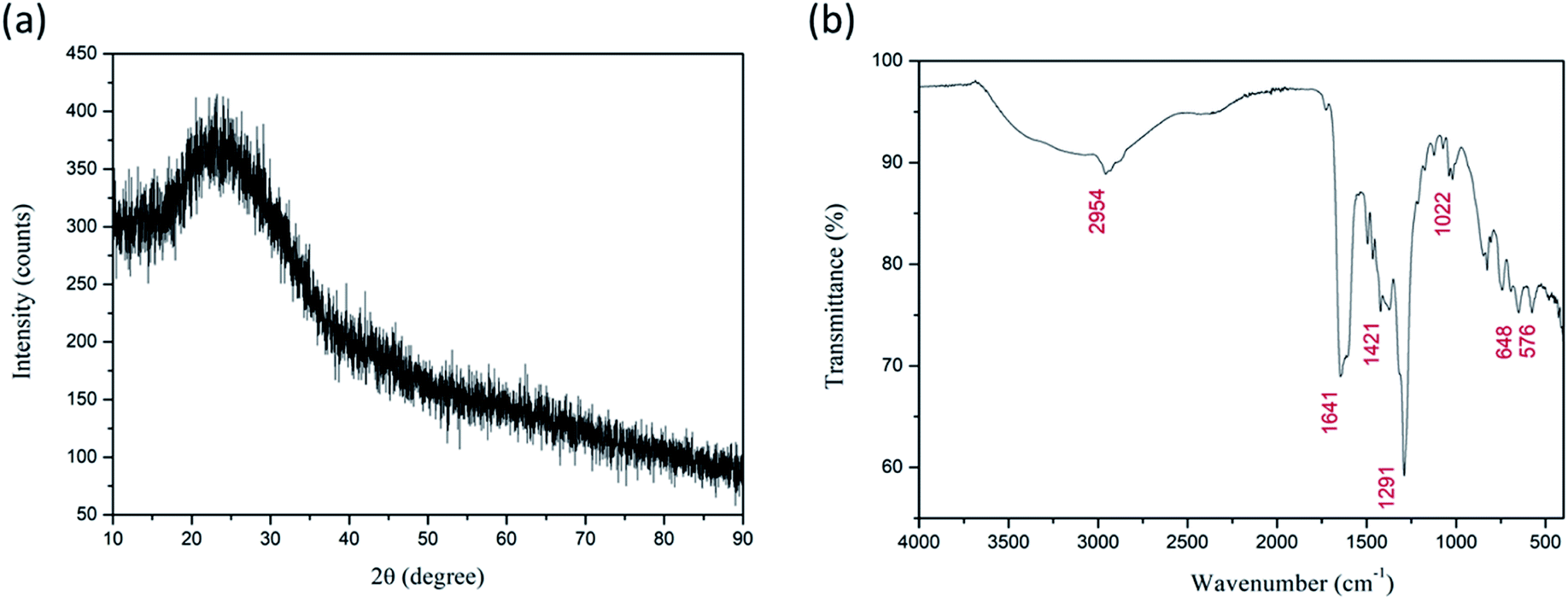 | ||
| Fig. 2 XRD diffractogram (a) and FT-IR spectrum (b) of as-spun Fe2TiO5 fibers. | ||
Analysis of the measured FT-IR transmittance of the as-spun fibers (Fig. 2b) confirmed the above results and showed the presence of bands originating from functional groups presented in PVP, DMF, and the Fe(NO3)3·9H2O and Ti[OCH(CH3)2]4 precursors as observed before for as-spun fibers with PVP, DMF, and metal nitrates or Ti[OCH(CH3)2]4.42,43,50 The wideband in the region 3500–3000 cm−1 represents stretching vibrations of the OH group associated with absorbed moisture (water). Water molecules are also strongly bonded inside the iron nitrate precursor.51 The marked band at ≈2954 cm−1 represents C–H stretching vibrations of CH2.42,44 The strongly marked band at ≈1641 cm−1 represents the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, while the band at ≈1421 cm−1 represents the bending vibration of the CH2 group.44,50 IR active anti-symmetric stretching vibration of the iron nitrate precursor salt can be expected in the region 1450–1270 cm−1, and the band at ≈1374 cm−1 can be attributed to this.52 The strong band at ≈1291 cm−1 represents the C–N stretching vibrations of PVP.47,50 The bands at ≈1071 and ≈1022 cm−1 correspond to symmetric stretching vibrations of the nitrate group.42,52 The band at ≈826 cm−1 can be attributed to out-of-plane deformation of vibrational modes of the nitrate group, while the band at ≈691 cm−1 can be assigned to Fe–OH groups in the iron nitrate precursor salt and also T–O–Ti vibrations of the titanium isopropoxide precursor that have been noted at 820 and 700 cm−1.51–53 The two bands at ≈648 and 576 cm−1 can be associated with metal–oxygen bonds, Ti–O and Fe–O bonds, respectively.42,44,47
O stretching vibration, while the band at ≈1421 cm−1 represents the bending vibration of the CH2 group.44,50 IR active anti-symmetric stretching vibration of the iron nitrate precursor salt can be expected in the region 1450–1270 cm−1, and the band at ≈1374 cm−1 can be attributed to this.52 The strong band at ≈1291 cm−1 represents the C–N stretching vibrations of PVP.47,50 The bands at ≈1071 and ≈1022 cm−1 correspond to symmetric stretching vibrations of the nitrate group.42,52 The band at ≈826 cm−1 can be attributed to out-of-plane deformation of vibrational modes of the nitrate group, while the band at ≈691 cm−1 can be assigned to Fe–OH groups in the iron nitrate precursor salt and also T–O–Ti vibrations of the titanium isopropoxide precursor that have been noted at 820 and 700 cm−1.51–53 The two bands at ≈648 and 576 cm−1 can be associated with metal–oxygen bonds, Ti–O and Fe–O bonds, respectively.42,44,47
Calcined nanofibers
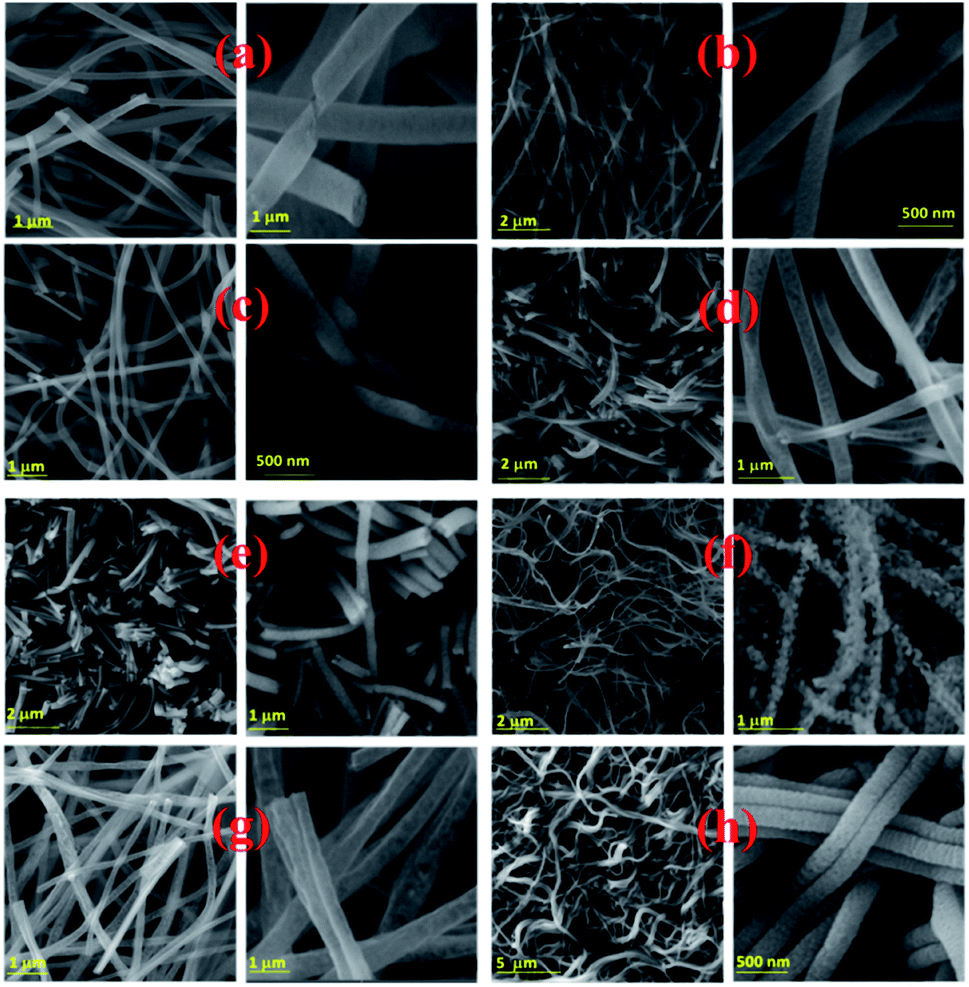 | ||
| Fig. 3 Electrospun PSB nanofibers after calcination at 500 °C_3 h (a), 550 °C_3 h (b), 600 °C_3 h (c), 650 °C_3 h (d), 700 °C_3 h (e), 750 °C_3 h (f), 550 °C_4 h (g), 500 °C_6 h (h). | ||
The phase composition of calcined NFs was further determined by XRD analysis. As shown in Fig. 4, calcination at 500 °C for 3 h resulted in the start of noticeable pseudobrookite formation, with wide crystalline peaks corresponding to Fe2TiO5 (JCPDS 76-1158): 25.5° (110) and 32.6° (023) with an orthorhombic Cmcm crystalline lattice structure. The presence of hematite according to JCPDS 89-8104 is at ≈33.2° (104) followed by the next highest peak intensity at ≈35.7° (110). The sample calcined at 550 °C for 3 h showed more crystalline peaks of hematite and Fe2TiO5 in the diffractogram. An increase in calcination temperature to 600 °C, leads to enhancement of Fe2TiO5 formation, resulting in an almost pure phase with traces of hematite. An increase in calcination temperature (650, 700, and 750 °C), while maintaining the duration at 3 h, resulted again in a mixed structure with a noticeable presence of hematite and traces of rutile (space group P42/mnm, JCPDS 89-4920). An increase in the calcination duration at temperatures lower than 600 °C, namely 550 °C for 4 h and 500 °C for 6 h resulted in phase-pure Fe2TiO5, showing that the duration of the calcination process, besides the temperature, had a direct influence on phase formation.
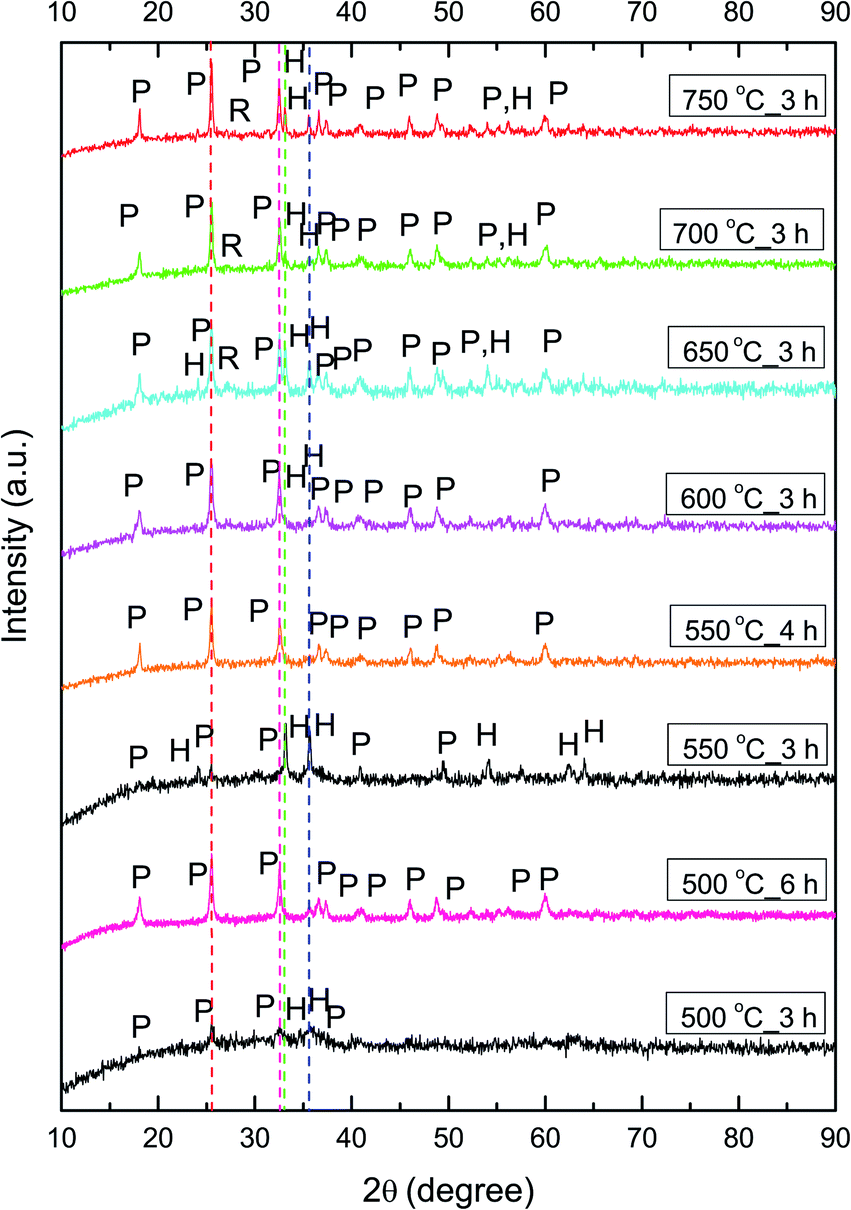 | ||
| Fig. 4 XRD diffractograms of Fe2TiO5 fibers calcined at 500–750 °C, peaks marked as P – Fe2TiO5 and H – hematite, R – rutile red. Pink lines denote 25.5° and 32.5° peaks of Fe2TiO5, while green and blue lines denote 33.2 and 35.7°peaks of hematite (α-Fe2O3). | ||
The average crystallite size was determined from the XRD pattern using Scherrer's equation.56 The crystallite size of samples calcined at 600, 650, 700, and 750 °C was 24, 25, 30, and 33 nm, respectively, showing that an increase in calcination temperature leads to an increase in crystallite size. Increasing the calcination duration at lower temperatures showed that small crystallites of 24 nm were obtained with calcination at 500 °C for 6 h, while calcination at 550 °C for 4 h resulted in slightly larger 30 nm crystallites. We determined the lattice parameters (Table 1), atomic positions, and ion occupancy by Rietveld refinement (Fig. S1 and Table S1†), for fibers calcined at 500 °C for 6 h, 550 °C for 4 h and 600 °C for 3 h. We used the values previously determined for Fe2TiO5 nanocrystalline powder obtained by a modified sol–gel method, as starting cell unit parameters for Fe2TiO5.19
| Sample | Fe2TiO5 | |||
|---|---|---|---|---|
| Calc. temp | a (Å) | b (Å) | c (Å) | Crystallite size (nm) |
| 500 °C_6 h | 3.7322(4) | 9.8052(17) | 9.9690(13) | 24 |
| 550 °C_4 h | 3.73330(23) | 9.7893(11) | 9.9607(9) | 30 |
| 600 °C_3 h | 3.73490(31) | 9.7987(16) | 9.9618(11) | 24 |
The lattice parameters varied slightly with calcination temperature and duration and were in agreement with values previously obtained for Fe2TiO5.10,19 Refinement of ion occupancy showed that in Fe2TiO5 nanofibers the preference of iron ions for 4c sites was 80%, while 60% iron ions are on 8f sites. This is higher than the value of 75% noted by Rodriguez et al.,57 and 72% for Fe2TiO5 obtained using a modified sol–gel method19 or the fully disordered structure for Fe2TiO5 obtained by solid-state synthesis.10
In FT-IR spectra of calcined NFs (Fig. 5) bands originating from metal–oxygen vibrations at ≈430, 615 and 800 cm−1 are noted, in accordance with literature data,19,58 representing vibrations of Fe–O, Ti–O and Fe–O–Ti bonds in Fe2TiO5. In the case of powders calcined at 650–750 °C the vibration band at ≈515 cm−1 is noted that can also be associated with Fe–O bonds,59 due to both Fe2TiO5 and hematite present in the samples, which is in accordance with XRD analysis (Fig. 4).
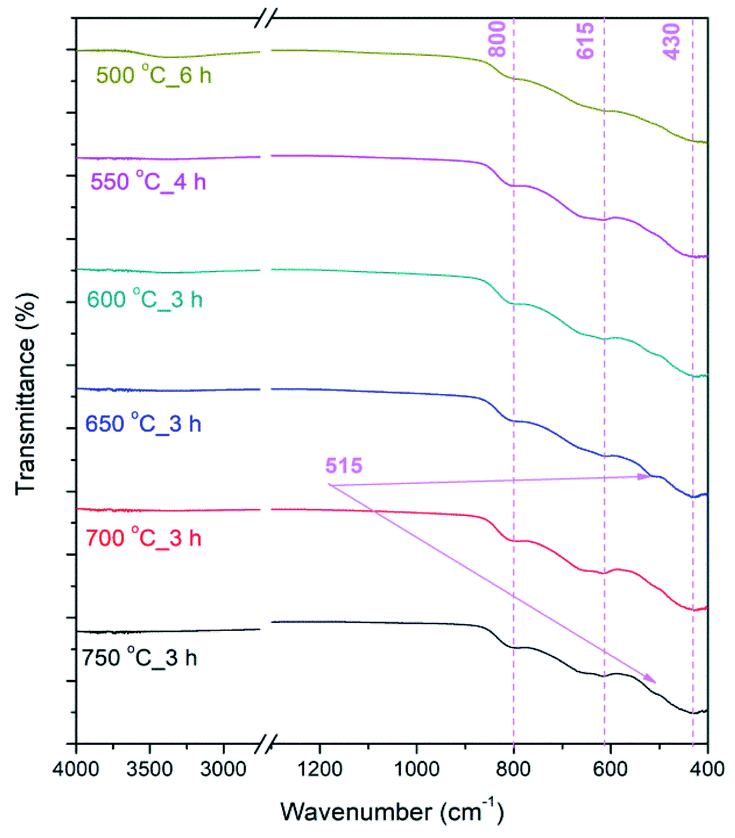 | ||
| Fig. 5 FTIR spectra of calcined PSB NFs measured in the range 4000–400 cm-1; transmittance showed in % in the range 0–100%. | ||
The crystal structure and morphology of the PSB 500 °C_6 h, PSB 550 °C_4 h and PSB 600 °C_3 h NFs were further analyzed by TEM and HRTEM, as shown in Fig. 6. The investigation further revealed the average particle size distribution similar to the crystallite size determined by Scherrer's equation. Smaller single-crystalline rhombohedral-shaped particles form long fibers. Small residuals in the vicinity of the fibers arose as a consequence of the TEM sample preparation technique which includes ultra-sonication. Small particles are stacked on top of each other making structural determination a bit difficult especially by selected area diffraction (SAED). At the edge of the fiber HRTEM and the corresponding FFT plot of PSB 500 °C_6 h and PSB 550 °C_4 h shows the lattice spacing of 0.49 nm (Fig. 6b and d), which corresponds to the (020) crystalline plane of an orthorhombic Cmcm (Fe2TiO5) nanostructure (JCPDS 76-1158) and is in accordance with XRD analysis (Fig. S1 and Table S1†). This particular structure can also be identified and resolved in the sample PSB 600 °C where selected area diffraction reveals a particle oriented along the 111 direction (Fig. 6f).
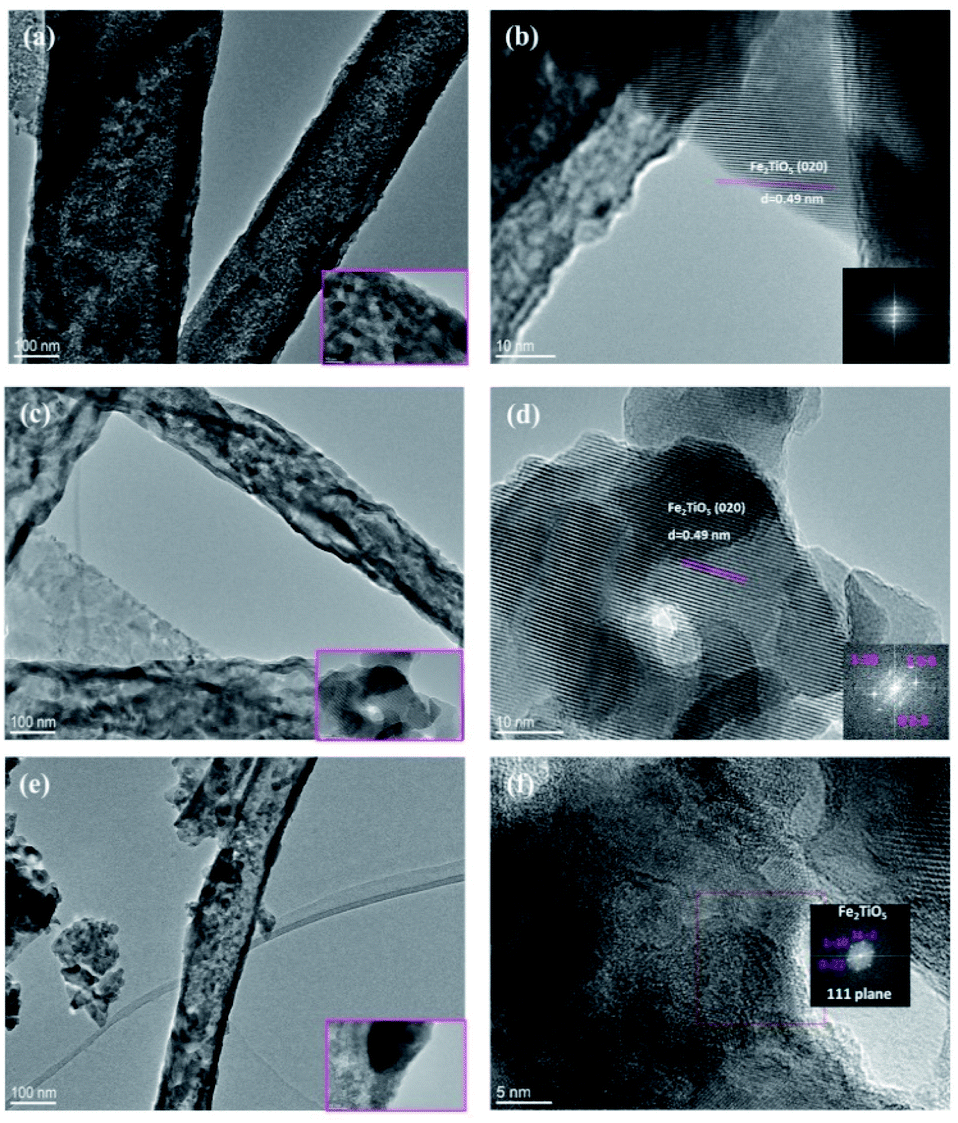 | ||
| Fig. 6 TEM images (a, c and e) and HRTEM images (b, d and f) of PSB 500 °C_6 h, PSB 550 °C_4 h and PSB 600 °C_3 h. The insets of HRTEM images (b, d and f) show FFT images. | ||
The surface chemical composition was further investigated by the XPS technique. The XPS spectra of samples 500 °C_6 h and 550 °C_4 h are presented in Fig. 7. The elements Fe, Ti, O, and C were identified in these spectra. Sample 500 °C_6 h consists of 54.8 at% of O, 20.9 at% of Fe, 15.7 at% of C, and 8.6 at% of Ti. Similar surface composition was found on sample 550 °C_4 h, i.e. 52.5 at% of O, 20.3 at% of Fe, 18.4 at% of C, and 8.8 at% of Ti. One can observe that the ratio of Fe/Ti cations on the surface of both samples is roughly 2, which reflects the expected stoichiometry. The carbon signal and part of the oxygen signal detected by XPS on the sample surface may be due to surface contamination. High energy resolution XPS spectra were recorded to get more details about the oxidation states of elements on the surface.
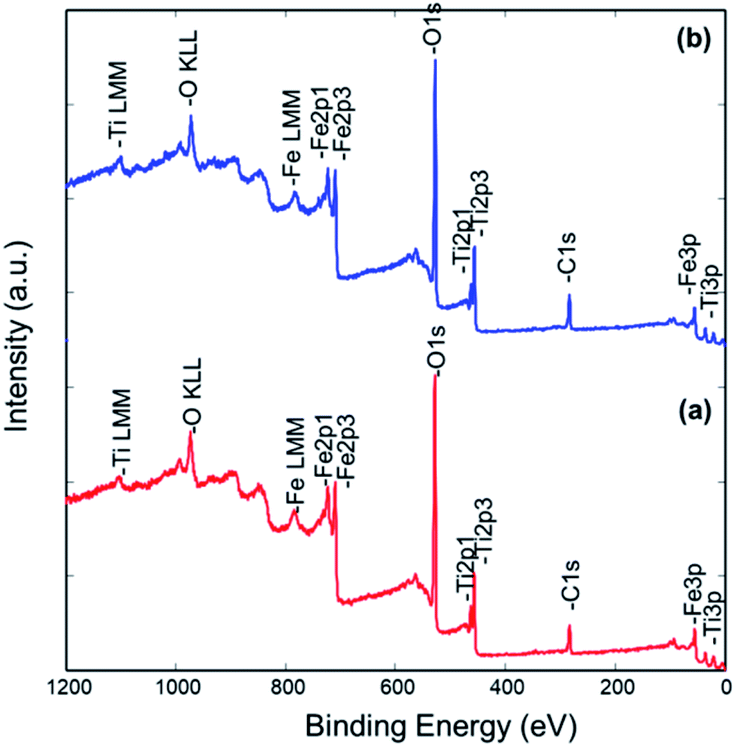 | ||
| Fig. 7 XPS survey spectra from samples 500 °C_6 h (a) and 550 °C_4 h (b). | ||
Ti 2p, Fe 2p, and O 1s spectra from both samples are shown in Fig. 8. All the spectra are very similar indicating that the same oxidation states of Ti, Fe, and O atoms are present on both types of samples. The Ti 2p spectrum consists of the Ti 2p3/2 peak at 458.5 eV and Ti 2p1/2 peak at 464.2 eV, which is assigned to the Ti4+ oxidation state, as expected for the Fe2TiO5 composition.60 No other oxidation state of Ti was identified. The Fe 2p spectrum consists of the Fe 2p3/2 peak at 711.1 eV, Fe 2p3/2 peak at 713.6 eV, and a plasmon peak at 719.3 eV. Corresponding peaks for the Fe 2p1/2 orbital are also present as Fe 2p1/2 peak at 724.6 eV, Fe 2p1/2 peak at 728.1 eV, and a plasmon peak at 733.0 eV. The binding energy of the main Fe 2p3/2 peak and the plasmonic peak indicates that Fe on the surface of both samples is in the Fe3+ oxidation state.61,62 The O 1s spectrum was deconvoluted into two peaks: the larger peak at 530.0 eV can be attributed to lattice oxygen (O2− ions of Fe–O and Ti–O) and a smaller peak at 531.5 eV is related with OH and other C–O based groups possibly adsorbed on the surface.60,61,63 Together with XRD analysis, these results confirmed the formation of Fe2TiO5 nanostructures.
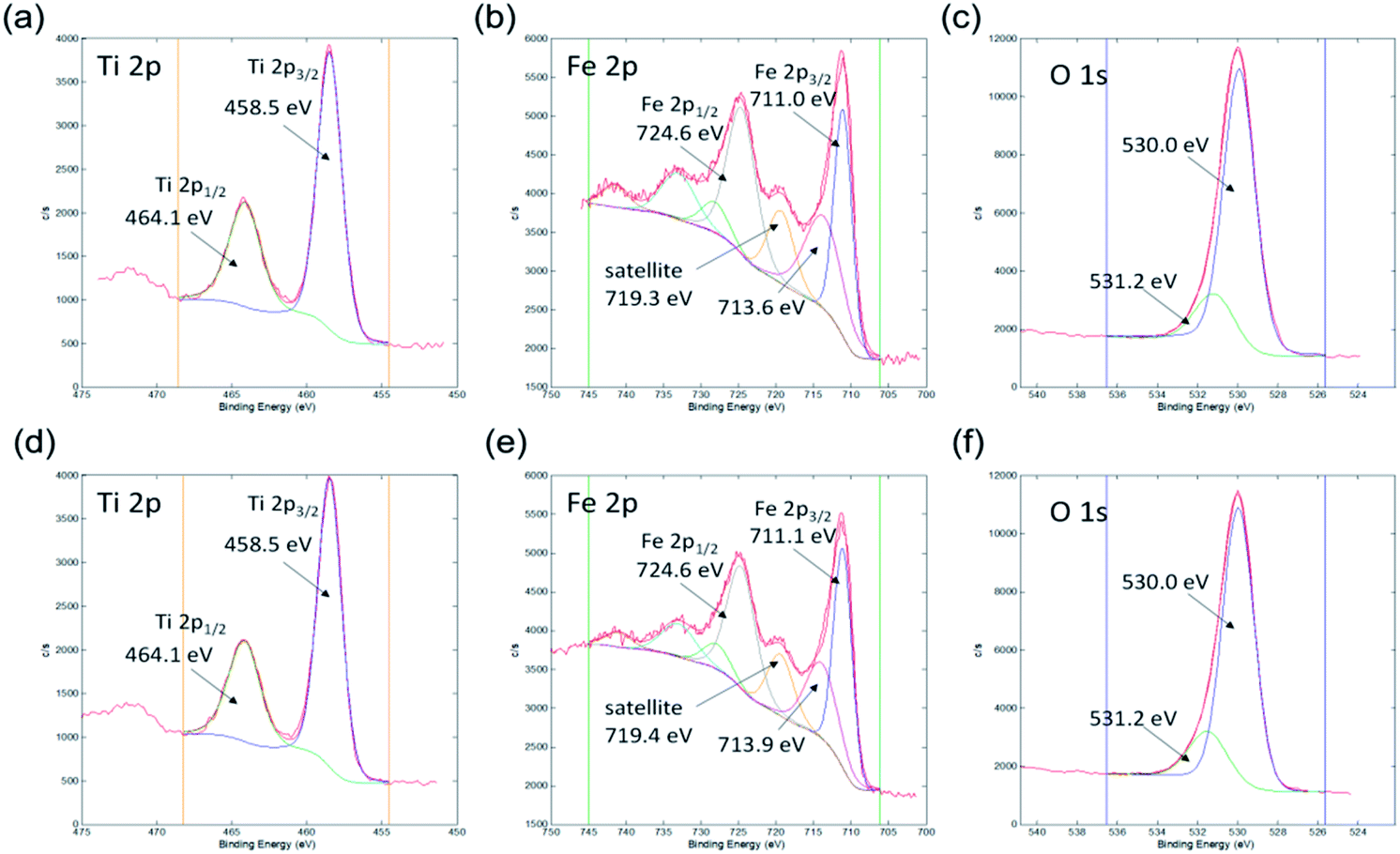 | ||
| Fig. 8 XPS high energy resolution spectra Ti 2p (a and d), Fe 2p (b and e) and O 1s (c and f) from samples 500 °C_6 h (a–c) and 550 °C_4 h (d–f). | ||
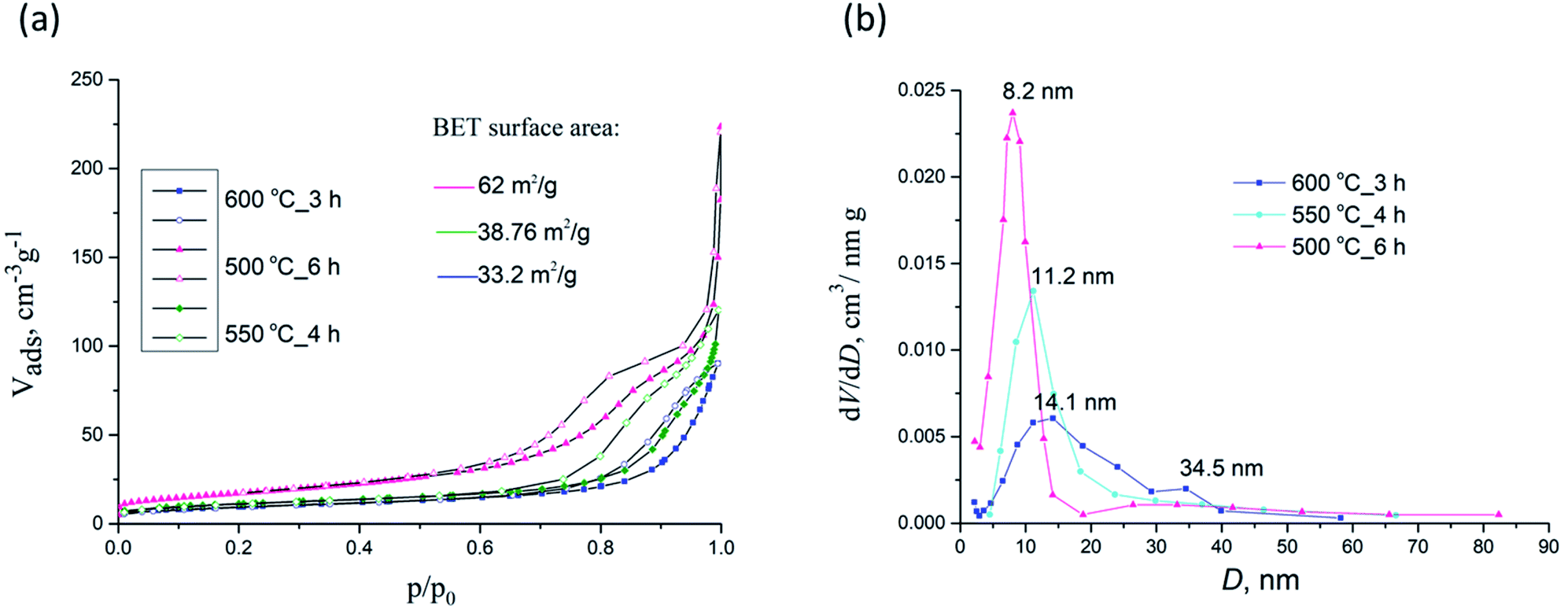 | ||
| Fig. 9 N2 adsorption–desorption isotherms (a) and pore size distributions (b) for samples 500 °C_6 h, 550 °C_4 h and 600 °C_3 h. | ||
The corresponding Barrett–Joyner–Halenda (BJH) pore size distribution plots for samples 500 °C_6 h, 550 °C_4 h and 600 °C_3 h are given in Fig. 9b whilst textural properties of calcined NFs are given in Table 2. PSB 600 °C_3 h exhibited a heterogeneous wide pore size distribution centered at 14.1 nm and 34.5 nm. For sample 500 °C_6 h pores 6–8 nm (with a mean size of 8.2 nm) were noted, with a small maximum at about 30 nm, to some extent similar to PSB 600 °C_3 h, but less expressed. For sample 550 °C_4 h only pores having a mean size of 11.2 nm were seen. SBET of 500 °C_6 h, 550 °C_4 h and 600 °C_3 h samples were estimated to be 64.4, 38.8, and 33.2 m2 g−1, respectively. As depicted in Table 2, SBET was reduced with an increase in calcination temperature from 550 to 750 °C, while the corresponding pore size diameter increased. These may be attributed to the growth of Fe2TiO5 crystallites at higher temperatures.
| Sample | Sp, m2 g−1 | Vmeso, cm3 g−1 | Vmicro, cm3 g−1 | Dmax, nm |
|---|---|---|---|---|
| 550 °C_3 h | 55.5 | 0.164 | 0.017 | 8.6 |
| 550 °C_4 h | 38.8 | 0.169 | 0.013 | 11.2 |
| 600 °C_3 h | 33.2 | 0.132 | 0.010 | 14.1 |
| 650 °C_3 h | 23.8 | 0.130 | 0.007 | 18.2–43.8 |
| 700 °C_3 h | 12.5 | 0.085 | 0.006 | 22.8 |
| 750 °C_3 h | 9.1 | 0.030 | 0.003 | 29.0–37.0 |
| 500 °C_6 h | 64.4 | 0.180 | 0.095 | 8.3 |
| (eqn 1) αhν = A(hν − Eg)m | (1) |
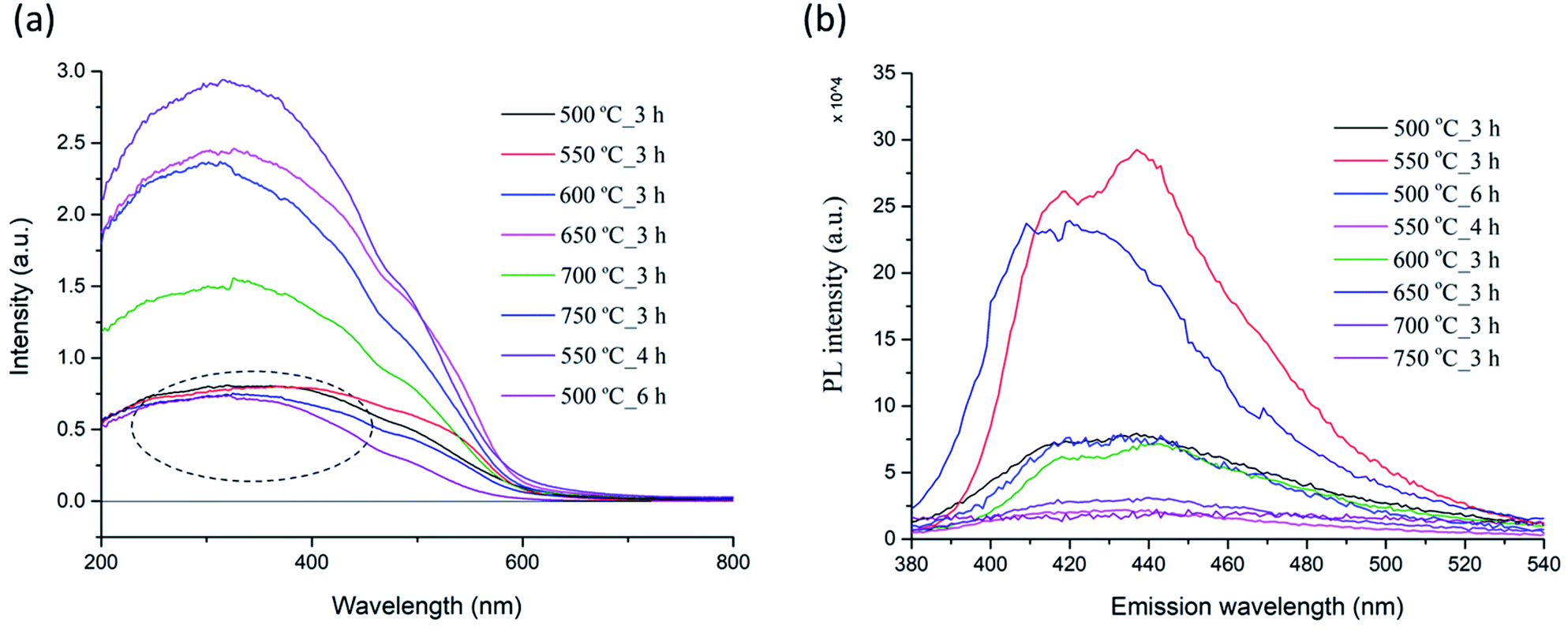 | ||
| Fig. 10 Optical absorbance (a) and photoluminescence spectra (b) of PSB NFs synthesized at different temperatures. | ||
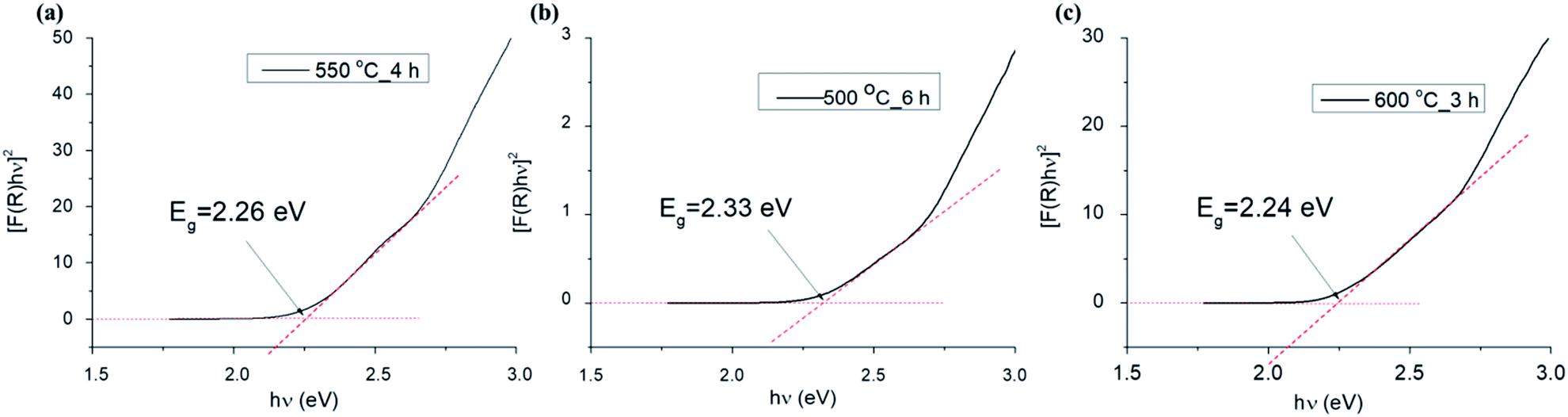 | ||
| Fig. 11 Plots of [F(R)hν]2 versus photon energy (hν) for samples 550 °C_4 h (a), 500 °C_6 h (b) and 600 °C_3 h (c). | ||
| Sample | Direct band gap (eV) | Indirect band gap (eV) |
|---|---|---|
| 500 °C_3 h | 2.30 | 1.80 |
| 550 °C_3 h | 2.20 | 1.93 |
| 600 °C_3 h | 2.25 | 1.94 |
| 650 °C_3 h | 2.20 | 1.94 |
| 700 °C_3 h | 2.23 | 1.94 |
| 750 °C_3 h | 2.16 | 1.90 |
Photoluminescence (PL) spectroscopy was used to evaluate the recombination of minority carriers (the photo-generated electron and hole pairs in a photochemical process), after an excitation (λ = 360 nm) at room temperature. The PL spectra of the PSB nanofibers after calcination at different temperatures (500 °C to 750 °C_3 h and 550 °C_4 h and 500 °C_6 h) are shown in Fig. 10b. A broad luminescence band ranging from 380 nm to 520 nm was observed in all the samples. Two small shoulders at around 420 nm and 440 nm were observed in the PL spectrum for the PSB nanofibers calcined at different temperatures with variations in the intensity of PL emission, which is mainly consistent with the XRD analysis and the occurrence of secondary phases such as hematite and rutile. Additionally, the PL emission spectra have changed over the prolonged duration of the calcination process (500 °C_6 h and 550 °C_4 h) in agreement with the XRD analysis where a pure Fe2TiO5 orthorhombic phase was obtained. The PL intensities of PSB_550 °C_4 h and of PSB_750 °C_3 h were the smallest. A lower PL intensity indicates a lower recombination rate of the photoexcited electron–hole pairs and higher photocatalytic activities are to be expected.17,19
It is known that besides crystallite size, other properties of nanofibers (such as surface area, phase structure, and composition, surface and lattice defects, etc.) also influence the PL spectra.75 The photoluminescence is generally a surface phenomenon, and the observed differences in the PL intensity could be explained by differences in the surface properties of nanofibers at various calcination temperatures. Literature data have shown that the PL intensity of TiO2 samples changed with increasing calcination temperatures, and also in the case when composites with Fe2O3 formed.76–78 According to Bhoi et al. the PL intensity decreases in order from TiO2, α-Fe2O3 to Fe2TiO5.79 It has been reported that the PL intensity of anatase samples increased with a rise of the specific surface specific area, which could be explained by the enhanced formation of surface defects and oxygen vacancies in the TiO2 structures.76 The same authors reported that the PL intensity of rutile TiO2 samples showed a more complicated behavior compared to anatase. The phase transformation of anatase and brookite to rutile favors higher calcination temperatures. The anatase to rutile phase transformation does not have a set temperature but occurs in the temperature interval 400–1000 °C. Previous research has shown that the presence of Fe (or hematite) accelerates the anatase to rutile transformation, so it completed at 850 °C, compared to 950 °C needed for pure powder and was accompanied with the formation of orthorhombic PSB.80
Conclusion
In summary, we report on the synthesis of Fe2TiO5 nanofibers via the simple electrospinning technique, followed by calcination (heat treatment) at different temperatures. The effect of calcination temperature (500–750 °C) and its duration on the morphology, structural, textural, and optical properties of as-synthesized nanofibers was evaluated. It was shown that the morphology was greatly affected by the calcination temperature. FESEM and TEM analysis revealed highly porous nanofibers with an orthorhombic structure. XRD and XPS analysis confirmed the formation of pure phase Fe2TiO5 at 500 °C_6 h and 550 °C_4 h whilst small traces of hematite were found in sample 600 °C_3 h. FTIR analysis showed complete degradation of PVP via the calcination step. All samples exhibited a slit-shaped mesoporous structure. Although sample PSB_500 °C_6 h exhibited a mesoporous structure, the presence of interparticle voids was noted. The superior specific surface area 64.4 m2 g−1 was obtained for the fibers calcined at 500 °C for 6 h and the surface area values of other calcined nanofibers decreased when the calcination temperature was increased. Further research will be focused on investigating the application of PSB nanofibers in chemical sensors, photoelectrochemical water splitting, or photocatalysis.Author contributions
Zorka Ž. Vasiljević: synthesis, UV-vis characterization, writing – original draft, writing – review & editing, visualization. Milena P. Dojčinović: synthesis, writing – review & editing. Jelena D. Vujančević: FESEM analysis, writing – review & editing. Matjaž Spreitzer: TEM and HRTEM analyis, data interpretation and visualisation. Janez Kovač: XPS analysis, data interpretation and visualisation. Dragana Bartolić: photoluminescence analysis, data interpretation and visualisation. Smilja Marković: TG/DTA measurements, writing – review & editing. Ivona Janković-Čaštvan: BET measurements, data visualization. Nenad B. Tadić: X-ray diffraction scattering measurements. Maria Vesna Nikolić: FTIR spectroscopy measurements, writing – review & editing, data interpretation and visualization.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to express their gratitude to Jugoslav B. Krstic (Institute of Chemistry, Technology and Metallurgy, Department of Catalysis and Chemical Engineering) for help and advice in relation to nitrogen adsorption–desorption isotherms. This research was funded by the Ministry for Education, Science and Technology Development under the contact 451-03-9/2021-14/200053 (ZZV, MPD, DB, MVN); and 451-03-9/2021-14/200175 (JDV, SM).References
- P. Sáenz-Trevizo, P. Amézaga-Madrid, B. Pizá-Ruiz, P. G. Monárrez-Cordero, W. Hernández-Salcedo, C. Antúnez-Flores, O. Ornelas-Gutiérrez, C. Solís-Canto, M. Leyva-Porras and M. Miki-Yoshida, Functional nanostructured oxides: synthesis, properties, and applications, in Micro and Nano Technologies, Emerging Applications of Nanoparticles and Architecture Nanostructures, ed. A. Barhoum and A. H. SalamMakhlouf, Elsevier, 2018, ch. 2, pp. 29–69 Search PubMed.
- U. P. M. Ashik, S. Kudo and J.-i. Hayashi, An Overview of Metal Oxide Nanostructures, in Micro and Nano Technologies, Synthesis of Inorganic Nanomaterials, ed. S. M. Bhagyaraj, O. S. Oluwafemi, N. Kalarikkal and S. Thomas, Woodhead Publishing, 2018, ch. 2, pp. 19–57 Search PubMed.
- S. S. Kumar, P. Venkateswarlu and V. R. Rao, et al., Int. Nano Lett., 2013, 3, 30 CrossRef.
- Q. Zhang, K. Zhang, D. Xu, G. Yang, H. Huang, F. Nie, C. Liu and S. Yang, Prog. Mater. Sci., 2014, 60, 208–337 CrossRef CAS.
- V. V. Pokropivny and V. V. Skorokhod, Mater. Sci. Eng., C, 2007, 27, 990–993 CrossRef CAS.
- J. Xu, Z. Xue, N. Qin, Z. Cheng and Q. Xiang, Sens. Actuators, B, 2017, 242, 148–157 CrossRef CAS.
- M. Khatami, H. Q. Alijani, M. S. Nejad and R. S. Varma, Appl. Sci., 2018, 8, 411 CrossRef.
- W. Fu, Z. Zhou and A. Hicks, Environ. Sci. Technol., 2019, 53, 4078–4087 CrossRef PubMed.
- Y. Gao, Y. Li, G. Yang, S. Li, N. Xiao, B. Xu, S. Liu, P. Qiu, S. Hao and L. Ge, ACS Appl. Mater. Interfaces, 2018, 10, 39713–39722 CrossRef CAS PubMed.
- M. V. Nikolic, Z. Z. Vasiljevic, M. D. Lukovic, V. P. Pavlovic, J. Vujancevic, M. Radovanovic, J. B. Krstic, B. Vlahovic and V. B. Pavlovic, Sens. Actuators, B, 2018, 277, 654–664 CrossRef CAS.
- E. Courtin, G. Baldinozzi, M. T. Sougrati, L. Stievano, C. Sanchez and C. Laberty-Robert, J. Mater. Chem. A, 2014, 2, 6567–6577 RSC.
- P. S. Bassi, S. Y. Chaim, J. Barber and L. H. Wong, ACS Appl. Mater. Interfaces, 2014, 6, 22490–22495 CrossRef CAS PubMed.
- M. Waquas, Y. Wei, D. Mao, J. Qi, Y. Yang and D. Wang, Nano Res., 2017, 10, 3920–3928 CrossRef.
- P. Zhang, X. F. Lu, D. Luau and X. W. Lou, Angew. Chem., 2020, 132, 8205–8209 CrossRef.
- D. Lee, V. U. Baltzar, T. J. Smart, Y. Ping and K. S. Choi, ACS Appl. Mater. Interfaces, 2020, 12, 29275–29284 CAS.
- O. S. Aleksic, Z. Z. Vasiljevic, M. Vujkovic, M. Nikolic, N. Labus, M. D. Lukovic and M. V. Nikolic, J. Mater. Sci., 2017, 52, 5938–5953 CrossRef CAS.
- Z. Lou, Y. Li, H. Song, Z. Ye and L. Zhu, RSC Adv., 2016, 6, 45343–45348 RSC.
- Q. Zhao, G. Feng, F. Jiang, S. Lan, J. Chen, F. Zhang, Z. Huang, H. Pan, J. Liu, Q. Hu and W. Jiang, Mater. Des., 2020, 194, 108928 CrossRef CAS.
- Z. Z. Vasiljevic, M. P. Dojcinovic, J. D. Vujancevic, I. Jankovic-Castvan, M. Ognjanovic, N. B. Tadic, S. Stojadinovic, G. O. Brankovic and M. V. Nikolic, R. Soc. Open Sci., 2020, 7, 200708 CrossRef CAS PubMed.
- Y. P. Bhoi, F. Fang, X. Zhou, Y. Li, X. Sun, J. Wang and W. Huang, Appl. Surf. Sci., 2020, 525, 140571 CrossRef.
- R. Yu, Z. Li, D. Wang, X. Lai, C. Xing, M. Yang and X. Xing, Scr. Mater., 2010, 63, 155–158 CrossRef CAS.
- J. Wang, L. Jiang, L. Zhao, F. Liu, R. You, Z. Yang, J. He, T. Liu, C. Zhang, C. Wang, X. Liang, P. Sun and G. Lu, Sens. Actuators, B, 2020, 321, 128489 CrossRef CAS.
- S. Wang, N. Wu, Y. Liu, W. Liu and J. Liu, J. Alloys Compd., 2017, 714, 583–592 CrossRef CAS.
- H. Zhang, J. H. Kim and J. S. Lee, Adv. Funct. Mater., 2017, 27, 1702428 CrossRef.
- P. S. Kumar, J. Sundaramurthy, S. Sundarrajan, V. J. Babu, G. Singh, S. I. Allakhverdiev and S. Ramakrishna, Energy Environ. Sci., 2014, 7, 3192–3222 RSC.
- L. A. Mercante, V. P. Scagion, F. L. Migliorini, L. H. C. Mattoso and D. S. Corre, Trends Anal. Chem., 2017, 91, 91–103 CrossRef CAS.
- H. Mei, S. Zhou, M. Lu, Y. Zhao and L. Cheng, Ceram. Int., 2020, 46, 18675–18682 CrossRef CAS.
- F. Kayaci, C. O. Akgun, I. Donmez, N. Biyikli and T. Uyar, ACS Appl. Mater. Interfaces, 2012, 4, 6185–6194 CrossRef CAS PubMed.
- A. Stafiniak, B. Boratyński, A. Baranowska-Korczyc, A. Szyszka, M. Ramiączek-Krasowska, J. Prażmowska, K. Fronc, D. Elbaum, R. Paszkiewicz and M. Tłaczała, Sens. Actuators, B, 2011, 160, 1413–1418 CrossRef CAS.
- P. Karuppuswamy, J. R. Venugopal, B. Navaneethan, A. L. Laiva and S. Ramakrishna, Mater. Lett., 2015, 141, 180–186 CrossRef CAS.
- X. Feng, J. Li, X. Zhang, T. Liu, J. Ding and X. Chen, J. Controlled Release, 2019, 302, 19–41 CrossRef CAS PubMed.
- J. Bhagwan, N. Kumar, K. L. Yadav and Y. Sharma, Solid State Ionics, 2018, 321, 75–82 CrossRef CAS.
- R. Kang, W. Q. Zhu and S. Li, et al., Rare Met., 2021, 40, 2424–2431 CrossRef CAS.
- M. V. Bhute and S. B. Kondawar, Solid State Ionics, 2019, 333, 38–44 CrossRef CAS.
- H. Liu, Z.-G. Zhang, X.-X. Wang, G.-D. Nie, J. Zhang, S.-X. Zhang, N. Cao, S.-Y. Yan and Y.-Z. Long, J. Phys. Chem. Solids, 2018, 121, 236–246 CrossRef CAS.
- M. Imran, S. S. A. A. H. Rashid, Y. Sabri, N. Motta, T. Tesfamichael, P. Sonar and M. Shafiei, J. Mater. Chem. C, 2019, 7, 2961–2970 RSC.
- J. Zhang, J. Xin, C. Shao, X. Li, X. Li, S. Liu and Y. Liu, J. Colloid Interface Sci., 2019, 550, 170–179 CrossRef CAS PubMed.
- B. Pant, G. P. Ojha, Y.-S. Kuk, O. H. Kwon, Y. W. Park and M. Park, Nanomaterials, 2020, 10, 1960 CrossRef CAS PubMed.
- X. Shi, W. Zhou, D. Ma, Q. Ma, D. Bridges, Y. Ma and A. Hu, J. Nanomater., 2015, 140716, 20 Search PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 44, 544–549 CrossRef.
- F. Yang, X. J. Yu, T. Zhang, J. F. Niu, J. Li, J. K. Nie, J. P. Li and B. H. Yao, IOP Conf. Ser. Earth Environ. Sci., 2019, 344, 012096 CrossRef.
- S. Maensiri, M. Sangmanee and A. Wiengmoon, Nanoscale Res. Lett., 2009, 4, 221–228 CrossRef CAS PubMed.
- C. Li, C. Feng, F. Qu, J. Liu, L. Zhu, Y. Liu, Y. Wang, F. Li, J. Zhou and S. Ruan, Sens. Actuators, B, 2015, 207, 90–96 CrossRef CAS.
- M. V. Someswararo, R. S. Dubey and P. S. V. Subbarao, Results Phys., 2018, 11, 223–231 CrossRef.
- W. Nuansing, S. Niumuang, W. Jarenboon, S. Maensiri and S. Seraphin, Mater. Sci. Eng., B, 2006, 131, 147–155 CrossRef CAS.
- I. Shepa, E. Mudra, M. Vojtko, O. Milkovic, Z. Donikova, V. Antal, A. Annusova, E. Majkova and J. Dusza, Results Phys., 2019, 13, 102243 CrossRef.
- W. Chang, F. Xu, X. Mu, L. Ji, G. Ma and J. Nie, Mater. Res. Bull., 2013, 48, 2661–2668 CrossRef CAS.
- D. A. Chalkias, D. I. Giannopoulos, E. Kollia, A. Petala, V. Kostopoulos and G. C. Papanicolau, Electrochim. Acta, 2018, 271, 632–640 CrossRef CAS.
- K. M. Anilkumar, B. Jinisha, M. Manoj and S. Jayalekshmi, Eur. Polym. J., 2017, 89, 249–262 CrossRef CAS.
- D. Vu, X. Li, Z. LI and C. Wang, J. Chem. Eng. Data, 2013, 58, 71–77 CrossRef CAS.
- M. F. B. Stadt, M. Gonchikzhapov, T. Kasper, U. Fritsching and J. Kiefer, Phys. Chem. Chem. Phys., 2019, 21, 24793 RSC.
- F. Zapata and C. Garcia-Ruiz, Spectrochim. Acta, Part A, 2018, 189, 535–542 CrossRef CAS PubMed.
- A. Jedsejczak, D. Batory, M. Prowizor, M. Dominik, M. Smietana, M. Cichomski, A. Kisielewska, W. Szymanski, W. Kozlowski and M. Dudek, Thin Solid Films, 2020, 693, 137697 CrossRef.
- A. G. Wee, H. Z. Soh, Y. L. Cheah, S. G. Mhaisalkar and M. Srinivasan, J. Mater. Chem., 2010, 20, 6720–6725 RSC.
- Y. Zhang, S. Liu, Y. Li, D. Deng, X. Si, Y. Ding, H. He, L. Luo and Z. Wang, Biosens. Bioelectron., 2015, 66, 308–315 CrossRef CAS PubMed.
- B. E. Warren, X-Ray Diffraction, Dover Publications Inc., 1969, New York Search PubMed.
- J. E. F. S. Rodriguez, W. S. Rosa, M. M. Ferrer, T. R. Cunha and M. J. M. Zapata, et al., J. Alloys Compd., 2019, 799, 563–572 CrossRef.
- S. Li, J. Zhong, Z. Cui, Q. Zhang, M. Sun and Y. Wang, J. Mater. Chem. C, 2019, 7, 13829 RSC.
- B. Pal, M. Sharon and G. Nogami, Mater. Chem. Phys., 1999, 59, 254–261 CrossRef CAS.
- M. Wang, X. Wu, K. Huang, Y. Sun, Y. Zhang, H. Zhang, J. He, H. Chen, J. Ding and S. Feng, Nanoscale, 2018, 10, 6678 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Physical Electronics Inc., Eden Prairie, Minnesota, 1995 Search PubMed.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- J. Wang, L. Jiang, L. Zhao, F. Liu, R. You, Z. Yang, J. He, T. Liu, C. Zhang, C. Wang, X. Liang, P. Sun and G. Lu, Sens. Actuators, B, 2020, 321, 128489 CrossRef CAS.
- J. Liu, J. Meeprasert, S. Namuangruk, K. Zha, H. Li, L. Huang, P. Maitarad, L. Shi and D. Zhang, J. Phys. Chem. C, 2017, 121, 4970–4979 CrossRef CAS.
- B. Pant, G. P. Ojha, Y.-S. Kuk, O. H. Kwon, Y. W. Park and M. Park, Nanomaterials, 2020, 10, 1960 CrossRef CAS PubMed.
- B. K. Kang, M. H. Woo, J. Lee, Y. H. Song, Z. Wang, Y. Guo, Y. Yamauchi, J. H. Kim, B. Lima and D. H. Yoon, J. Mater. Chem. A, 2017, 5, 4320 RSC.
- X. Dong, Y. Yu, Y. Zhang, Z. Xu, H. Jiang, C. Meng and C. Huang, Electrochim. Acta, 2021, 380, 138225 CrossRef CAS.
- M. Sharma, J. Singh, S. Hazra and S. Basu, Microchem. J., 2019, 145, 105–112 CrossRef CAS.
- M. Laurenti, A. Lamberti, G. G. Genchi, I. Roppolo, G. Canavese, C. Vitale-Brovarone, G. Ciofani and V. Cauda, ACS Appl. Mater. Interfaces, 2019, 11, 449–456 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- S. Guo, S. Wang, N. Wu, J. Liu, Y. Ni and W. Liu, RSC Adv., 2015, 5, 103767–103775 RSC.
- R. Alcántara, J. Navas, C. Fernández-Lorenzo, J. Martín, E. Guillén and J. A. Anta, Phys. Status Solidi C, 2011, 8, 1970–1973 CrossRef.
- J. Tauc, A. Menth and D. L. Wood, Phys. Rev. Lett., 1970, 25, 749–752 CrossRef CAS.
- A. Simpraditpan, T. Wirunmongkol, S. Pavasupree and W. Pecharapa, Mater. Res. Bull., 2013, 48, 3211–3217 CrossRef CAS.
- J. Liqiang, Q. Yichun, W. Baiqi, L. Shudan, J. Baojiang, Y. Libin and S. Jiazhong, Sol. Energy Mater. Sol. Cells, 2006, 90, 1773–1787 CrossRef.
- L. Kernazhitsky, V. Shymanovska, T. Gavrilko, V. Naumov, L. Fedorenko, V. Kshnyakin and J. Baran, J. Lumin., 2014, 146, 199–204 CrossRef CAS.
- Q. Mei, F. Zhang, N. Wang, Y. Yang, R. Wu and W. Wang, RSC Adv., 2019, 9, 22764–22771 RSC.
- Y. Q. Cao, T. Q. Zi and X. R. Zhao, et al., Sci. Rep., 2020, 10, 13437 CrossRef CAS PubMed.
- Y. P. Bhoi, F. Fang, X. Zhou, Y. Li, X. Sun, J. Wang and W. Huang, Appl. Surf. Sci., 2020, 525, 146571 CrossRef CAS.
- Z. Z. Djuric, O. S. Aleksic, M. V. Nikolic, N. Labus, M. Radovanovic and M. D. Lukovic, Ceram. Int., 2014, 40, 15131–15141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05748k |
| This journal is © The Royal Society of Chemistry 2021 |