
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical constituents from Limonium tubiflorum and their in silico evaluation as potential antiviral agents against SARS-CoV-2†
Ahmed R. Hassan *a,
Ibrahim M. Sanada,
Ahmed E. Allam*b,
Mohamed E. Abouelelab,
Ahmed M. Sayedc,
Shalabia S. Emama,
Salah M. El-Kousyd and
Kuniyoshi Shimizue
*a,
Ibrahim M. Sanada,
Ahmed E. Allam*b,
Mohamed E. Abouelelab,
Ahmed M. Sayedc,
Shalabia S. Emama,
Salah M. El-Kousyd and
Kuniyoshi Shimizue
aMedicinal and Aromatic Plants Department, Desert Research Center, El-Matariya, 11753, Cairo, Egypt. E-mail: ahmedhasan_81@yahoo.com
bDepartment of Pharmacognosy, Faculty of Pharmacy, Al-Azhar University, Assiut, 71524, Egypt. E-mail: aallam81@yahoo.co.uk
cDepartment of Pharmacognosy, Faculty of Pharmacy, Nahda University, Beni-Suef 62513, Egypt
dChemistry Department, Menoufia University, Shebin El-Kom 32861, El-Menoufia, Egypt
eDepartment of Agro-Environmental Sciences, Graduate School of Bioresource and Bioenvironmental Sciences, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
First published on 30th September 2021
Abstract
Wild plants growing in the Egyptian deserts are facing abiotic stress, which can lead to interesting & safe natural products possessing potential chemical profiles. Consequently, our study was designed to assess the phytochemical composition of the aerial parts of Limonium tubiflorum (family Plumbaginaceae) growing wild in Egypt for the first time. In addition, in silico screening and molecular dynamic simulation of all isolated phytoconstituents were run against the main protease (Mpro) and spike glycoprotein SARS-CoV-2 targets which displayed a crucial role in the replication of this virus. Our findings showed that the phytochemical investigation of 70% ethanol extract of L. tubiflorum aerial parts afforded six known flavonoids; myricetin 3-O-(2′′-galloyl)-β-D-galactopyranoside (1), myricetin 3-O-(2′′-galloyl)-α-L-rhamnopyranoside (2), myricetin 3-O-(3′′-galloyl)-α-L-rhamnopyranoside (3), myricetin 3-O-β-D-galactopyranoside (5), apigenin (6), myricetin (7), along with two known phenolic acid derivatives; gallic acid (4) and ethyl gallate (8). Docking studies revealed that compounds (1) & (2) were the most effective compounds with binding energies of −17.9664 & −18.6652 kcal mol−1 against main protease and −18.9244 & −18.9272 kcal mol−1 towards spike glycoprotein receptors, respectively. The molecular dynamics simulation experiment agreed with the docking study and reported stability of compounds (1) and (2) against the selected targets which was proved by low RMSD for the tested components. Moreover, the structure–activity relationship revealed that the presence of the galloyl moiety is necessary for enhancement of the activity. Overall, the galloyl substructure of myricetin 3-O-glycoside derivatives (1 and 2) isolated from L. tubiflorum may be a possible lead for developing COVID-19 drugs. Further, in vitro and in vivo assays are recommended to support our in silico studies.
1. Introduction
COVID-19, a disease induced by SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus-2), has been the cause of a worldwide pandemic, which is a global rapid spreading disease with an increased rate of complication and mortality.1,2 According to the world health organization (WHO), from December 2019 to now, there have been about 200 million confirmed cases and about 4.3 million deaths all over 220 countries and areas. The infection and replication cycle of SARS-CoV-2 begins with the binding of its S protein to the angiotensin-converting enzyme 2 (ACE2) receptor on a human cell surface, followed by a structural change of the S protein that enables the fusion of the viral membrane and the cell membrane. Then, the viral genes can enter the host cell to be replicated, producing more viruses for further viral shedding.3,4So far, there has been no effective treatment of COVID-19. Several potential drug candidates, including lopinavir/ritonavir (Kaletra), nucleoside analogs, neuraminidase inhibitors, remdesivir, umifenovir (arbidol), DNA synthesis inhibitors (such as tenofovir disoproxil, and lamivudine), chloroquine, and Chinese traditional medicine (such as Shu Feng Jie Du or Lianhuaqingwen capsules), have been proposed.5
Several drug discovery approaches like quantitative structure–activity relationship (QSAR), virtual screening (VS), artificial intelligence and drug repositioning, are strongly required to help in discovering a treatment of the uncontrolled pandemic caused by SARS-CoV-2.6 Numerous molecular studies were addressed to figure out the active sites of the SARS-CoV-2.7,8 Main protease (Mpro) of SARS-CoV-2 is some of the decisive factors in the infectious route of the virus; they have been reported as important targets for therapeutic strategies.9
Because of discovery of new drugs require long time and expense, searching for new compounds from natural sources known with its high safety and applicability will be a good avenue to treat SARS-CoV-2. In order to quickly discovered lead compounds especially from the promising wild plants for clinical trials, virtual screening study was initiated to identify new drug targeting SARS-CoV-2. From which Limonium tubiflorum (Delile) Kuntze (Plumbaginaceae) have been shown to possess important medicinal properties.10,11 In terms of its biological activity, family Plumbaginaceae has been reported to have medicinal plants with immunomodulatory activities.12 Researches on species of this genus revealed important bioactivities, including antimicrobial, antioxidant, anti-inflammatory and anticancer properties, with phenolics (epigallocatechin gallate, gallic acid, trans 3-hydroxycinnamic acid, myricetin and isorhamnetin) and triterpenoids (mainly limonoids) being considered as the main actives.11 Other important pharmacological effects of Limonium species include the antiviral effect against herpes simplex type 1, HSV-1 and influenza viruses.13,14
The pleiotropic activities of flavonoids such as their antioxidant and anti-microbial functions, to inhibit key proteins involved in the coronavirus infective cycle, as well as lack of their systemic toxicity, led to encourage the researchers to study their possible potential role against COVID-19 by in silico analysis.15 Thus, several studies represented that some flavonoids and phenolic acids having a role against coronavirus infection.15–19 For instance, myricetin was suggested to be a potential drug for anti-virus and symptomatic treatment of COVID-19.18 Also, gallic acid derivatives such as epigallocatechin gallate and (−)-epicatechin 3-O-(3′-O-methyl) gallate proposed to have inhibitory activity against coronavirus proteins.15,20
Herein, our work is considered as the first study aims to identify the chemical constituents of L. tubiflorum aerial parts (leaves & stem) growing in the Northwestern coast, Egypt as well as its isolated phytochemicals (especially those have galloyl substructure of myricetin 3-O-glycoside derivatives) were assessed for the binding affinity with an in silico study against two important SARS-CoV-2 protein targets; main protease (Mpro) and Spike Glycoprotein.
The main protease (Mpro, also known as 3CLpro), is one of the coronavirus nonstructural proteins (Nsp5) designated as a potential target for drug development.21,22 Mpro cleaves the viral polyproteins, generating 12 nonstructural proteins (Nsp4–Nsp16), including the RNA-dependent RNA polymerase (RdRp, Nsp12) and the helicase (Nsp13). Inhibition of Mpro would prevent the virus from replication and therefore constitutes one of the potential anticoronaviral strategies.21–23
Spike glycoprotein is crystal structure receptor-binding domain which bounds with ACE2 (PDB ID: 6M0J, resolution = 2.45 Å) and forms large trimeric structures that are essential for entry into host cells upon receptor binding and membrane fusion. Spike proteins are targeted by host neutralizing antibodies.24,25
Molecular docking provides a feasible strategy for exploring the basis and mechanism of the phytocompounds.26 This study utilized SARS-CoV-2 Mpro and spike glycoprotein as receptors, and molecular docking of the two was performed to select potential antiviral active ingredients for the development of effective and quick-acting chemical components that can resist COVID-19.
2. Results and discussion
2.1. Identification of the isolated compounds
The phytochemical studies on 70% ethanol extract of L. tubiflorum aerial parts led to the isolation of eight known phenolic compounds (1–8) using extensive chromatographic techniques for the first time (Fig. 1). The structures of the isolated compounds were elucidated by comprehensive 1D (1H- and 13C-NMR) and 2D NMR (COSY, HSQC and HMBC) spectroscopy in CD3OD & DMSO-d6, and compared with the previously published data. The known compounds were identified as myricetin 3-O-(2′′-galloyl)-β-D-galactopyranoside (1),27 myricetin 3-O-(2′′-galloyl)-α-L-rhamnopyranoside (2), myricetin 3-O-(3′′-galloyl)-α-L-rhamnopyranoside (3),28,29 gallic acid (4),27,28 myricetin 3-O-galactopyranoside (5),28 apigenin (6),27,30 myricetin (7),27,29 and ethyl gallate (8).31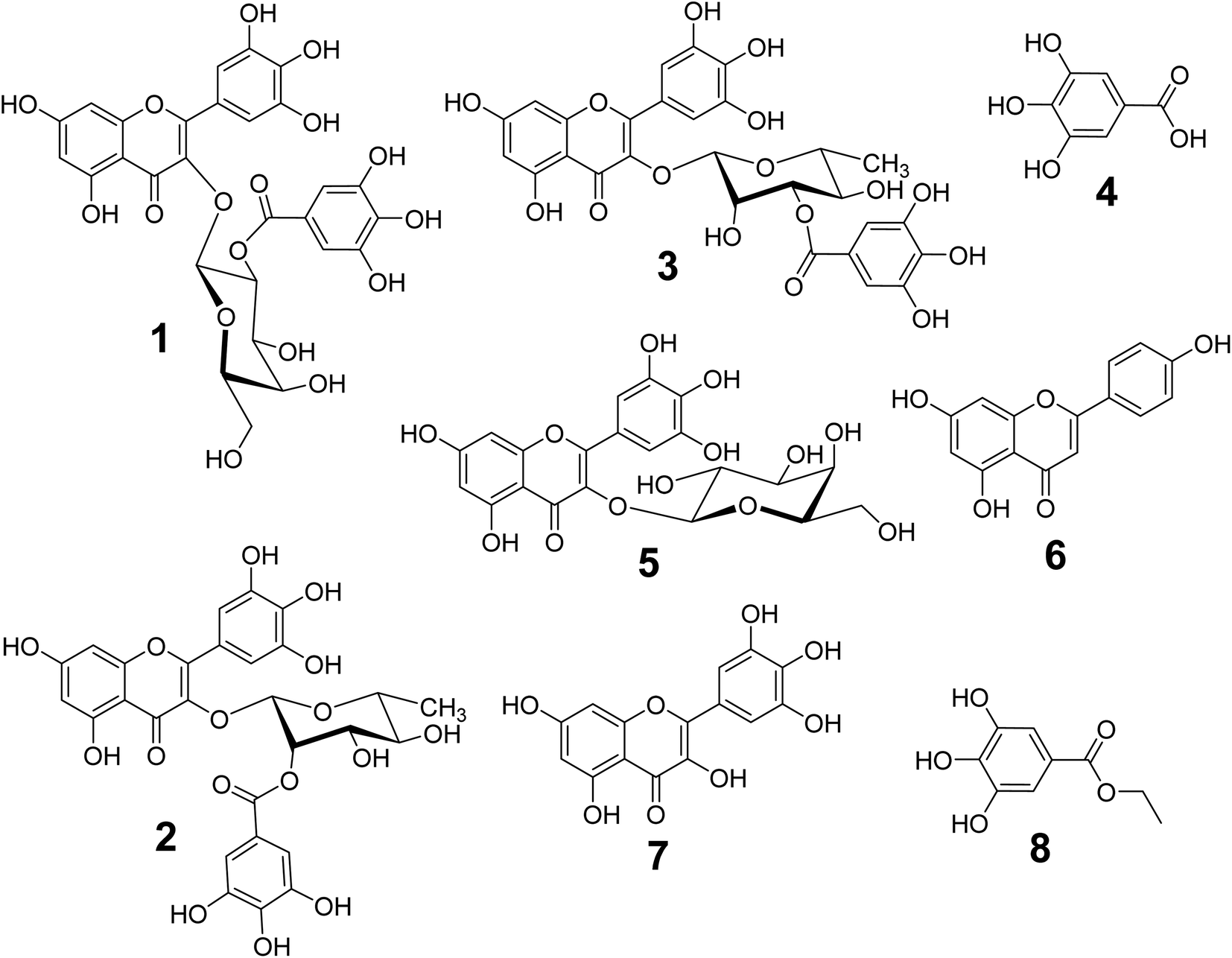 | ||
| Fig. 1 Chemical structures of the isolated compounds (1–8) from the 70% EtOH extract of L. tubiflorum aerial parts (stem & leaves). | ||
2.2. Molecular docking results
The interacting group and hydrogen bonds formed with the group interaction atoms of the corresponding amino acids are shown in Fig. 2–4. Interaction binding score of compounds (Table 1) with the receptors revealed the presence of variation ranged from −18.6652 kcal mol−1 to −9.6228 in which compounds 1–3 and 5–7 showed potent binding affinity with pose scores −17.9664 (RMSD = 1.49 Å), −18.6652 (RMSD = 1.4 Å), −16.6877 (RMSD = 1.3 Å), −12.7774 (RMSD = 1.4 Å), −10.5906 (RMSD = 0.93 Å) and −13.8128 (RMSD = 1.2 Å) kcal mol−1, respectively in comparison with remdesivir (score = −10.0932, RMSD = 1.5 Å).
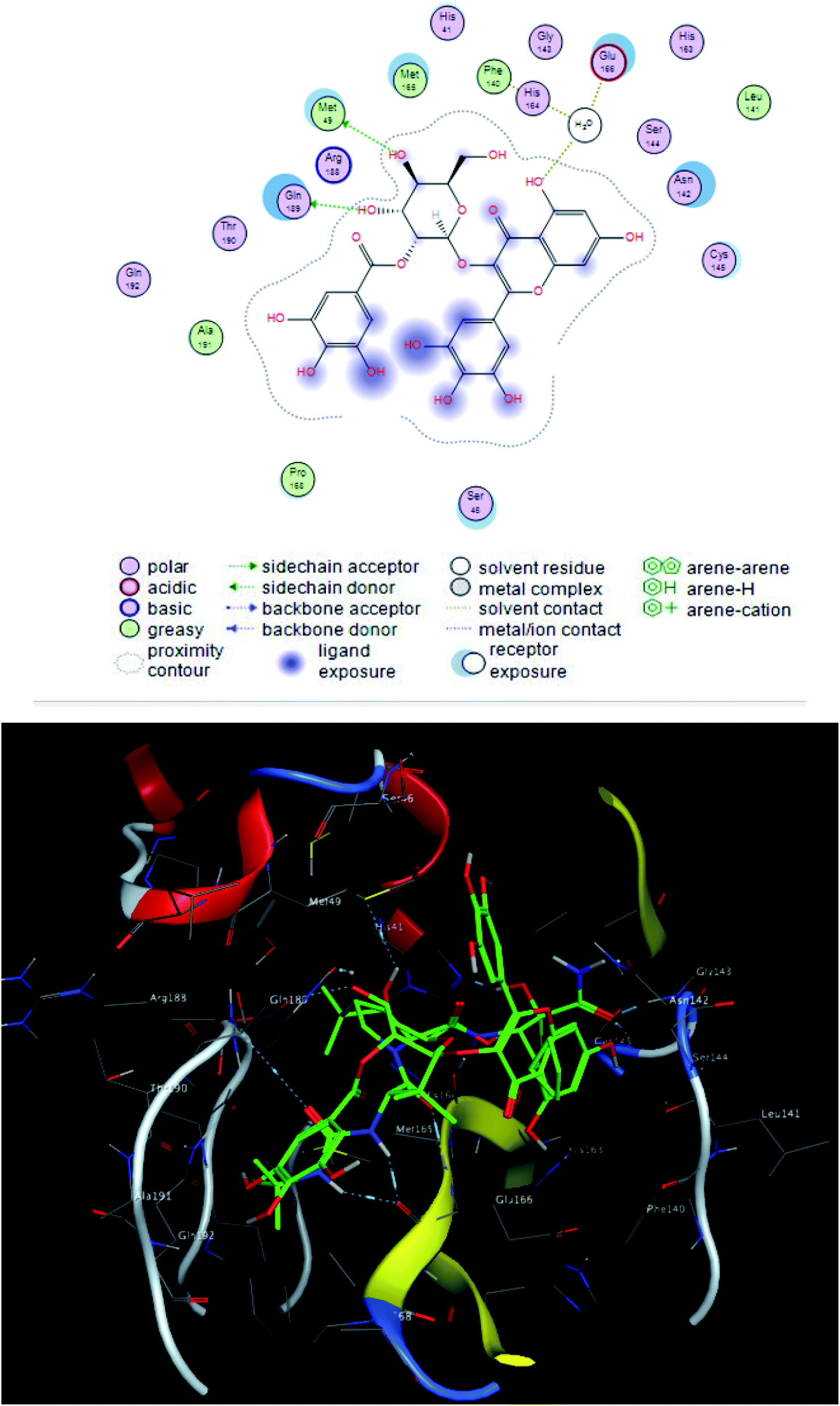 | ||
| Fig. 2 2D and 3D ligand interactions of compound 1 with main protease receptor. | ||
| No. | Sa kcal mol−1 | RMSD_refineb |
|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket.b RMSD_refine: the root-mean-squared-deviation (RMSD) between the predicted pose and those of the crystal one (after and before refinement process, respectively). | ||
| 1 | −17.9664 | 1.49 |
| 2 | −18.6652 | 1.44 |
| 3 | −16.6877 | 1.33 |
| 4 | −9.54093 | 0.91 |
| 5 | −12.7774 | 1.45 |
| 6 | −10.5906 | 0.93 |
| 7 | −13.8128 | 1.21 |
| 8 | −9.62281 | 0.75 |
| Remdesivir | −10.0932 | 1.57 |
Compound 2 was the most potent compound −18.6652 (RMSD = 1.4 Å) Fig. 3, showed hydrogen bond formation with Glu168, Met165 and Phe 140 amino acid residues of the receptor. In addition, the hydrophobic interaction of the compound with the amino acid residues of receptor pocket were included Ala 191, Met 49, Leu 167, Pro 168 and Leu 141. The other interactions are shown in Fig. 2, 4 and S25–S29 (ESI†), compared with that of remdesivir (ESI; Fig. S30†).
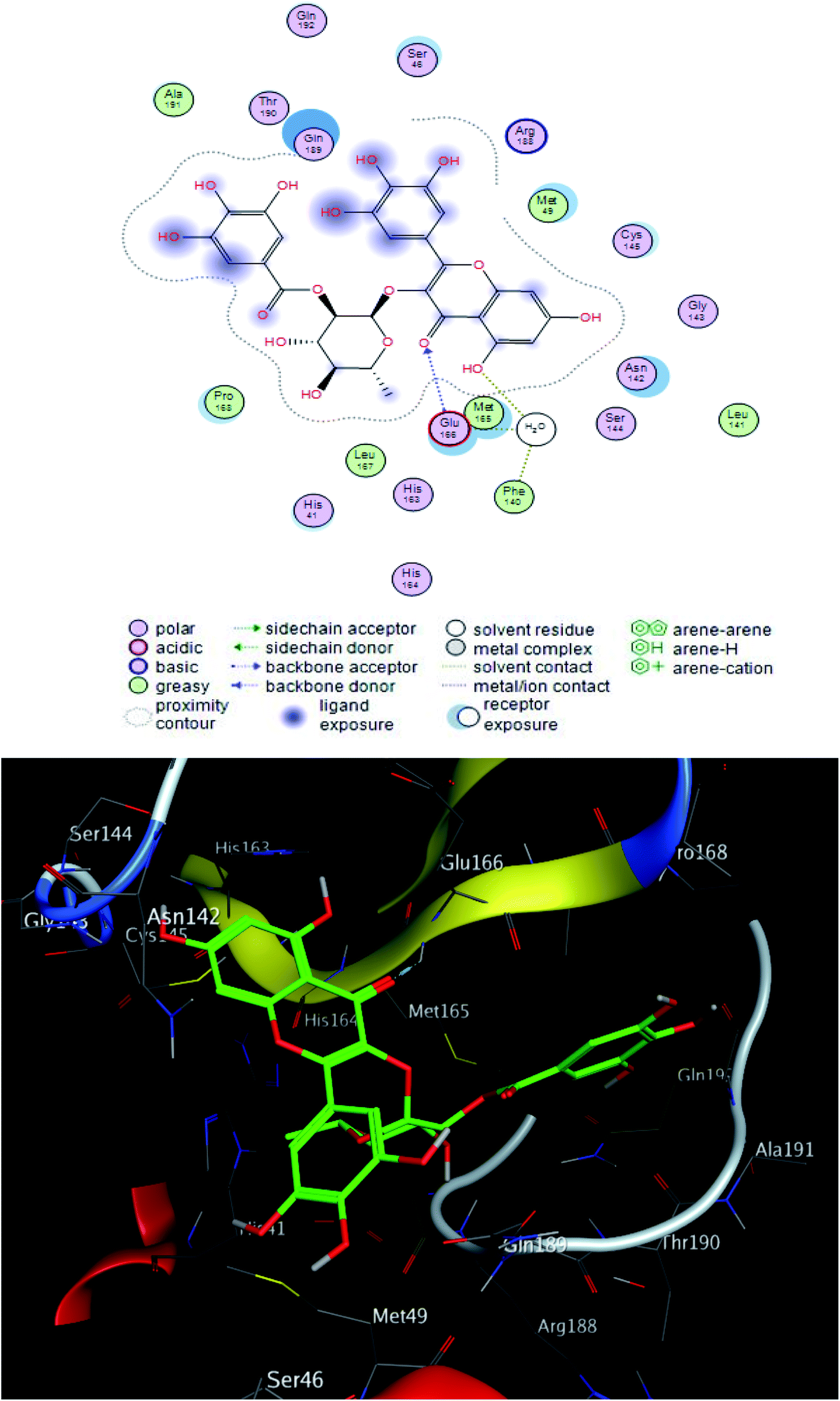 | ||
| Fig. 3 2D and 3D ligand interactions of compound 2 with main protease receptor. | ||
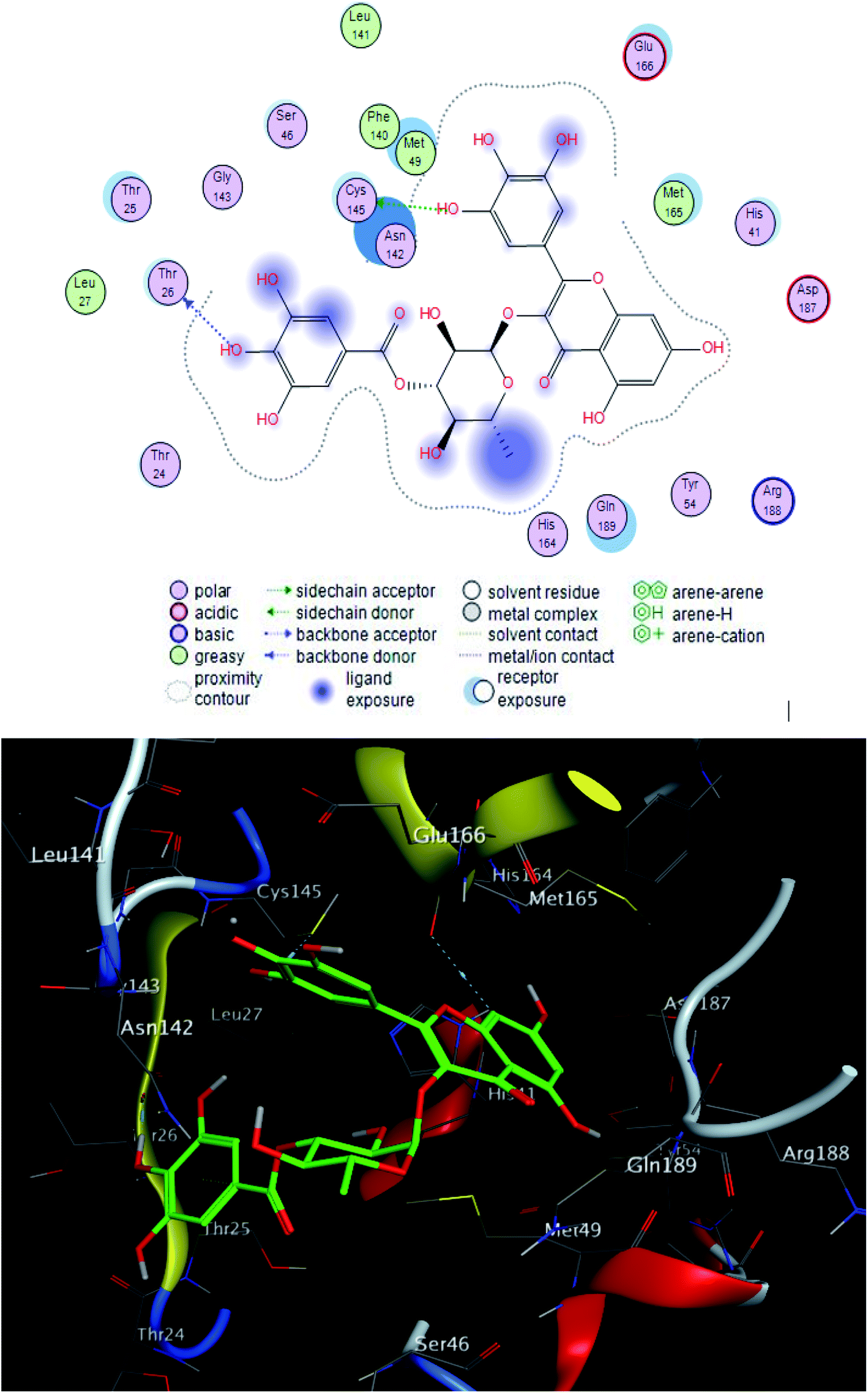 | ||
| Fig. 4 2D and 3D ligand interactions of compound 3 with main protease receptor. | ||
| No. | Sa kcal mol−1 | RMSD_refineb |
|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket.b RMSD_refine: the root-mean-squared-deviation (RMSD) between the predicted pose and those of the crystal one (after and before refinement process, respectively). | ||
| 1 | −18.9244 | 1.02 |
| 2 | −18.9272 | 1.61 |
| 3 | −16.6887 | 1.45 |
| 4 | −9.60763 | 0.90 |
| 5 | −15.6998 | 1.07 |
| 6 | −9.98701 | 0.82 |
| 7 | −14.4476 | 1.00 |
| 8 | −10.0071 | 0.98 |
| Remdesivir | −14.2976 | 1.49 |
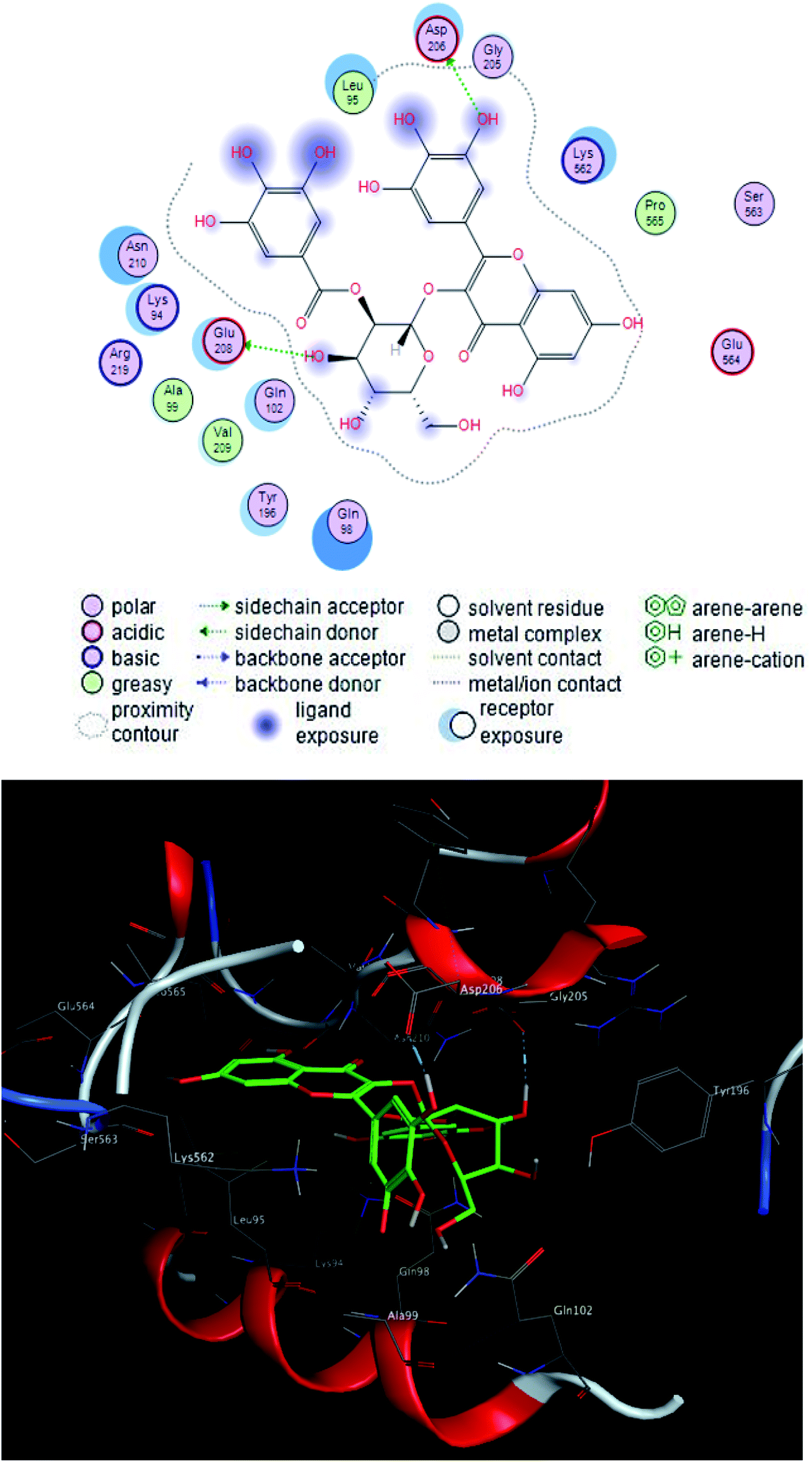 | ||
| Fig. 5 2D and 3D ligand interactions of compound 1 with spike glycoprotein. | ||
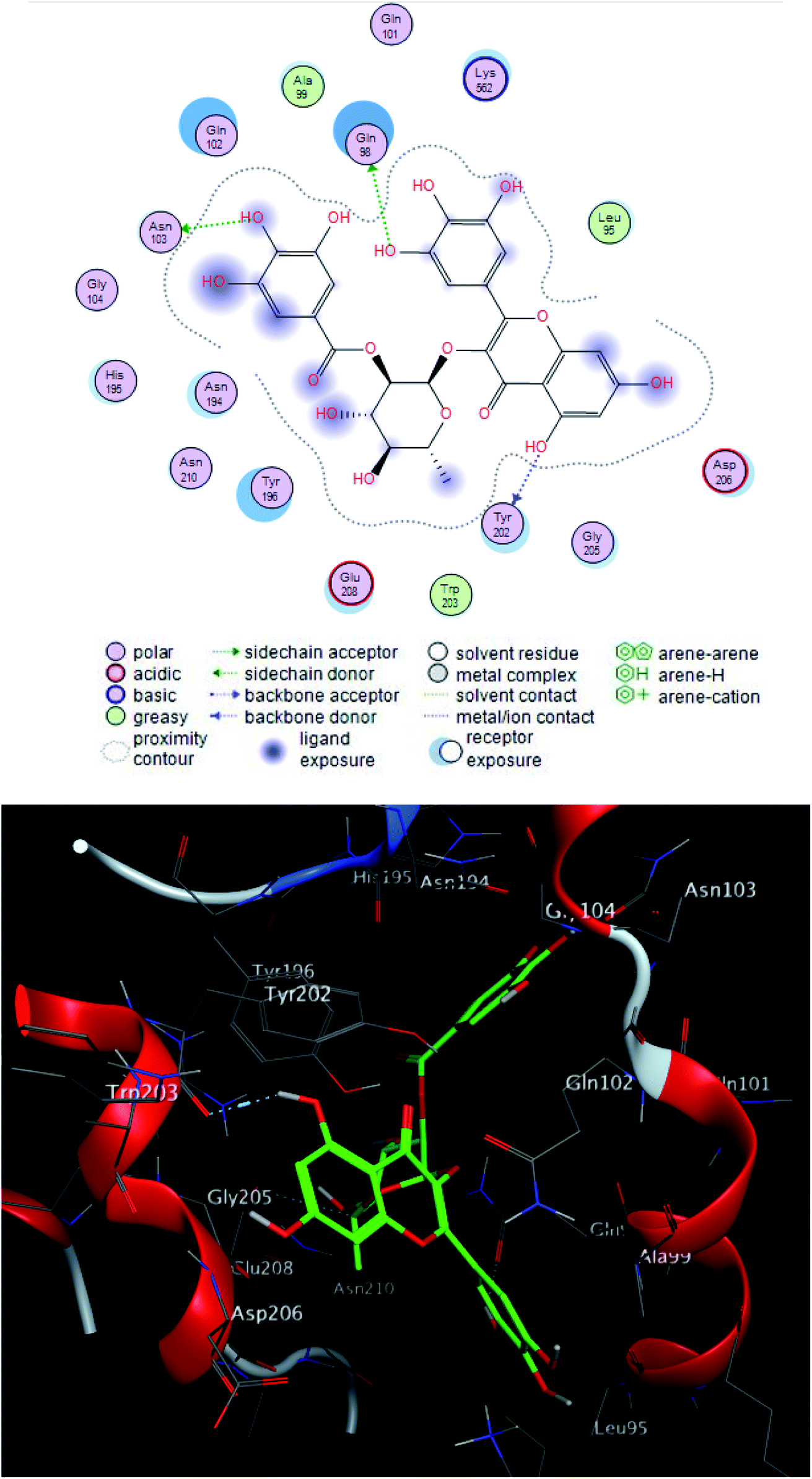 | ||
| Fig. 6 2D and 3D ligand interactions of compound 2 with spike glycoprotein. | ||
Finally, compounds 1–3, 5 and 7 could be used for development of new treatment for SARS CoV-2.
2.3. Molecular dynamic simulation
Since, the molecular docking of the drug molecule with the receptor gives important information about drug–receptor interactions and is commonly used to find out the binding orientation of drug candidates to their protein targets in order to predict the affinity and activity.32 So, depending on the binding affinity score that suggested the predominance of the galloyl substructure of myricetin 3-O-glycoside derivatives (1, 2) over other tested compounds (3–8).To validate the docking experiments, we conducted a number of molecular dynamic simulation experiments along with binding free energy (ΔG) calculations. As depicted in Fig. 7, compound 2 was apparently more stable than compound 1 inside the active site of Mpro (their average RMSDs were 1.2 and 4.4 Å, respectively). Accordingly, the binding free energy (ΔGbinding) of compound 2 was lower than that of compound 1 (ΔGbinding = −9.1 and −5.2 kcal mol−1, respectively).
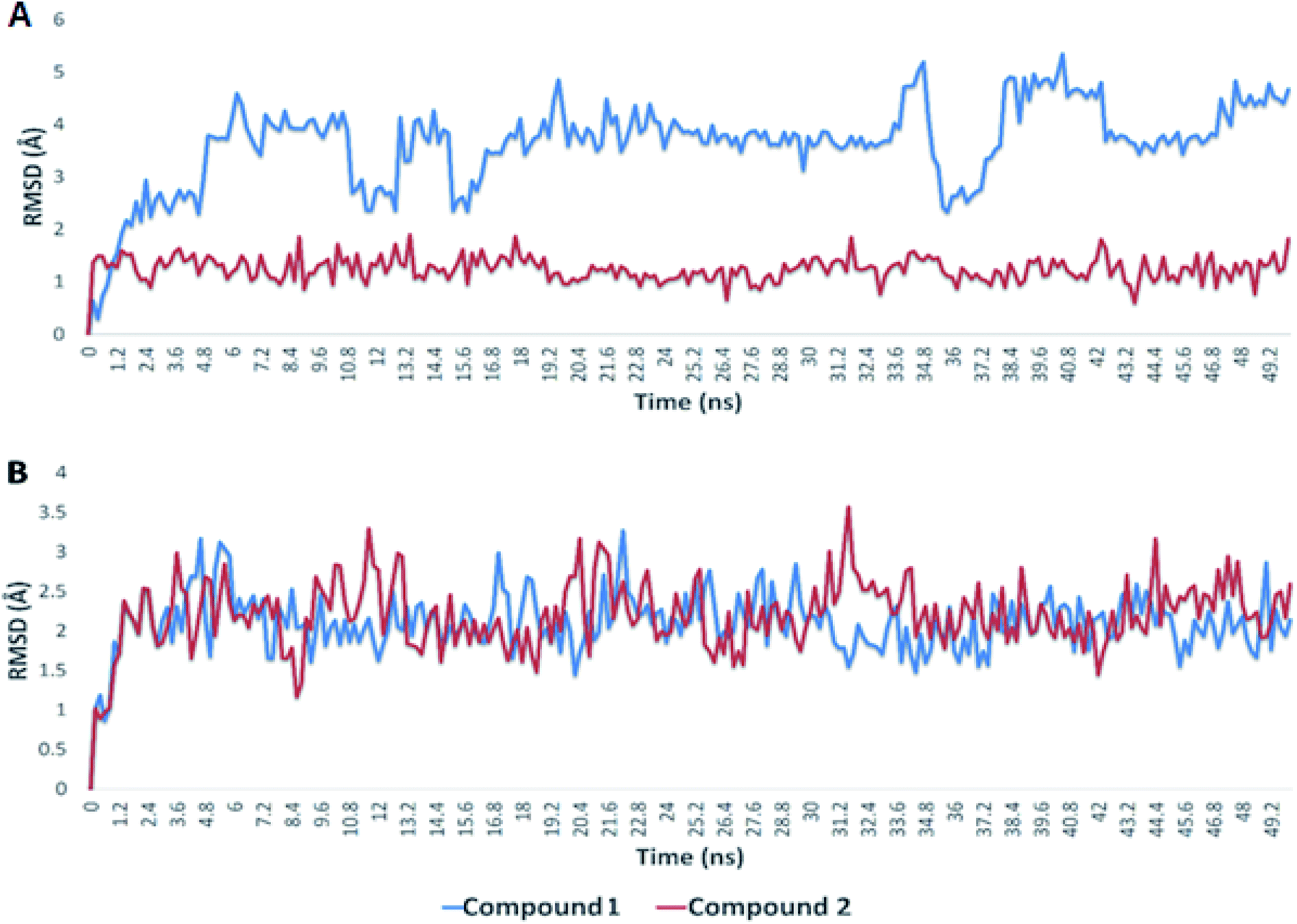 | ||
| Fig. 7 RMSDs of compounds 1 and 2 inside the binding sites of Mpro and S-protein over 50 ns of MDS. | ||
In regard to the viral S-protein, both compound 1 and 2 achieved stable binding with the binding site, and didn't deviate significantly from the starting docking pose throughout the course of MDS (their average RMSD was 2.3 Å). Hence, their ΔGbinding were almost the same (−8.1 and −8.0 kcal mol−1, respectively). According to these in silico dynamic and free energy studies, compound 2 is suggested to be a very good candidate for discovering COVID-19 drugs. Additional in vitro and in vivo investigations are needed to assist our primary in silico findings.
2.4. Structure–activity relationship (SAR) for the isolated predominance myricetin derivatives
Inspection of the structure–activity relationship for the isolated myricetin derivatives from L. tubiflorum aerial parts was deduced based on the previous reported studies which represented that myricetin has a potent SARS-CoV-2 inhibition activity.15,18 The identified compounds; myricetin 3-O-(2′′-galloyl)-β-D-galactopyranoside (1), myricetin 3-O-(2′′-galloyl)-α-L-rhamnopyranoside (2) and myricetin 3-O-(3′′-galloyl)-α-L-rhamnopyranoside (3) exhibited potential SARS-CoV-2 inhibition activity more than the other myricetin derivatives deprived from galloyl and/or sugar moieties (5 and 7). Hence, it can be suggested that presence of 2′′-galloyl or 3′′-galloyl linked to sugar moieties of myricetin 3-O-glucoside in compounds (1–3) is important for enhancement of SARS-CoV-2 inhibition activity. Moreover, the attachment of galloyl moiety at position 2′′ of the sugar moiety is more favorable rather than position 3′′ for increasing this inhibition activity. This suggestion was confirmed from the in silico study, where it was noticed that compounds (1 and 2) are best fitting in receptor pocket active site and receptor exposure increases its affinity.3. Experimental
3.1. Reagents and apparatus
Whatman No. 1 and 3 paper chromatography (PC) were purchased from Sigma Aldrich (Darmstadt, Germany). Analytical pre-coated normal phase silica gel 60 GF245 TLC plates were obtained from Merck (Darmstadt, Germany) and the spots were visualized using UV light and by exposure of PC and TLC with ammonia vapor. Also, the TLC plates were sprayed with 5% sulfuric acid in methanol followed by heating at 105 °C for 3 min. Column chromatography (CC) was carried out on polyamide 6S and Sephadex LH-20 (Riedal-De Haen Ag, Sellze-Hannover, Germany). All the organic solvents used were of the highly analytical grade. NMR experiments were recorded on Bruker DRX 400 and 600 MHz NMR spectrometer instruments (Bruker Daltonics Inc., MA, USA) in methanol-d4 and DMSO-d6. LC-MS-ESI of the two compounds; 1 and 2 was carried out on Waters Acquity UPLC with ESI-detector based on a UPLC-MS equipped with C18 column detect the compounds at both positive and negative ionization modes.3.2. Plant material
The aerial parts of Limonium tubiflorum (Delile) Kuntze var tubiflorum were collected from the Wadi Habis at Mersa matrouh governorate (Northwestern coast, Egypt) in April 2017 (geographic position coordinates N: 31°22′81′′; E: 27°3′54′′). The plant sample was kindly authenticated by Prof. Dr Azza El Hadidy, Professor of plant taxonomy and flora, Botany Department, Faculty of Science, Cairo University, Egypt. A voucher specimen (18/4/2017-H) of the plant has been deposited at the Herbarium of Faculty of Science, Cairo University, Egypt.3.3. Preparation of total extract
The aerial parts (leaves & stem) of L. tubiflorum plant were air-dried in shade then ground to a fine powder (1.2 kg) and was extracted using 70% ethanol (70% EtOH, 3 × 3 L, each 48 h). The obtained extracts were combined then concentrated by rotary evaporator at 40 °C to afford a residue of 98 g, which was stored at 4 °C for further investigations.3.4. Fractionation and isolation
88 g from the dry extract (98 g) was desalted in absolute ethanol. The desalted extract was concentrated to give 150 mL and pre-adsorbed on small amount of polyamide 6S for column chromatography then evaporated under reduced pressure. The mixture was fractionated on a polyamide column. Elution was started with distilled water (H2O) followed by H2O–EtOH stepwise until finally pure EtOH was used. The obtained fractions from the column (500 mL each) were examined on paper chromatography (PC) and thin layer chromatography (TLC) using the systems; n-butanol–acetic acid–water (BAW) (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5 v/v/v) upper phase, and 15% acetic acid for paper chromatography PC and ethyl acetate–methanol–water (30:5:4), ethyl acetate–glacial acetic acid–formic acid–water (100:11:11:26), and dichloromethane–methanol (9:1 and 8:2) for TLC and observed under ultraviolet light. Similar fractions were pooled together, which were combined into five major collective fractions (F1–F5), and then evaporated under vacuum. Fractions; F2 to F5 were the promising for our study based on TLC and PC detections. Fraction F2 (2 g) which obtained from the polyamide column using 30% EtOH was chromatographed over a Sephadex LH-20 column eluted by n-butanol (BuOH) saturated with water (upper phase) to give a promising subfraction (0.2 g) which was further re-chromatographed on Sephadex LH-20 column using the mobile phase of EtOH–H2O (30–70%) to afford pure compound (1, 60 mg). 3 g of fraction F3 (7.3 g) resulted from the polyamide column using 50% EtOH was first applied to preparative paper chromatography (PPC) using system BAW (4:1:5, upper phase) yielded three bands, then purification of each band on Sephadex LH-20 using EtOH–H2O (30–70%) affording three pure compounds (2, 50 mg), (3, 70 mg) and (4, 45 mg). Fraction F4 (3.5 g) obtained from the polyamide column by 70% EtOH was subjected to a Sephadex LH-20 column using BuOH saturated with water (upper phase) to give two corresponding fractions. The two fractions; F4a (230 mg) and F4b (95 mg) were further separated on Sephadex LH-20 column using EtOH–H2O (40–100%) to afford compounds (5, 66 mg) and (6, 40 mg), respectively. Finally, 2 g of fraction F5 (5.6 g) resulted from the polyamide column using the eluent pure EtOH, was subjected to PPC using BAW (4:1:5, upper phase) solvent system to yield two major bands. The two corresponding bands were further separated on Sephadex LH-20 column using EtOH–H2O (50–100%) to give the two compounds; (7, 47 mg) and (8, 45 mg). The structures of the isolated compounds were elucidated by 1D and 2D NMR experiments (ESI; Tables 1–8 and Fig. S1–S25†).
:1:5 v/v/v) upper phase, and 15% acetic acid for paper chromatography PC and ethyl acetate–methanol–water (30:5:4), ethyl acetate–glacial acetic acid–formic acid–water (100:11:11:26), and dichloromethane–methanol (9:1 and 8:2) for TLC and observed under ultraviolet light. Similar fractions were pooled together, which were combined into five major collective fractions (F1–F5), and then evaporated under vacuum. Fractions; F2 to F5 were the promising for our study based on TLC and PC detections. Fraction F2 (2 g) which obtained from the polyamide column using 30% EtOH was chromatographed over a Sephadex LH-20 column eluted by n-butanol (BuOH) saturated with water (upper phase) to give a promising subfraction (0.2 g) which was further re-chromatographed on Sephadex LH-20 column using the mobile phase of EtOH–H2O (30–70%) to afford pure compound (1, 60 mg). 3 g of fraction F3 (7.3 g) resulted from the polyamide column using 50% EtOH was first applied to preparative paper chromatography (PPC) using system BAW (4:1:5, upper phase) yielded three bands, then purification of each band on Sephadex LH-20 using EtOH–H2O (30–70%) affording three pure compounds (2, 50 mg), (3, 70 mg) and (4, 45 mg). Fraction F4 (3.5 g) obtained from the polyamide column by 70% EtOH was subjected to a Sephadex LH-20 column using BuOH saturated with water (upper phase) to give two corresponding fractions. The two fractions; F4a (230 mg) and F4b (95 mg) were further separated on Sephadex LH-20 column using EtOH–H2O (40–100%) to afford compounds (5, 66 mg) and (6, 40 mg), respectively. Finally, 2 g of fraction F5 (5.6 g) resulted from the polyamide column using the eluent pure EtOH, was subjected to PPC using BAW (4:1:5, upper phase) solvent system to yield two major bands. The two corresponding bands were further separated on Sephadex LH-20 column using EtOH–H2O (50–100%) to give the two compounds; (7, 47 mg) and (8, 45 mg). The structures of the isolated compounds were elucidated by 1D and 2D NMR experiments (ESI; Tables 1–8 and Fig. S1–S25†).
3.5. Molecular docking study
The 3D X-ray crystal structure of two SARS-CoV-2 receptors; main protease (PDB ID: 6ZRU, resolution = 2.10 Å)9 and spike glycoprotein crystal structure receptor-binding domain bound with ACE2 (PDB ID: 6M0J, resolution = 2.45 Å),33 were obtained from the RCSB Protein Data Bank (www.rcsb.org/pdb). The adopted docking procedure followed the standard protocol implemented in MOE 2014.0901 and the geometry of the resulting complexes was studied using MOE's Pose Viewer utility.34In order to prepare the protein for docking studies, the proteins obtained from the Protein Data Bank was prepared for docking as follows: first, the enzyme was 3D protonated, where hydrogen atoms were added at their standard geometry then the partial charges were computed, minimized and the system was optimized. Second, the unbound water molecules which aren't involved in the interaction were deleted and the binding pocket had been defined before docking. The receptor active site for SARS-CoV-2 main protease was determined by complexed ligand while spike glycoprotein receptor active site was determined by MOE site finder function used to calculate and predict possible active potential site of selected proteins for ligand binding in the receptor.
The 2D structure of the ligand compounds identified from the total extract of L. tubiflorum aerial parts (stem & leaves) was drawn using the ChemDraw software 17.0.0.206 then converted to a 3D structure and protonated at pH 7, GB/VI as electrostatics parameter and dielectric points was adjusted at 2. The partial charges of the compounds were adjusted and its energy was minimized using Merck Molecular Forcefield (MMFF94s) method to a gradient 0.05.
Flexible ligand-rigid receptor docking was done with MOE-DOCK by running the docking and defining the receptor atoms as receptor + solvent, site of placement was defined as MDB file that contain the conformers of ligands and placement method was adjusted to triangle matcher.
The obtained poses were subjected to London dG as the scoring function and forcefield refinement using the same scoring function. The interaction between the ligands and receptors binding site were generated and the results of docking were recorded as pose score (S) and Root Mean Square Deviation (RMSD).
3.6. Molecular dynamic simulation
Molecular dynamic simulations (MDS) for the generated ligand–enzyme complexes were carried out by the Nanoscale Molecular Dynamics (NAMD) 2.6 software,35 using the CHARMM27 force field.36 Hydrogen atoms were added to the protein structures using the psfgen plugin including the Visual Molecular Dynamic (VMD) 1.9 software.37 Then, the whole generated systems were solvated using water molecules (TIP3P) and 0.15 M NaCl. At first, the total energy of the generated systems was minimized and gradually heated to reach 300 K and equilibrated for 10 ns. Afterwards, the MDS was continued for 50 ns, and the trajectory was stored every 0.1 ns and further analyzed with the VMD 1.9 software. The MDS output was sampled every 0.1 ns to calculate the root mean square deviation (RMSD). The parameters of compounds (1 and 2) were prepared using the online software the VMD force field ToolKit (ffTK).37 Binding free energies (ΔGbinding) were calculated using the free energy perturbation (FEP) method.38 The web-based software Absolute Ligand Binder was applied to generate the input files for NAMD software which was performed the simulations required for ΔGs calculations.4. Conclusion
In conclusion, this study provides the first report regarding the metabolic profile of L. tubiflorum aerial parts. The total 70% ethanol plant extract was subjected to chromatographic and spectroscopic methods to isolate and characterize of two gallic acid derivatives (4 and 8) together with apigenin (6) and five flavonoid derivatives of myricetin (1–3, 5 and 7) for the first time from the plant under investigation. The results showed that L. tubiflorum aerial parts are a rich source of myricetin derivatives. Based on the in silico molecular docking and dynamic studies of the isolated plant components against the main protease (Mpro) and spike glycoprotein SARS-CoV-2 receptors, it can be suggested that myricetin 3-O-(2′′-galloyl)-glycoside derivatives (1 and 2) in L. tubiflorum aerial parts could be as potential COVID-19 drug candidates. Hence, this finding proves that the value of this screening study can lead to the rapid discovery of drugs from natural sources and prioritized them a potential multitarget acting on pandemic COVID-19. More in vitro and in vivo biological studies are necessary to strengthen our in silico results.Conflicts of interest
We declare that we have no conflict of interest.Acknowledgements
The authors would like to thank Prof. Dr Azza El-Hadidy, Botany Department, Faculty of Science, Cairo University, Egypt, for authenticating the plant material. In addition, we gratefully acknowledge the Research and Education Support Center of the faculty of Agriculture, Kyushu University for supporting NMR and MS facilities.References
- L. Caly, J. D. Druce, M. G. Catton, D. A. Jans and K. M. Wagstaff, Antiviral Res., 2020, 178, 104787, DOI:10.1016/j.antiviral.2020.104787.
- W. R. Ferraz, R. A. Gomes, A. L. S. Novaes and G. H. Goulart Trossini, Future Med. Chem., 2020, 12(20), 1815–1828, DOI:10.4155/fmc-2020-0165.
- S. S. El Hawary, A. R. Khattab, H. S. Marzouk, A. S. El Senousy, M. G. Alex, O. M. Aly and U. R. Abdelmohsen, RSC Adv., 2020, 10(70), 43103–43108, 10.1039/d0ra08997d.
- B. Luan, T. Huynh, X. Cheng, G. Lan and H. R. Wang, J. Proteome Res., 2020, 19(11), 4316–4326, DOI:10.1021/acs.jproteome.0c00430.
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res., 2020, 30(3), 269–271, DOI:10.1038/s41422-020-0282-0.
- A. E. Allam, H. K. Assaf, H. A. Hassan, K. Shimizu and Y. A. Elshaier, RSC Adv., 2020, 10(50), 29983–29998, 10.1039/d0ra05265e.
- G. Mariano, R. J. Farthing, S. L. Lale-Farjat and J. R. Bergeron, Front. Mol. Biosci., 2020, 7, 344, DOI:10.3389/fmolb.2020.605236.
- D. Singh and V. Y. Soojin, Exp. Mol. Med., 2021, 53(4), 537–547, DOI:10.1038/s12276-021-00604-z.
- R. Oerlemans, A. J. Ruiz-Moreno, Y. Cong, N. D. Kumar, M. A. Velasco-Velazquez, C. G. Neochoritis, J. Smith, F. Reggiori, M. R. Groves and A. Dömling, RSC Med. Chem., 2021, 12(3), 370–379, 10.1039/d0md00367k.
- D. Geng, X. Chi, Q. Dong and F. Hu, Ind. Crops Prod., 2015, 67, 492–497, DOI:10.1016/j.indcrop.2015.01.063.
- A. de Oliveira Caleare, A. Hensel, J. C. P. Mello, A. B. Pinha, G. P. Panizzon, M. Lechtenberg, F. Petereit and C. V. Nakamura, Fitoterapia, 2017, 118, 87–93, DOI:10.1016/j.fitote.2017.03.002.
- C. J. Ugwah-Oguejiofor and I. M. Adebisi, Ife J. Sci., 2021, 23(1), 161–194, DOI:10.4314/ijs.v23i1.16.
- F. Medini, S. Bourgou, K. Lalancette, M. Snoussi, K. Mkadmini, I. Coté, C. Abdelly, J. Legault and R. Ksouri, S. Afr. J. Bot., 2015, 99, 158–164, DOI:10.1016/j.sajb.2015.04.007.
- Y. C. Kuo, L. C. Lin, W. J. Tsai, C. J. Chou, S. H. Kung and Y. H. Ho, Antimicrob. Agents Chemother., 2002, 46(9), 2854–2864, DOI:10.1128/AAC.46.9.2854-2864.2002.
- M. Russo, S. Moccia, C. Spagnuolo, I. Tedesco and G. L. Russo, Chem.–Biol. Interact., 2020, 328, 109211, DOI:10.1016/j.cbi.2020.109211.
- A. da Silva Antonio, L. S. Wiedemann and V. F. Veiga-Junior, RSC Adv., 2020, 10(39), 23379–23393, 10.1039/d0ra03774e.
- A. G. Omokhua-Uyi and J. Van Staden, S. Afr. J. Bot., 2021, 139, 386–398, DOI:10.1016/j.sajb.2021.03.012.
- T. Xiao, M. Cui, C. Zheng, M. Wang, R. Sun, D. Gao, J. Bao, S. Ren, B. Yang, J. Lin, X. Li, D. Li, C. Yang and H. Zhou, Front. Pharmacol., 2021, 12, 669642, DOI:10.3389/fphar.2021.669642.
- S. S. El-Hawary, M. A. Rabeh, M. A. Raey, E. M. El-Kadder, M. Sobeh, U. R. Abdelmohsen, A. Albohy, A. M. Andrianov, I. P. Bosko, M. M. Al-Sanea and D. G. El-Kolobby, J. Biomol. Struct. Dyn., 2021, 1–3, DOI:10.1080/07391102.2021.1885494.
- H. I. Umar, B. Siraj, A. Ajayi, T. O. Jimoh and P. O. Chukwuemeka, J. Genet. Eng. Biotechnol., 2021, 19(1), 1–4, DOI:10.1186/s43141-021-00120-7.
- R. Hilgenfeld, FEBS J., 2014, 281(18), 4085–4096, DOI:10.1111/febs.12936.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S. H. Jung, J. Med. Chem., 2016, 59(14), 6595–6628, DOI:10.1021/acs.jmedchem.5b01461.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368(6489), 409–412, DOI:10.1126/science.abb3405.
- F. Li, W. Li, M. Farzan and S. C. Harrison, Science, 2005, 309(5742), 1864–1868, DOI:10.1126/science.1116480.
- G. Lu, Y. Hu, Q. Wang, J. Qi, F. Gao, Y. Li, Y. Zhang, W. Zhang, Y. Yuan, J. Bao and B. Zhang, Nature, 2013, 500(7461), 227–231, DOI:10.1038/nature12328.
- N. Hirayama, Drug Metab. Pharmacokinet., 2017, 32(1), 31–39, DOI:10.1016/j.dmpk.2016.10.002.
- Z. G. Yang, L. N. Jia, Y. Shen, A. Ohmura and S. Kitanaka, Molecules, 2011, 16(10), 8305–8318, DOI:10.3390/molecules16108305.
- L. C. Lin and C. J. Chou, Planta Med., 2000, 66(04), 382–383, DOI:10.1055/s-2000-8547.
- Y. C. Kuo, L. M. Yang and L. C. Lin, Planta Med., 2004, 70(12), 1237–1239, DOI:10.1055/s-2004-835859.
- K. S. Kumar, V. Sabu, G. Sindhu, A. A. Rauf and A. Helen, Int. Immunopharmacol., 2018, 59, 157–167, DOI:10.1016/j.intimp.2018.04.004.
- A. Ooshiro, S. Hiradate, S. Kawano, T. Takushi, Y. Fujii, M. Natsume and H. Abe, Weed Biol. Manage., 2009, 9(2), 169–172, DOI:10.1111/j.1445-6664.2009.00335.x.
- H. K. Kumara, R. Suhas, D. S. Vardhan, M. Shobha and D. C. Gowda, RSC Adv., 2018, 8(19), 10644–10653, 10.1039/c8ra00531a.
- M. A. Hussien and A. E. Abdelaziz, Netw. Model. Anal. Health Inform. Bioinform., 2020, 9(1), 1–18, DOI:10.1007/s13721-020-00263-6.
- B. Drung, C. Scholz, V. A. Barbosa, A. Nazari, M. H. Sarragiotto and B. Schmidt, Bioorg. Med. Chem. Lett., 2014, 24(20), 4854–4860, DOI:10.1016/j.bmcl.2014.08.054.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26(16), 1781–1802, DOI:10.1002/jcc.20289.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102(18), 3586–3616, DOI:10.1021/jp973084f.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29(11), 1859–1865, DOI:10.1002/jcc.20945.
- S. Jo, W. Jiang, H. S. Lee, B. T. Roux and W. Im, J. Chem. Inf. Model., 2013, 53(1), 267–277, DOI:10.1021/ci300505n.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05927k |
| This journal is © The Royal Society of Chemistry 2021 |