
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn aerobic oxidation of alcohols into carbonyl synthons using bipyridyl-cinchona based palladium catalyst†
Ravi Kumar Cheedarala *a,
Ramasamy R. Chidambaramb,
Ayyanar Siva*b and
Jung Il Song*a
*a,
Ramasamy R. Chidambaramb,
Ayyanar Siva*b and
Jung Il Song*a
aResearch Institute of Mechatronics, Department of Mechanical Engineering, Changwon National University, Changwon City, Republic of Korea. E-mail: ravi@changwon.ac.kr; jisong@changwon.ac.kr
bSupramolecular and Organometallic Chemistry Lab, Department of Inorganic Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai 625021, Tamil Nadu, India. E-mail: drasiva@gmail.com
First published on 6th October 2021
Abstract
We have reported an aerobic oxidation of primary and secondary alcohols to respective aldehydes and ketones using a bipyridyl-cinchona alkaloid based palladium catalytic system (PdAc-5) using oxygen at moderate pressure. The PdAc-5 catalyst was analysed using SEM, EDAX, and XPS analysis. The above catalytic system is used in experiments for different oxidation systems which include different solvents, additives, and bases which are cheap, robust, non-toxic, and commercially available on the industrial bench. The obtained products are quite appreciable in both yield and selectivity (70–85%). In addition, numerous important studies, such as comparisons with various commercial catalysts, solvent systems, mixture of solvents, and catalyst mole%, were conducted using PdAc-5. The synthetic strategy of oxidation of alcohol into carbonyl compounds was well established and all the products were analysed using 1H NMR, 13CNMR and GC-mass analyses.
1. Introduction
Palladium-catalysed aerobic transformation has been one of the most challenging topics within organometallic chemistry. This system is widely used in oxidation, involving alcohols, alkenes, oxidative coupling and so on, and has triggered commercial interest in the above-mentioned chemistry. Generally, an alcohol oxidation reaction plays an important role in functional group manipulation and heteroatom incorporation in the synthesis of chemically and biologically active compounds. Thus, developing a new process in both academic and industrial sectors to beat conventional systems1 becomes a bit challenging for chemists. These challenges force researchers to come up with novel catalysis design systems for the above-mentioned transformation.2 For key factors like stoichiometric oxidants, their versatility, expense, and environmental impact need to be considered. A viable and attractive approach is the use of metal-catalysed oxidation in the presence of molecular oxygen as an oxidant.3In the past 4–5 decades, there have been significant advances in the development of new transition metal catalysed oxidations using improved catalytic systems4 with increased turnover numbers. The use of highly stable ligands has brought about these advances, thus initiating the search for new catalytic systems. The development of ligand-modulated catalytic systems, which do not require the use of a co-catalyst, greatly simplified the catalysis and has allowed for an elucidation of the mechanistic features of these reactions. A significant number of mechanistic studies5,6 have been performed by various research groups over the years and more are on their way. Recently, our group have reported a ligand-free recyclable palladium catalyst for (1) the oxidation of alcohols into carbonyl compounds, (2) the highly selective α-alkylation of ketones with alcohols and imines, and (3) the production of secondary amines by coupling benzyl alcohols and primary amines.7 However, these studies exhibited both important improvements and the current limitations of palladium oxidase reactions, which illustrate the challenges of developing new more efficient direct molecular oxygen-coupled processes.8 Based on these previous points, we concentrate on developing a Pd-based chiral organometallic catalyst for the aerobic oxidation reaction under mild reaction conditions.
2. Experimental section
2.1 Materials and methods
5,5-Dimethyl-2,2-bipyridine, AIBN, (+)-cinchonine, and N-bromosuccinimide were purchased from Sigma-Aldrich. 2-Methyl benzyl alcohol, 4-methyl benzyl alcohol, 2-methoxy benzyl alcohol, 4-methoxy benzyl alcohol, 3-methoxy 4-hydroxy benzyl alcohol, 2-fluoro benzyl alcohol, 3-fluoro benzyl alcohol, 4-fluro benzyl alcohol, 2-chloro benzyl alcohol, 4-chloro benzyl alcohol, 2,3-dichloro benzyl alcohol, 2-nitro benzyl alcohol, 3-nitro benzyl alcohol, 4-tert-butyl benzyl alcohol, mesitaldehyde, 2,6-dimethyl benzyl alcohol, 3,5-dibromo-2,4,6-trimethyl benzyl alcohol, 1-phenyl-1 ethanol, 1-phenyl-1 propanol, diphenyl methanol, cyclohexyl methanol, cyclohexanol, 1,4-dihydroxy cyclohexane, cyclododecanol, piperonyl alcohol, 4-acetamido benzyl alcohol, 4-trifluromethyl benzyl alcohol, 2,4-trifluromethyl benzyl alcohol, 4-phenyl benzyl alcohol, pyridine-2-yl methanol, pyridine-3-yl methanol, pyridine-4-yl methanol, naphthyl-1-methanol, and naphthyl-2-methanol were purchased from various sources as technical grade. All the solvents for the reactions were obtained as laboratory grade. All the solvents for analysis were purchased as analytical grade.The melting points were measured in open capillary tubes and are uncorrected. The 1H, and 13C NMR spectra were recorded on a Bruker (Avance) 400 MHz NMR instrument using TMS as an internal standard, and CDCl3 and DMSO as solvents. Standard Bruker software was used throughout. Chemical shifts are given in parts per million (δ-scale) and the coupling constants are given in Hertz. Silica gel-G plates (Merck) were used for TLC analysis with a mixture of n-hexane and ethyl acetate as an eluent. Column chromatography was carried out in silica gel (60–120 mesh) using n-hexane, dichloro methane (DCM), methanol, and ethyl acetate as eluents. Pressure reactions were carried out using a PREMEX A-Line Avalon Instrument. Distillation of the products was carried out using a glass oven – B585 Kugelrohr (Buchi made) instrument. Electrospray Ionization Mass Spectrometry (ESI-MS) analyses were recorded in an LCQ Fleet, Thermo Fisher Instruments Limited, US. ESI-MS was performed in positive ion mode. The collision voltage and ionization voltage were −70 V and −4.5 kV, respectively, using nitrogen as atomization and desolvation gas. The desolvation temperature was set at 300 °C. The relative amount of each component was determined from the LC-MS chromatogram, using the area normalization method. GC was recorded using an Agilent GC 6890 N, with split ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. GCMS was recorded using a 5977B GC/MSD single quadrupole analyser using helium as carrier gas.
:100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. GCMS was recorded using a 5977B GC/MSD single quadrupole analyser using helium as carrier gas.
3. Experimental section
3.1 Preparation of cinchona based palladium catalyst (PdAc-5)
:4) giving a white powder. Yield 63%. Mp: 193–194 °C. 1H NMR (300 MHz, CDCl3) δ 8.67 (d, J = 3 Hz, 2H), 8.39 (d, J = 9 Hz, 2H), 7.87–7.84 (m, 2H), 4.53 (s, 4H). 13C NMR (75 MHz, CDCl3) δC 149.78, 143.67, 131.82, 128.19, 115.44, 23.74. ESI-MS (M+); 342.88.:ethyl acetate) as an eluent or by distillation using a Kugelrohr instrument. The purified product was then analysed by NMR and mass spectroscopy to confirm the product.4. Characterization of products
4.1 2-Methylbenzaldehyde (7b)
Colourless liquid. Yield: 84.0%. GC purity: 94.6%. 1H NMR (400 MHz, CDCl3) δH 10.26 (s, 1H), 7.79 (dd, J = 7.6, 1.5 Hz, 1H), 7.47 (td, J = 7.5, 1.6 Hz, 1H), 7.36 (td, J = 7.5, 1.2 Hz, 1H), 7.28–7.22 (m, 1H), 2.67 (s, 3H); 13C NMR (100 MHz, CDCl3) δC 192.73, 140.53, 134.07, 133.58, 131.98, 131.70, 126.25, 19.52.4.2 4-Methylbenzaldehyde (8b)
Colourless liquid. Yield: 89.5%. GC purity: 93.7%. 1H NMR (400 MHz, CDCl3) δH 9.96 (s, 1H), 7.77 (d, J = 8.1 Hz, 2H), 7.33 (d, J = 7.8 Hz, 2H), 2.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δC 191.94, 145.49, 134.13, 129.71, 21.83.4.3 2-Methoxybenzaldehyde (9b)
Pale yellow liquid. Yield: 80.5%. GC purity: 94.2%. 1H NMR (400 MHz, CDCl3) δH 10.45 (s, 1H), 7.80 (dd, J = 7.7, 1.8 Hz, 1H), 7.52 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.08–6.90 (m, 2H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δC 189.62, 161.75, 135.83, 128.38, 124.80, 120.55, 111.58, 55.53.4.4 4-Methoxybenzaldehyde (10b)
Pale brown oil. Yield: 86.6%. GC purity: 99.10%, 1H NMR (400 MHz, CDCl3) δH 9.88 (s, 1H), 7.88–7.86 (d, J = 8 Hz, 2H), 7.18–7.16 (d, J = 8 Hz, 2H), 3.83 (s, 3H), 13C NMR (100 MHz, CDCl3) δC 55.8, 114.8, 129.2, 132.3, 166.4, 191.0.4.5 3-Methoxy-4-hydroxybenzaldehyde (11b)
Off-white solid. Yield: 73.5%. GC purity: 99.62%, 1H NMR (400 MHz, CDCl3) δH 9.81 (s, 1H), 7.43–7.41 (d, J = 8 Hz, 1H), 7.05–7.03 (d, J = 8 Hz, 1H), 6.75 (s, 1H), 5.65 (s, 1H), 3.93 (s, 3H), 4.22 (s, 1H), 3.83 (s, 3H), 13C NMR (100 MHz, CDCl3) δC 191.16, 151.93, 147.30, 129.73, 127.58, 114.55, 108.96, 56.08.4.6 2-Fluorobenzaldehyde (12b)
Yellow liquid. Yield: 61.99%. GC purity: 98.25%. 1H NMR (400 MHz, CDCl3) δH 10.37 (d, J = 0.8 Hz, 1H), 7.88 (td, J = 7.4, 1.9 Hz, 1H), 7.61 (dddd, J = 8.4, 7.3, 5.4, 1.9 Hz, 1H), 7.33–7.09 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 187.12, 166.34, 162.92, 136.29, 128.64, 124.58, 116.45.4.7 3-Fluorobenzaldehyde (13b)
Yellow liquid. Yield: 66.58%. GC purity: 97.10%. 1H NMR (400 MHz, CDCl3) δH 10.00 (d, J = 1.9 Hz, 1H), 7.69 (dt, J = 7.5, 1.3 Hz, 1H), 7.61–7.48 (m, 2H), 7.39–7.26 (m, 1H); 13C NMR (101 MHz, CDCl3) δC 190.82, 164.69, 161.38, 138.36, 130.74, 126.00, 121.52, 115.27.4.8 4-Fluorobenzaldehyde (14b)
Yellow liquid. Yield: 76.58%. GC purity: 90.56%. 1H NMR (400 MHz, CDCl3) δH 9.98 (s, 1H), 7.92 (dd, J = 8.7, 5.4 Hz, 2H), 7.22 (t, J = 8.5 Hz, 2H); 13C NMR (101 MHz, CDCl3) δC 190.45, 168.16, 164.76, 132.91, 132.18, 116.30.4.9 2-Chlorobenzaldehyde (15b)
Light brown liquid. Yield: 69.6%. GLC purity: 92.11%. 1H NMR (400 MHz, CDCl3) δH 10.47 (d, J = 0.8 Hz, 1H), 7.91 (dd, J = 7.7, 1.8 Hz, 1H), 7.53 (ddd, J = 8.0, 7.1, 1.8 Hz, 1H), 7.48–7.32 (m, 2H). 13C NMR (100 MHz, CDCl3) δC 189.67, 137.81, 135.03, 132.32, 130.49, 129.24, 127.18.4.10 4-Chlorobenzaldehyde (16b)
Light brown solid. Yield: 78.5%. GLC purity: 97.7%. Mp: 46–50 °C (lit. 47–51 °C). 1H NMR (400 MHz, CDCl3) δH 9.98 (s, 1H), 7.82 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 190.75, 140.81, 134.61, 130.80, 129.34.4.11 2,3-Dichlorobenzaldehyde (17b)
Pale yellow solid. Yield: 82.6%. GC purity: 90.15% (decomposes upon storage), Mp: 62–65 °C (64–67 °C, lit.). 1H NMR (400 MHz, CDCl3) δH 10.47 (s, 1H), 7.84–7.82 (d, J = 8 Hz, 1H), 7.71–7.69 (d, J = 8 Hz, 1H), 7.33–7.37 (t, J = 6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δC 189.34, 136.02, 135.59, 134.33, 134.20, 127.42, 127.60.4.12 2-Nitrobenzaldehyde (18b)
Brown solid. Yield: 88%. GLC purity: 95.89%. Mp: 42–44 °C (reported: 42–44 °C); 1H NMR (400 MHz, CDCl3): δH 10.41 (s, 1H), 8.12–8.10 (m, 1H), 7.95–7.93 (m, 1H), 7.81–7.73 (m, 2H); 13C NMR (100 MHz, CDCl3): δC 188.31, 149.69, 134.22, 133.84, 131.45, 129.75, 124.62.4.13 3-Nitrobenzaldehyde (19b)
Pale yellow solid. Yield: 58.6%. GC purity: 92.64%, Mp 57.5–58.5 °C (lit. 57–58.5 °C). 1H NMR (400 MHz, CDCl3) δH 10.14 (s, 1H), 8.72 (s, 1H), 8.51–8.49 (d, J = 8 Hz, 1H), 8.27–8.25 (d, J = 8 Hz, 1H), 7.82–7.78 (t, J = 6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δC 189.83, 148.76, 137.38, 134.74, 130.44, 128.59, 124.40.4.14 4-t-Butylbenzaldehyde (20b)
Colourless liquid. Yield: 85.5%. GC purity: 90.41%, 1H NMR (400 MHz, CDCl3) δH 9.88 (s, 1H), 7.83–7.81 (d, J = 8 Hz, 2H), 7.48–7.46 (d, J = 8 Hz, 2H), 1.35 (s, 9H), 13C NMR (100 MHz, CDCl3) δC 31.3, 34.2, 125.5, 129.5, 133.8, 157.1, 193.0.4.15 2,6-Dimethylbenzaldehyde (21b)
Pale brown liquid. Yield: 78.66%. GC purity: 96.36%. 1H NMR (400 MHz, CDCl3) δH 10.62 (s, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.09 (d, J = 7.6 Hz, 2H), 2.61 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.39, 140.99, 132.85, 132.30, 129.58, 20.34.4.16 Mesitaldehyde (22b)
Colourless liquid. Yield: 80.14%. GC purity: 95.87%. 1H NMR (400 MHz, CDCl3) δH 10.53 (s, 1H), 6.87 (s, 2H), 2.56 (s, 6H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.76, 143.66, 141.31, 130.38, 129.82, 21.31, 20.33.4.17 3,5-Dibromo-2,4,6-trimethylbenzaldehyde (23b)
Colourless solid. Yield: 68.8%. GC purity: 95.45%. Mp: 162–164 °C (lit. 160 °C, decomposes); 1H NMR (CDCl3, 400 MHz) δH 10.47 (s, 1H), 2.74 (s, 3H), 2.57 (s, 6H); 13C NMR (CDCl3, 100 MHz) δC 194.0, 142.5, 137.6, 135.0, 127.7, 26.9, 20.4.4.18 1-Phenylethan-1-one (24b)/(acetophenone)
Colourless oil. Yield: 87.55%. GC purity: 99.73%. 1H NMR (400 MHz, CDCl3) δH 7.94–7.57 (m, 5H), 2.52 (s, 3H). 13C NMR (100 MHz, CDCl3) δC 197.11, 133.14, 128.80, 128.64, 26.45.4.19 Propiophenone (25b)
Colourless liquid. Yield: 82.33%. GLC purity: 98.8%. 1H NMR (400 MHz, CDCl3): δH 7.94–7.91 (m, 2H), 7.53–7.48 (m, 1H), 7.43–7.39 (m, 2H), 2.95 (q, J1 = J2 = 7.2 Hz, 2H), 1.19 (t, J1 = J2 = 2.4 Hz). 13C NMR (100 MHz, CDCl3): δC 200.79, 136.90, 132.86, 128.54, 127.95, 31.75, 8.22.4.20 Benzophenone (26b)
Colourless solid. Yield: 88.12%. GLC purity: 97.32%. Mp: 48–50 °C (reported: 47–51 °C); 1H NMR (400 MHz, CDCl3): δH 7.81–7.79 (m, 2H), 7.60–7.56 (m, 1H), 7.50–7.46 (m, 2H); 13C NMR (100 MHz, CDCl3): δC 196.89, 137.72, 132.53, 130.17, 128.39.4.21 Cyclohexanecarbaldehyde (27b)
Pale yellow liquid. Yield: 83%. GLC purity: 95.23%. 1H NMR (400 MHz, CDCl3): δH 9.59 (s, 1H), 2.24–2.19 (m, 1H), 1.90–1.20 (m, 10H); 13C NMR (100 MHz, CDCl3): δC 205.17, 50.07, 26.08, 26.02, 25.12.4.22 Cyclohexanone (28b)
Colourless liquid. Yield: 81%. 1H NMR (400 MHz, CDCl3): δH 4.49–4.39 (m, 1H), 3.33–2.90 (m, 1H), 2.41–2.12 (m, 3H), 2.11–1.61 (m, 4H); 13C NMR (100 MHz, CDCl3): δC 211.52, 42.00, 27.11, 25.07.4.23 Cyclohexane-1,4-dione (29b)
Colourless solid. Yield: 77%. Mp: 75–77 °C (reported: 77–78.5 °C); 1H NMR (400 MHz, CDCl3): δ 2.67 (s, 4H); 13C NMR (100 MHz, CDCl3): δ 208.51, 36.73.4.24 Cyclododecanone (30b)
Colourless solid. Yield: 70.6%. GC purity: 99.76%. Mp 57–59 °C (reported 58–60 °C). 1H NMR (400 MHz, CDCl3): δH 2.48–2.45 (m, 4H), 1.74–1.68 (m, 4H), 1.28–1.30 (m, 14H). 13C NMR (100 MHz, CDCl3) δC 212.88, 40.25, 24.73, 24.58, 24.20, 24.20, 22.54, 22.31.4.25 Piperonyl aldehyde (31b)
Light yellow waxy solid. Yield: 86.25%. GC purity: 96.78%. Mp: 35–39 °C (reported: 37–39 °C). 1H NMR (400 MHz, CDCl3) δH 10.30 (s, 1H), 7.16 (dd, J = 8.1, 1.7 Hz, 1H), 7.14 (d, J = 1.7 Hz, 1H), 6.62 (d, J = 8.1 Hz, 1H), 5.99 (s, 2H); 13C NMR (101 MHz, CDCl3) δC 186.81, 152.20, 151.48, 146.02, 128.14, 107.47, 105.06, 103.89.4.26 4-Acetamidobenzaldehyde (32b)
Brown solid (slight decomposition observed in storage). Yield: 78.99%. GC purity: 98.25%. Mp: 146–152 °C (reported: 148–152 °C). 1H NMR (400 MHz, CDCl3) δH 9.91 (s, 1H), 8.00 (s, 1H), 7.8–7.79 (m, 2H), 7.71 (d, J = 8.4 Hz, 2H), 2.23 (s, 3H); 13C NMR (101 MHz, CDCl3) δC 191.14, 168.86, 143.62, 132.14, 131.14, 119.22, 24.76.4.27 4-(Trifluoromethyl)benzaldehyde (33b)
Light brown liquid. Yield: 74.52%. GC purity: 92.9%. 1H NMR (400 MHz, CDCl3) δH 10.09 (s, 1H), 8.00 (dt, J = 7.9, 0.9 Hz, 2H), 7.80 (d, J = 8.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δC 191.07, 138.63, 135.79, 135.36, 129.89, 126.11, 121.60, 117.99.4.28 2,4-Bis(trifluoromethyl)benzaldehyde (34b)
Yellow liquid. Yield: 58.23%. GC purity: 87.25%. 1H NMR (400 MHz, CDCl3) δH 10.04 (d, J = 0.5 Hz, 1H), 8.06 (d, J = 1.5 Hz, 1H), 7.90 (dd, J = 7.5, 0.5 Hz, 1H), 7.72 (dd, J = 7.5, 1.5 Hz, 1H); 13C NMR (101 MHz, CDCl3) δC 189.42, 134.81, 134.80, 134.74, 133.42, 133.17, 129.59, 129.57, 129.56, 129.54, 126.95, 126.92, 126.88, 126.85, 125.22, 125.19, 125.16, 125.13, 124.91, 123.66, 122.76.4.29 4-Biphenylcarboxaldehyde (35b)
Yellow solid. Yield: 86.54%. GC purity: 98.66%. Mp: 58–62 °C (reported: 57–59 °C). 1H NMR (400 MHz, CDCl3) δH 10.06 (s, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.68–7.60 (m, 2H), 7.53–7.36 (m, 3H); 13C NMR (101 MHz, CDCl3): δC 191.84, 147.13, 139.65, 135.15, 130.21, 128.96, 128.42, 127.63, 127.3.4.30 2-Pyridinecarboxaldehyde (36b)
Brown liquid. Yield: 82.77%. GC purity: 96.35%. 1H NMR (400 MHz, CDCl3): δ 10.09 (s, 1H), 8.81 (dt, J = 4.8, 1.3 Hz, 1H), 7.98 (dt, J = 7.8, 1.2 Hz, 1H), 7.91 (td, J = 7.6, 1.6 Hz, 1H), 7.57 (ddd, J = 7.4, 4.8, 1.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δC 193.20, 152.60, 150.02, 136.89, 127.70, 121.51.4.31 4-Pyridinecarboxaldehyde (37b)
Yellow liquid. Yield: 76.25%. GC purity: 91.25%. 1H NMR (400 MHz, CDCl3) δH 9.98 (s, 1H), 8.77 (d, J = 6.0 Hz, 2H), 7.60 (d, J = 6.0 Hz, 2H); 13C NMR (101 MHz, CDCl3) δC 191.29, 150.97, 141.15, 121.85.4.32 3-Pyridinecarboxaldehyde (38b)
Light brown liquid (darkens upon storage). Yield: 68.5%. GC purity: 89.23%. 1H NMR (400 MHz, CDCl3) δH 10.04 (s, 1H), 8.99 (d, J = 2.1 Hz, 1H), 8.75 (dd, J = 4.9, 1.8 Hz, 1H), 8.08 (dt, J = 7.9, 2.0 Hz, 1H), 7.41 (dd, J = 7.9, 4.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δC 190.60, 154.55, 151.85, 135.58, 131.19, 123.87.4.33 5-Nitrofuran-2-carbaldehyde (39b)
Colourless solid. Yield: 68%. Mp: 38–40 °C (reported: 37–39 °C). 1H NMR (400 MHz, CDCl3): δH 9.82 (s, 1H), 7.41 (d, J = 4 Hz, 1H), 7.34 (d, J = 3.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 178.45, 151.09, 118.92, 111.86.4.34 1-Napthaldehyde (40b)
Light brown liquid. Yield: 86.23%. GC purity: 96.18%, 1H NMR (400 MHz, CDCl3) δH 10.38 (s, 1H), 9.26 (dd, J = 8.6, 1.1 Hz, 1H), 8.08 (dt, J = 8.2, 1.0 Hz, 1H), 8.01–7.83 (m, 2H), 7.79–7.44 (m, 3H); 13C NMR (101 MHz, CDCl3) δC 193.45, 136.60, 135.19, 133.60, 131.27, 130.41, 128.97, 128.38, 126.86, 124.77.4.35 2-Naphthaldehyde (41b)
Pale yellow solid. Yield: 83.5%. GC purity: 94.68%. Mp: 59–63 °C (reported: 59–61 °C). 1H NMR (400 MHz, CDCl3) δH 10.16 (s, 1H), 8.34 (s, 1H), 8.05–7.84 (m, 4H), 7.62 (dddd, J = 17.6, 8.2, 6.9, 1.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δC 192.23, 136.42, 134.5, 134.08, 132.61, 129.50, 129.08, 128.05, 127.07, 122.74.5. Results and discussion
Compound 2 was synthesised by the reaction between 1 and NBS in CCl4 using AIBN as a catalyst in 80% yield. The resulting 2 was reacted with cinchona alkaloid (free base) (3) to obtain the bipyridyl-cinchona ligand (4) in 96% yield.8 It was then converted into its palladium acetate derivative (PdAc-5), as shown in Scheme 1.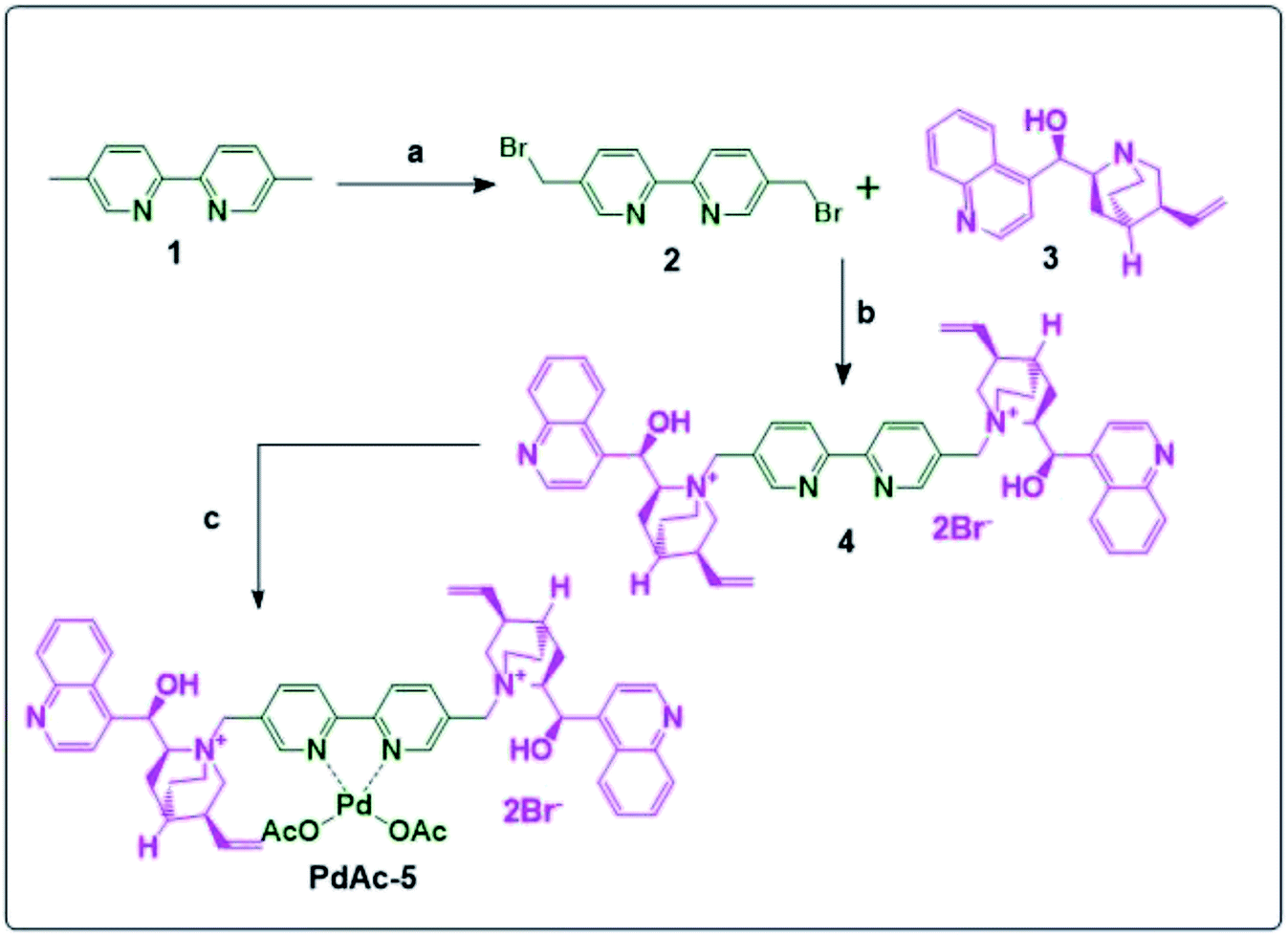 | ||
| Scheme 1 Synthesis of PdAc-5; reagents and conditions: (a): NBS, AIBN, CCl4, reflux 4 h; (b): THF, reflux, 16 h; and (c): palladium acetate, DMF, 16 h, 100 °C. | ||
The morphological evidence for PdAc-5 was confirmed by SEM and EDAX analysis, as presented in Fig. 1.9 The SEM image of PdAc-5 showed the presence of highly crystalline structures (Fig. 1a), which confirmed the incorporation of palladium metal in the catalytic system of PdAc-5. Further, the atomic weight (%) of PdAc-5 is 29.4%, measured by atomic absorption spectroscopy.10
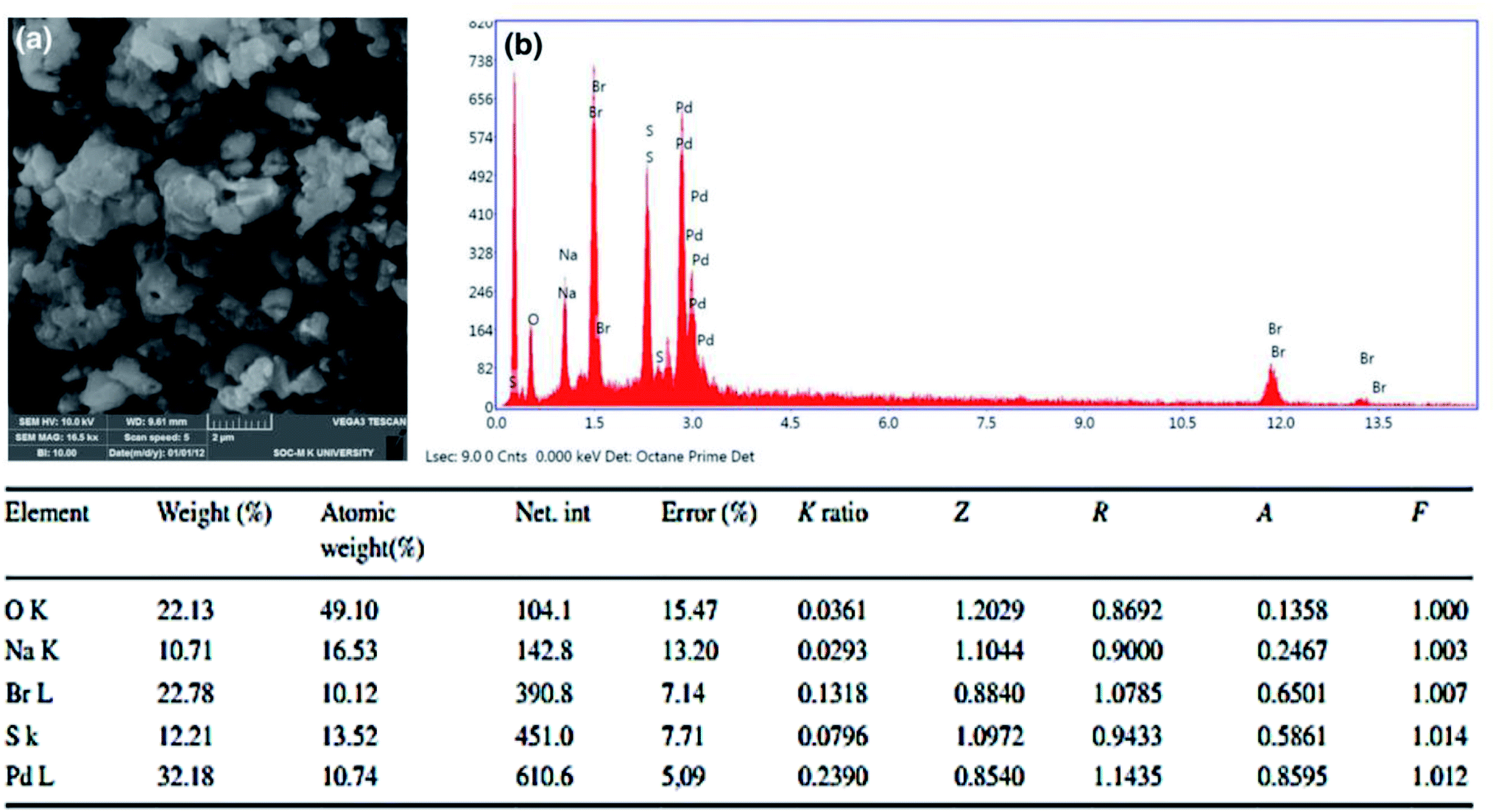 | ||
| Fig. 1 SEM (a) and EDAX (b) spectrum of PdAc-5. | ||
To investigate the oxidation state of palladium, the binding affinity between the metal and cinchonine-based bipyridine ligand was confirmed by X-ray photoelectron spectroscopy (XPS), as shown in Fig. 2. PdAc-5 showed two binding energy values that appeared at 337 and 342.42 eV. These were attributed to Pd 3d5/2 and Pd 3d3/2, which were assigned to palladium being present in +2 oxidation states. In addition to that, the free palladium acetate binding energy value that appeared at 338.4 eV for our catalyst 5 showed a 1.24 eV negative shift, which is consistent with previously reported literature. This negative shift indicates that palladium acetate coordinated with the 2,2′-bipyridine nitrogen atom. Furthermore, the N 1s spectrum (Fig. 2b) shows an asymmetric and wide peak, which was fitted with three components at 397.8, 399.3, and 401.02 eV. The components were assigned to the quinoline nitrogen, pyridinic nitrogen, and quaternary nitrogen, respectively. Generally, the reported pyridine nitrogen (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) binding energy values appear at 398.2 eV, but we observed an N 1s core level binding energy of 399.3 eV, so the pyridine unit N 1s core level of PdAc-5 is slightly positively shifted by 1.1 eV. Finally, these results clearly showed that Pd(OAc)2 was strongly coordinated with the bipyridine nitrogen atom as well as the palladium being present in +2 oxidation states. In addition to that, Pd was incorporated with the bipyridine ligand, which was easily identified by the 1H NMR and 13C NMR spectra of ligand 4 and PdAc-5. Here, ligand 4 and PdAc-5 bipyridine ligand ‘N’ adjacent to the aromatic proton appeared at 9.12–9.18 ppm, and also the acetate carbonyl and methyl protons appeared at 182.01 and 2.73 ppm in 1H NMR and 13C NMR spectra, respectively (Table S1†).11
) binding energy values appear at 398.2 eV, but we observed an N 1s core level binding energy of 399.3 eV, so the pyridine unit N 1s core level of PdAc-5 is slightly positively shifted by 1.1 eV. Finally, these results clearly showed that Pd(OAc)2 was strongly coordinated with the bipyridine nitrogen atom as well as the palladium being present in +2 oxidation states. In addition to that, Pd was incorporated with the bipyridine ligand, which was easily identified by the 1H NMR and 13C NMR spectra of ligand 4 and PdAc-5. Here, ligand 4 and PdAc-5 bipyridine ligand ‘N’ adjacent to the aromatic proton appeared at 9.12–9.18 ppm, and also the acetate carbonyl and methyl protons appeared at 182.01 and 2.73 ppm in 1H NMR and 13C NMR spectra, respectively (Table S1†).11
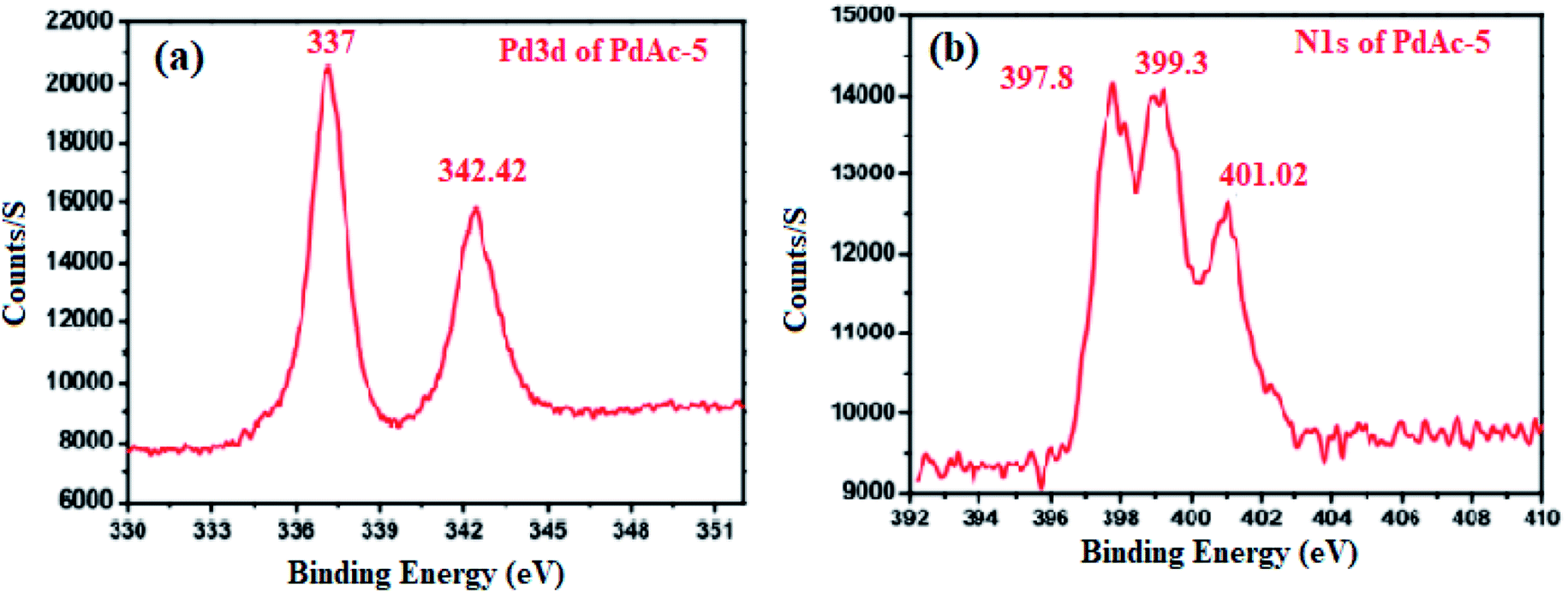 | ||
| Fig. 2 (a) X-ray photo-electron spectrum of P 3d; and (b) N 1s core level spectrum of PdAc-5. | ||
To start with, we scrutinised various oxidation condition reactions with PdAc-5 and oxygen gas using different solvents and additives.
The initial entries in Table 1 (entries 1–4) reflect the fact that there is no product formation using toluene as a solvent in the presence of various additives. The solvent was shifted from toluene to polar entities like acetonitrile, a water–acetic acid mixture and water. The conversion are very poor using acetonitrile (entries 5 and 6) as a solvent in the presence of an additive (K2CO3 or CH3COONa), but appreciable conversion was observed using a water–acetic acid mixture (1:1) and sodium acetate as an additive (entry 7). The same system was explored using a reduced catalyst which resulted in poor conversion (entry 8). The above experiment was further explored using DTBP as a co-oxidant in the system, which resulted in the formation of acid 8 in substantial quantities apart from aldehyde 7 (entries 9 and 10).12–14
|
||||
|---|---|---|---|---|
| Entry | Solvent (mL) | Additive | Conversion, % | |
| (7)b | (8) | |||
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, 8 h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C.c Di-tertiary butyl peroxide (aqueous solution: 70%).d Reduced catalyst (5 mg per gram of substrate). |
||||
| 1 | Toluene | None | NA | NA |
| 2 | Toluene | DBU (10 mg) | NA | NA |
| 3 | Toluene | K2CO3 (10 mg) | NA | NA |
| 4 | Toluene | CH3COONa (10 mg) | NA | NA |
| 5 | Acetonitrile | K2CO3 (10 mg) | 12.85% | NA |
| 6 | Acetonitrile | CH3COONa (10 mg) | 27.83% | NA |
| 7 | Water:acetic acid (1:1) |
CH3COONa (10 mg) | 79.28% | 6.5% |
| 8 | Water:acetic acid (1:1) |
CH3COONa (10 mg) | 35.70%d | NA |
| 9 | Water:acetic acid (1:1) |
CH3COONa (10 mg)/DTBPc(50 mg) | 38.87% | 12.35 |
| 10 | Water:acetic acid (1:1) |
CH3COONa (10 mg)/DTBPc(200 mg) | 35.33% | 36.22% |
In Table 2 we have evaluated the solvent system to carry out the oxidation process.
|
||||
|---|---|---|---|---|
| Entry | Solvent (mL) | Catalyst | Additive | Conversion, % (7)b |
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, 8 h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. |
||||
| 1 | Water:acetic acid (1:1) |
1 mol% | CH3COONa (5 mol%) | 79.28% |
| 2 | Water:acetonitrile (1:1) |
1 mol% | CH3COONa (5 mol%) | 74.8% |
| 3 | Water | 1 mol% | CH3COONa (5 mol%) | 15.0% |
| 4 | Toluene | 1 mol% | CH3COONa (5 mol%) | 10.46% |
| 5 | Acetic acid:toluene (1:1) |
1 mol% | CH3COONa (5 mol%) | 27.93% |
| 6 | Water:acetic acid (1:1) |
Nil | CH3COONa (5 mol%) | NA |
In this case, we found that the aqueous mixtures of acetic acid and acetonitrile (Table 2, entries 1 and 2) both gave better results than other systems like water, toluene, or an acetic acid:toluene mixture (entries 3–5), which yielded very poor conversions. The system was conducted in a water–acetic acid mixture without a catalyst, which yielded no conversion of the substrate (entry 6). On the account of reactor stability, we opted to use a water–acetonitrile mixture as the solvent for further screening experiments rather than a water–acetic acid mixture (acetic acid corrodes the rubber gasket of the mini-clave at higher temperatures (pH < 3)).15–18
The screening process studied the influence of various additives in our oxidation system. From Table 3, it is evident that additives like sodium acetate (entry 1) or potassium carbonate (entry 4) show better conversion than the other bases. Potassium carbonate is preferred over the former because of its alkaline pH (9–10), which ensures the stability of the reaction mass during the process. From the above and Table 3 we concluded that potassium carbonate in a water–acetonitrile mixture gave better conversion than other systems. On this accord, we also screened the above system with different base quantities (Table 4) and aqueous solvent mixtures (Table 5) which are shown below.17,18
|
|||
|---|---|---|---|
| Entry | Solvent (mL) | Additive (5 mol%) | Conversion, (7)b |
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, 8 h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. |
|||
| 1 | Water:acetonitrile (1:1) |
NaOAC | 74.8% |
| 2 | Water:acetonitrile (1:1) |
NaOH | 40.0% |
| 3 | Water:acetonitrile (1:1) |
Na2CO3 | 67.1% |
| 4 | Water:acetonitrile (1:1) |
K2CO3 | 83.09% |
| 5 | Water:acetonitrile (1:1) |
DBU | 26.76% |
| 6 | Water:acetonitrile (1:1) |
Triethanolamine | 44.32% |
|
||||
|---|---|---|---|---|
| Entry | Solvent system (mL) | Catalyst | Additive | Conversion, (7)b |
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. |
||||
| 1 | Water:acetonitrile (1:1) |
2 mol% | K2CO3 (5 mol%) | 86.1% |
| 2 | Water:acetonitrile (1:1) |
1 mol% | K2CO3 (5 mol%) | 83.09% |
| 3 | Water:acetonitrile (1:1) |
0.5 mol% | K2CO3 (5 mol%) | 65.00% |
| 4 | Water:acetonitrile (1:1) |
1 mol% | K2CO3 (10 mol%) | 84.00% |
| 5 | Water:acetonitrile (1:1) |
— | K2CO3 (10 mol%) | No reaction |

|
||
|---|---|---|
| Entry | Solvent mixture | Conversion, (7)b |
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. |
||
| 1 | Water:acetonitrile (1:1) |
83.09% |
| 2 | Water | 12.08% |
| 3 | Water:THF (1:1) |
24.98% |
| 4 | Water:DMF (1:1) |
17.82% |
| 5 | Water:acetone (1:1) |
47.10% |

From Table 4 it is evident that a higher quantity of base (Table 4, entry 4) does not affect the conversion to a great extent. The same holds for an increase in the quantity of catalyst for the above reaction (entry 1). But conversion does drop with a lower catalyst loading (entry 3) which may be completed in a longer time period than usual.
From Table 5, we concluded that water–acetonitrile (Table 5, entry 1) works better than the other solvent mixtures (entries 2–4). The conversion is moderate in an acetone–water system (entry 5), but the rate of conversion is quite slow compared with a water–acetonitrile system. Last but not least, the reaction was also screened for its conversion rates at different temperatures as tabulated in Table 6.
|
||
|---|---|---|
| Entry | Temperature (°C) | Conversion, (7)b |
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, 8 h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. |
||
| 1 | 25 | 4.08% |
| 2 | 40 | 6.60% |
| 3 | 60 | 31.30% |
| 4 | 80 | 83.09% |
| 5 | 125 | 75.90% |

As shown in Table 6, conversion is poor at lower temperatures (entries 1–3), and better conversion is observed in the temperature range of 80–90 °C (entry 4). Also, experiments were performed at much higher temperatures at which product decomposition was observed, giving rise to impurities and thus lowering the product ratio in the mass (entry 5). This may be due to the decomposition of PdAc-5 along with the product.19
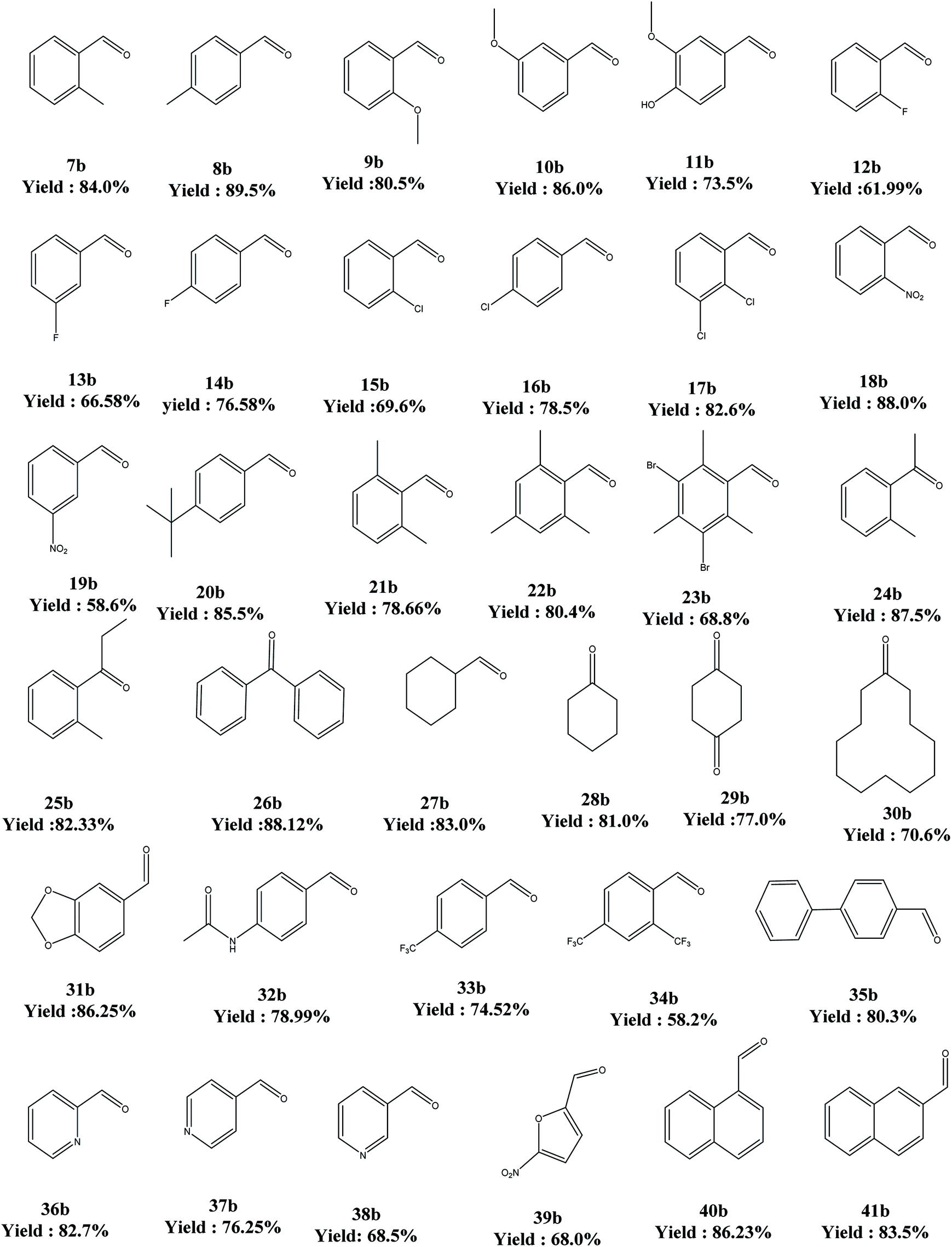
The above said catalyst was tried for different substrates for the oxidation reaction. In this regard, electron-donor substituents offered better yields but electron-withdrawing substituents ended up in poor conversions. As far as the secondary alcohols are concerned, the yields are better than for their primary counterparts (Table 7).
|
|---|
| a Reaction conditions: 4-butyl benzyl alcohol (6) (6.0 mmol), solvent (10 mL), PdAc-5 (1 mol%), additive (5 mol%), oxygen (5 bar), 80 °C, 8 h.b Agilent GLC-GCMS: 6890 N, split ratio: 1:100, 250 °C, FID: 300 °C, flow rate: nitrogen 30 mL min−1. Column: HP-5; 30 m × d: 0.32 mm × f: 0.25 mm, −60 to 325 °C. Yield of the product after column purification/distillation. All the products are matched against their reference standards. |
 |

6. Catalyst recycling
To check the catalyst performance, the catalyst was recovered from the reaction mass, filtered and dried at 70 °C in a glass oven under vacuum. The dried catalyst was reintroduced into the new reaction system and then processed for the catalyst performance. The yields are quite similar for initial the 5 runs with a catalyst recovery of 90% per run (Fig. 3).20,21 | ||
| Fig. 3 Yields for catalyst recycling. | ||
7. Scale-up study
The performance of the above-prepared catalyst on a commercial scale was checked using the oxidation of carveol (5-isopropenyl-2-methyl-cyclohexanol) to carvone (5-isopropenyl-2-methyl-cyclohexanone). As we know, there is a huge demand for synthetic carvone in the commercial market. The oxidation of carveol to carvone is currently performed using Oppenauer oxidation employing an aluminium isopropoxide and 2-butanone system on an industrial scale. We performed oxidation using PdAc-5 under pressurised conditions using an acetonitrile–water system with potassium carbonate as base (Fig. 4). The isolated yield was 74.5% with 98.25% purity. This can be further enhanced using process engineering and process chemistry techniques.22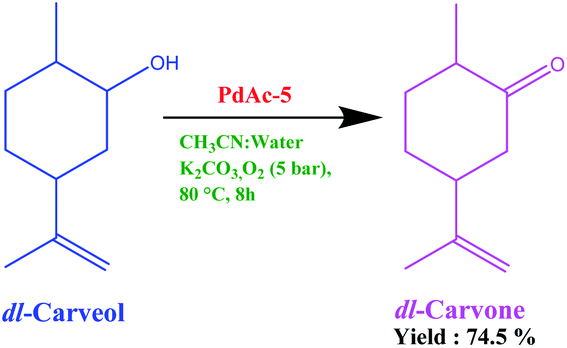 | ||
| Fig. 4 Oxidation of carveol to carvone using PdAc-5. | ||
8. Reaction mechanism
Based on the observed results, we proposed a suitable mechanism. The alcohol binds to the catalyst through oxidative addition, where it undergoes deprotonation followed by beta hydride elimination to yield the corresponding aldehyde/ketone. Thereafter, the catalyst undergoes a reductive elimination followed by redox transfer where it is regenerated for the next cycle (Scheme 2).23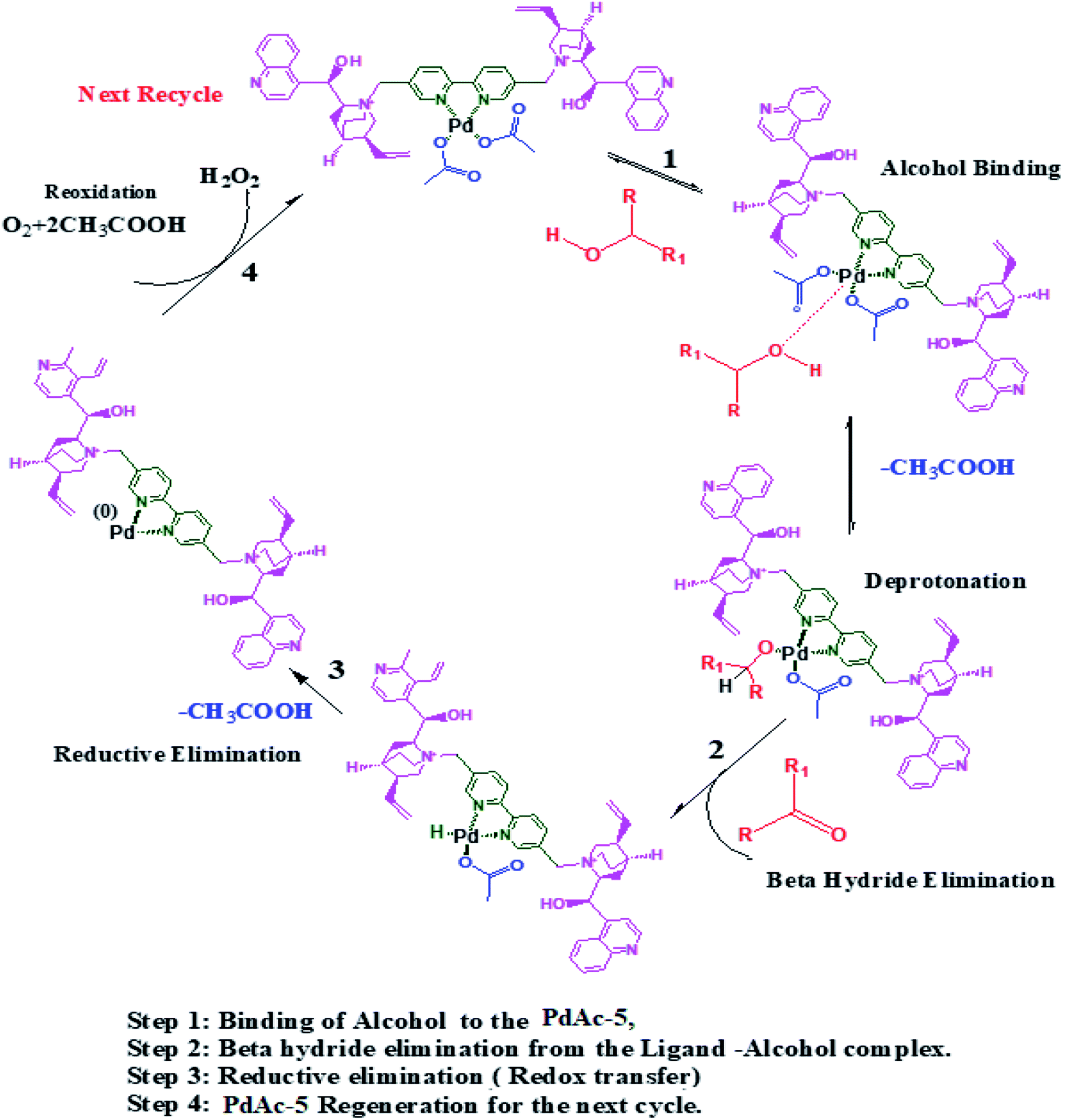 | ||
| Scheme 2 Plausible mechanism for the oxidation of alcohols using PdAc-5. | ||
9. Conclusions
In summary, the organo-palladium catalytic system of PdAc-5 has transformed primary and secondary alcohols into valuable aldehydes and ketones of industrial importance under an oxygen atmosphere with considerable yields. Despite our effort to achieve better results, some of the substrates were not good enough, and there is still much potential for the continued development of these reactions. The obtained products are quite appreciable in both yield and selectivity (70–85%). Concerning environmental and economic issues, the introduction of non-toxic, cheap, and at the same time efficient and universal catalyst systems, able to operate under mild reaction conditions in a highly selective manner and for a broad range of substrates, remains a challenge for future research.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) No. 2017R1A2B2011730, No. 2018R1A6A1A03024509 and 2019R1A2C1011113.References
- (a) R. A. Sheldon and J. K. Kochi, Metal Catalysed Oxidations of Organic Compound, Academic Press, New York, 1981 Search PubMed; (b) J. Mancuso and D. Swern, Synthesis, 1981, 1981(3), 165–185 CrossRef; (c) K. Bowden, I. M. Heilbron, E. R. H. Jones and B. C. L. Weedon, J. Chem. Soc., 1946, 3, 39–45 RSC; (d) Oxidation: Selectivity, Strategy & Efficiency in Modern Organic Chemistry, ed. B. M. Trost, Elsevier, 1992 Search PubMed; (e) Oxidation of Alcohols to Aldehydes and Ketones, A Guide to Current Common Practice, ed. G. Tojo and M. Fernández, Springer Science & Business Media, Inc., 2006 Search PubMed; (f) J. E. Backvall, Modern oxidation methods, Wiley-VCH, Weinheim, Germany, 2nd edn, 2010 CrossRef.
- (a) D. Daniel and C. Fabrizio, Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry, 2014, vol. 24 Search PubMed; (b) K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 14(26), 3854–3867 RSC; (c) J. Liqun and L. Aiwen, Sci. China: Chem., 2012, 55, 2027 Search PubMed.
- (a) S. D. Pike and A. S. Weller, Philos. Trans. R. Soc., A, 2018, 373, 2037 Search PubMed; (b) C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 13, 42 Search PubMed; (c) N. R. Conley, L. A. Labios, D. M. Pearson, C. C. L. McCrory and R. M. Waymouth, Organometallics, 2007, 26, 5447 CrossRef CAS; (d) V. R. Gangwal, B. G. M. Van, W. Kuster and J. C. Schouten, Chem. Eng. Sci., 2002, 57, 5051 CrossRef CAS; (e) D. C. Ebner, R. M. Trend, C. Genet, M. J. McGrath, P. O'Brien and B. M. Stoltz, Angew. Chem., Int. Ed., 2008, 47, 6367 CrossRef CAS PubMed.
- (a) I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507 CrossRef CAS PubMed; (b) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed; (c) P. Lahtinen, J. U. Ahmad, E. Lankinen, P. Pihko, M. Leskelä and T. Repo, J. Mol. Catal. A: Chem., 2007, 275, 228 CrossRef CAS; (d) C. Keresszegi, T. Mallat and A. Baiker, New J. Chem., 2001, 25, 1163 RSC; (e) C. Keresszegi, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 2002, 211, 244 CrossRef CAS; (f) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC; (g) N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2016, 6, 2664 CrossRef CAS PubMed; (h) J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247 CrossRef CAS.
- (a) D. Könning, F. D. Sypaseuth, C. C. Tzschucke and M. Christmann, Chem. Commun., 2014, 50, 5014 RSC; (b) G. Saravanan, N. Helfried, G. Dirk, T. Kerstin, J. Haijun and B. Matthias, Chem.–Eur. J., 2013, 19, 15979 CrossRef PubMed; (c) S. Abedi and A. Morsali, New J. Chem., 2017, 41, 5846 RSC; (d) Y. Li, J. Huang, X. Hu, F. Leung-YukLam, W. Wang and R. Luque, J. Mol. Catal. A: Chem., 2016, 425, 61–67 CrossRef CAS; (e) J. Muzart, Tetrahedron, 2003, 59, 5789 CrossRef CAS; (f) S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC; (g) G. Mayan and R. Neumann, Catal. Lett., 2008, 123, 41 CrossRef; (h) K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4353 CrossRef CAS PubMed; (i) R. DiCosimo and G. M. Whitesides, J. Phys. Chem., 1989, 93, 768 CrossRef CAS; (j) T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 196 CrossRef CAS PubMed.
- (a) B. Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917 CrossRef CAS; (b) B. A. Steinhoff and S. S. Stahl, Org. Lett., 2002, 4, 4179 CrossRef CAS PubMed; (c) B. A. Steinhoff, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2004, 126, 11268 CrossRef CAS PubMed; (d) R. J. Nielsen and W. A. Goddard III, J. Am. Chem. Soc., 2006, 128, 9651 CrossRef CAS PubMed; (e) H. Wu, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2005, 347, 1356 CrossRef CAS.
- (a) C. Xu, C. Zhang, H. Li, X. Zhao, L. Song and X. Li, Catal. Surv. Asia, 2016, 20, 13 CrossRef CAS; (b) S. Zavahir, Q. Xiao, S. Sarina, J. Zhao, S. Bottle, M. Wellard, J. Jia, L. Jing, Y. Huang, J. P. Blinco, H. Wu and H. Y. Zhu, ACS Catal., 2016, 6, 3580 CrossRef CAS; (c) P. A. Shapley, N. Zhang, J. L. Allen, D. H. Pool and H. C. Liang, J. Am. Chem. Soc., 2000, 122, 1079 CrossRef CAS; (d) T. Nishimura, T. Onoue, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 6750 CrossRef CAS PubMed; (e) T. S. T. Komanoya, Catal. Commun., 2009, 10, 1095 CrossRef; (f) N. Kakiuchi, Y. Maeda, T. Nishimura and S. Uemura, J. Org. Chem., 2001, 66, 6620 CrossRef CAS PubMed.
- M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Organomet. Chem., 2015, 63, 91–174 CrossRef CAS.
- R. A. Fernandes, A. K. Jha and P. Kumar, Catal. Sci. Technol., 2020, 10, 7448–7470 RSC.
- (a) B. Z. Zhan and A. Thompson, Arabian J. Chem., 2004, 60, 2917 CAS; (b) B. A. Steinhoff and S. S. Stahl, Org. Lett., 2002, 4, 4179 CrossRef CAS PubMed; (c) B. A. Steinhoff, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2004, 126, 11268 CrossRef CAS PubMed; (d) R. J. Nielsen and W. A. Goddard III, J. Am. Chem. Soc., 2006, 128, 9651 CrossRef CAS PubMed; (e) H. Wu, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2005, 347, 1356 CrossRef CAS.
- (a) G. J. Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636 CrossRef PubMed; (b) G. J. Brink, I. W. C. E. Arends and R. A. Sheldon, Adv. Synth. Catal., 2002, 344, 355 CrossRef; (c) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed.
- J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247 CrossRef CAS.
- G. Saravanan, N. Helfried, G. Dirk, T. Kerstin, J. Haijun and B. Matthias, Chem.–Eur. J., 2013, 19, 15979 CrossRef PubMed.
- N. Decharin, V. P. Brian and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 13268 CrossRef CAS PubMed.
- (a) M. S. Kwon, N. Kim, C. M. Park, J. S. Lee, K. Y. Kang and J. Park, Org. Lett., 2005, 7, 1077–1079 CrossRef CAS PubMed; (b) M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala and J. Park, Angew. Chem., Int. Ed., 2005, 44, 6913–6915 CrossRef CAS PubMed; (c) M. S. Kwon, S. Kim, S. Park, W. J. Bosco, R. K. Cheedrala and J. Park, J. Org. Chem., 2009, 74, 2877–2879 CrossRef CAS PubMed.
- (a) D. Daniel and C. Fabrizio, Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry, 2014, 24, p. 2005 Search PubMed; (b) K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 14, 3854 RSC; (c) J. Liqun and L. Aiwen, Sci. China: Chem., 2012, 55, 2027 Search PubMed.
- (a) P. K. Vijaya, S. Murugesan and A. Siva, Tetrahedron Lett., 2015, 56, 5209 CrossRef; (b) A. Siva, P. K. Vijaya, M. Mariyappan, V. Ashokkumar, V. Sadhasivam, S. Balakrishnan, C. Chinnadurai and S. Murugesan, New J. Chem., 2017, 41, 7980–7986 RSC; (c) C. Chinnadurai, P. K. Vijaya, M. Harikrishnan, N. Malini, M. Sepperumal and A. Siva, Res. Chem. Intermed., 2021, 47, 895–909 CrossRef.
- R. R. Chidambaram, V. Sadhasivam, M. Mariyappan and A. Siva, J. Iran. Chem. Soc., 2019, 16, 373–384 CrossRef CAS.
- D.-W. Tan, J.-B. Xie, Q. Li, H.-X. Li, J.-C. Li, H.-Y. Li and J.-P. Lang, Dalton Trans., 2014, 43, 14061–14071 RSC.
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC.
- G. Zhang, X. Xie, Y. Wang, X. Wen, Y. Zhao and C. Ding, Org. Biomol. Chem., 2013, 11, 2947–2950 RSC.
- A. M. Thomas, J. Peter, S. Nagappan, A. Mohan and C.-S. Ha, Nanomaterials, 2020, 10, 2051 CrossRef CAS PubMed.
- G. Zhang, X. Wen, Y. Wang, X. Han, Y. Luan, L. Zheng, C. Ding and X. Cao, RSC Adv., 2013, 3, 22918–22921 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05855j |
| This journal is © The Royal Society of Chemistry 2021 |