
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of rapid and selective epoxidation of α-pinene using single-step addition of H2O2 in an organic solvent-free process
Mohamad Faiz Mukhtar Gunam Resulab,
Abdul Rehman *ac,
Ana María López Fernándeza,
Valentine C. Ezea and
Adam P. Harveya
*ac,
Ana María López Fernándeza,
Valentine C. Ezea and
Adam P. Harveya
aSchool of Engineering, Newcastle University, Newcastle upon Tyne NE1 7RU, UK
bDepartment of Chemical and Environmental Engineering, Faculty of Engineering, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
cDepartment of Chemical and Polymer Engineering, University of Engineering and Technology Lahore, Faisalabad Campus, Pakistan. E-mail: a.rehman2@uet.edu.pk
First published on 7th October 2021
Abstract
This study reports substantial improvement in the process for oxidising α-pinene, using environmentally friendly H2O2 at high atom economy (∼93%) and selectivity to α-pinene oxide (100%). The epoxidation of α-pinene with H2O2 was catalysed by tungsten-based polyoxometalates without any solvent. The variables in the screening parameters were temperatures (30–70 °C), oxidant amount (100–200 mol%), acid concentrations (0.02–0.09 M) and solvent types (i.e., 1,2-dichloroethane, toluene, p-cymene and acetonitrile). Screening the process parameters revealed that almost 100% selective epoxidation of α-pinene to α-pinene oxide was possible with negligible side product formation within a short reaction time (∼20 min), using process conditions of a 50 °C temperature in the absence of solvent and α-pinene/H2O2/catalyst molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.01. A kinetic investigation showed that the reaction was first-order for α-pinene and catalyst concentration, and a fractional order (∼0.5) for H2O2 concentration. The activation energy (Ea) for the epoxidation of α-pinene was ∼35 kJ mol−1. The advantages of the epoxidation reported here are that the reaction could be performed isothermally in an organic solvent-free environment to enhance the reaction rate, achieving nearly 100% selectivity to α-pinene oxide.
:1:0.01. A kinetic investigation showed that the reaction was first-order for α-pinene and catalyst concentration, and a fractional order (∼0.5) for H2O2 concentration. The activation energy (Ea) for the epoxidation of α-pinene was ∼35 kJ mol−1. The advantages of the epoxidation reported here are that the reaction could be performed isothermally in an organic solvent-free environment to enhance the reaction rate, achieving nearly 100% selectivity to α-pinene oxide.
1 Introduction
Waste biomass-derived terpenes such as α-pinene are important sources of flavours, fragrances and pharmaceutical applications.1–7 α-Pinene is a central component of turpentine oil, a useful by-product of wood and paper industries. The sustainable utilisation of waste biomass-derived α-pinene is beneficial, as it decreases waste and maintains CO2 neutrality.8 The oxidation of α-pinene generates many valuable products (Fig. 1), such as α-pinene oxide, verbenol and verbenone,9,10 the last of which is an important intermediate in many processes and has many applications.11 The isomerisation of α-pinene oxide into products for instance campholenic aldehyde is useful for the production of fragrances.12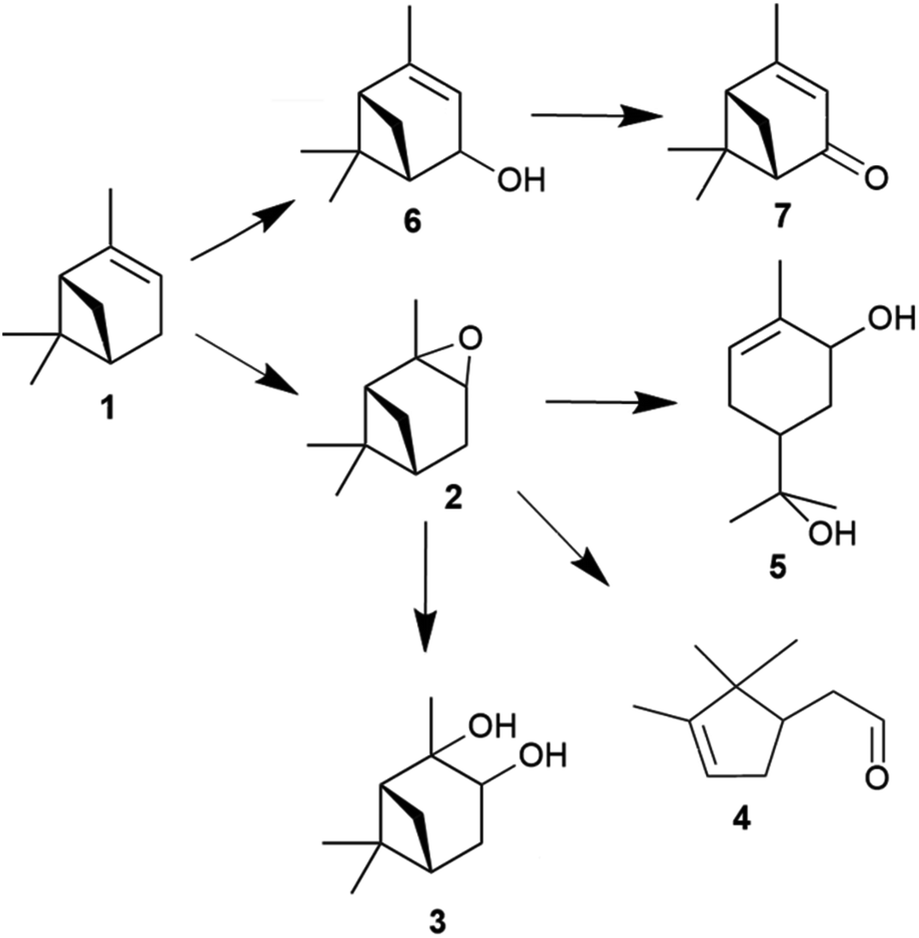 | ||
| Fig. 1 Products obtained from the epoxidation of α-pinene with hydrogen peroxide (H2O2) catalysed by tungsten-based polyoxometalates; α-pinene 1, α-pinene oxide 2, pinanediol 3, campholenic aldehyde 4, sobrerol 5, verbenol 6 and verbenone 7. | ||
The epoxidation of α-pinene (1) with H2O2 using tungsten-based polyoxometalates produced α-pinene oxide (2) as the main product, as the oxidant is attracted to the more-substituted π bonds. In the presence of H+ and H2O, the α-pinene oxide can undergo hydrolytic decomposition to form pinanediol (3) and sobrerol (5). α-Pinene oxide can also rearrange to form campholenic aldehyde (4) and experience allylic oxidation to produce verbenol (6), which can be further transformed to verbenone (7) by oxidative dehydrogenation. The dominance of epoxidation or allylic oxidation is due to the nature of the catalyst and the generation of radicals during the reaction.13 In principle, an active catalyst could also abstract H atoms from more than one position, and various hydroperoxides could be formed as intermediates, such as verbenyl hydroperoxides.14,15 In addition, α-pinene could isomerise to form many products, including β-pinene, limonene, terpineol, camphene and 3-carene.16–18 Many products, such as pinocarveol, isopinocamphone and trans-carveol, have been formed from the rearrangement process of α-pinene oxide.12,19 Moreover, polymeric compounds can also be formed during the oxidation process.
In recent decades, there is a lot of research in the epoxidation of α-pinene with hydrogen peroxide (H2O2).20–30 H2O2 is favoured over many types of oxidants, as it only yields H2O as a by-product.31,32 Developing a selective process for the epoxidation of α-pinene is challenging due to the tendency of α-pinene and its products to undergo rearrangement, hydrolysis, double bond migration and over-oxidation.33,34 The exothermic reaction usually necessitates the use of an excessive solvent or the drop-wise inclusion of an oxidant to mitigate the exotherm. This increases the reaction time, which in turn increases reactor size and consequently the capital and running costs. Among the several types of catalysts used for this reaction, a polytungstophosphate is effective with H2O2.35–39
In our previous work, we have developed a 100% selective method of limonene epoxidation with H2O2 in an organic solvent-free condition.40 In this work, we studied the possibility of developing a highly selective process using a more challenging terpene i.e., α-pinene. Hence, this work aimed to improve/intensify the α-pinene epoxidation process by removing the need for an organic solvent and to develop a process with 100% selectivity towards α-pinene oxide. This work explores the various conditions that affect the formation of many side products in α-pinene epoxidation. A kinetic study was performed to formulate an analytical kinetic model for the α-pinene epoxidation. The possibility of reducing reaction time while maintaining an isothermal condition was also studied.
2 Experimental
2.1 Materials
Following reagents were utilised in this study: tungstic acid sodium salt dihydrate (Na2WO4·2H2O), hydrogen peroxide (H2O2) (30% wt in H2O), phosphoric acid (H3PO4) 42.5%, sulphuric acid (H2SO4) 48.5%, 0.02 M potassium permanganate (KMNO4) solution (0.1 N), anhydrous sodium sulphate (Na2SO4, ≥99%), anhydrous sodium chloride (NaCl, ≥99%), α-pinene (98%), p-cymene (99%), acetonitrile (99%), toluene (99%), 1,2-dichloroethane (99%), Adogen 464® phase transfer catalyst. α-Pinene oxide (97%), pinanediol (99%), trans-sobrerol (99%), campholenic aldehyde (≥96%), verbenol (95%) and verbenone (≥99%) were used for product identification. All reagents were purchased from Merck, UK.2.2 Preparation of the oxidant
The polytungstophosphate catalyst ({PO4[WO(O2)2]43–) was prepared following an existing procedure.41 A tungsten source, Na2WO4·2H2O (0.4 g), was first added to a stirred H2O2 (30% wt, 122 mmol) solution at the desired temperature, typically 50 °C. A phosphate source, H3PO4 (42.5%, 0.06 mmol), was then added to the well-mixed solution, and the pH of the solution was adjusted to <1.0 with H2SO4 (48.5%, 4 mmol). A phase transfer catalyst, Na2SO4 (5.2 g), was later added and continuously stirred until fully dissolved. The addition of a saturated amount of Na2SO4 for the epoxidation of terpenes with H2O2 has been investigated in our previous work and was found to significantly inhibit hydrolysis of epoxides and thus increasing its selectivity.402.3 Epoxidation of α-pinene
A typical procedure used in this study for the epoxidation reaction is as follows: α-pinene (122 mmol) and Adogen 464® (1 g) as a phase transfer catalyst, were first added to a jacketed 150 mL flask. Solvent (100–500 mol%) was added to achieve isothermal conditions. The mixture was stirred vigorously (1250 rpm) at the desired temperature (30–70 °C). Temperature control was provided by a water bath (VWR MX7LR-20) and a thermocouple. The prepared oxidant (12.5 mL) was later added to the jacketed flask and allowed to react. About 1 mL of samples were collected by the reaction at selected periods of 5–120 min. The samples were analysed for α-pinene oxide content using a gas chromatography method. For purification, the organic phase was isolated from the aqueous phase using a separating funnel after the reaction was completed. To destabilise the phase transfer catalyst, 5 mL of 0.1 M NaCl was added with vigorous shaking before the phases were separated. The bulk organic layer obtained after the completion of the reaction was dried with anhydrous Na2SO4, and the unreacted α-pinene was separated from α-pinene oxide using a rotary evaporator coupled with a silicone oil bath at 150 °C and a reduced pressure of less than 50 mbar. The purity of the α-pinene oxide achieved after removal of unreacted α-pinene was ≥98%. All reactions performed were ensured to be conducted similarly with very minimal variations between the reactions. The same set of apparatus, heaters and stirrers were used to minimise the probability of other influencing factors.2.4 Kinetics of epoxidation of α-pinene and effects of reaction parameters
In this study, the kinetic parameters and reaction order was determined using an initial-rate method on the basis of pseudo-first-order conditions. The rate laws for all the reactions considered are shown in eqn (1)–(3), rdecomp, roxidative and repox are the rates of reaction for the H2O2 decomposition, the formation of oxidative species and α-pinene epoxidation, respectively, while kdecomp, koxidative and kepox are the rate constants for each rate law. The reaction order for the catalyst was studied using Na2WO4·2H2O concentrations in the range of 0.008–0.016 M, and fixed initial concentrations of α-pinene (1.25 M), H2O2 (1.25 M) and 500 mol% solvent (toluene). The study was performed using a lower concentration of catalyst (≤0.016 M), since higher metal loading is known to cause H2O2 decomposition.42 The reaction order for α-pinene was determined by varying the initial concentration of α-pinene from 0.25–1.25 M, while keeping the concentrations of H2O2 (1.25 M) and Na2WO4 (0.012 M) constant and in excess. Similarly, the reaction order for H2O2 was established by changing the initial concentration of H2O2 from 0.25–1.25 M, with the initial concentration of α-pinene (1.25 M) and Na2WO4 (0.012 M) being kept constant and in excess.| rdecomp = kdecomp[H2O2] | (1) |
| roxidative = koxidative[H+]([8H2O2][4WO42−][PO43−][3Q] − Keq−1[Q3POM][7H2O]) | (2) |
| repox = kepox[pinene][Q3POM] | (3) |
Effects of temperatures, oxidant and H+ concentrations, and solvent types on the epoxidation of α-pinene were also studied. The reaction temperatures in the range of 30–70 °C were studied, whereas the effect of oxidant amount on the α-pinene epoxidation was carried out by changing the quantity of H2O2 from 100–200 mol% for α-pinene. The effects of H+ on the epoxidation of α-pinene was investigated by varying the concentration of H2SO4 from 0.04–0.09 M, with the initial pH ≤ 1 in all cases as measured using a pH meter. The tungsten-based polyoxometalates were reported to be stable in an acidic environment.34,35,41,43,44 However, an acid-labile epoxide such as α-pinene oxide could undergo some competitive reactions in such an environment, hence the need for further investigations. The exothermic nature of the α-pinene epoxidation reaction with H2O2 is typically mitigated by the drop-wise addition of H2O2 and after adding an organic solvent. To mitigate the exothermic nature of the α-pinene epoxidation reaction with H2O2, a selection of organic solvents: 1,2-dichloroethane, toluene, p-cymene and acetonitrile were investigated for this application in the ranges of 100–500 mol% based on the α-pinene.
2.5 Product analysis (gas chromatography)
The product was analysed using a 5890 Hewlett Packard Series II gas chromatography (GC) provided with a CP Wax Capillary column (BPX70). About 40 μL of each sample was collected into 2 mL vials and mixed with 1.96 mL of chloroform. Naphthalene (10 mg) was used as an internal standard. About 0.5 μL of the product collected from the reaction was injected into the GC by an auto-sampler. The GC oven temperature was set as: an initial temperature of 80 °C was maintained for 4 min. The temperature was then ramped up by 15 °C min−1 to 260 °C, which was maintained for 10 min. The total running time was approximately 26 min per sample. The GC injector and flame ionisation detector (FID) temperatures were both set at 250 °C. The reaction conversions and yields were determined by GC analysis, following internal standard response factors obtained from calibration data using the commercial analytical grade α-pinene, α-pinene oxide, pinanediol, trans-sobrerol, campholenic aldehyde, verbenol and verbenone. The response factors were obtained by the plots of ratios of the GC peak areas of each analyte to the internal standard compared to mass ratios of the analyte. The GC analysis for all the analytes showed less than ∼2% error for the mean values taken from parallel experiments.The α-pinene conversion was determined using the expression in eqn (4), where C0 is the initial concentration of α-pinene, and Ci is the concentration of α-pinene in the sample. When H2O2 is the limiting reactant, the concentration of H2O2 and its conversion were determined by titration with 0.02 M potassium permanganate solution. The product yields were calculated using eqn (5), where Xi is the concentration of specific products in the sample as determined by GC.
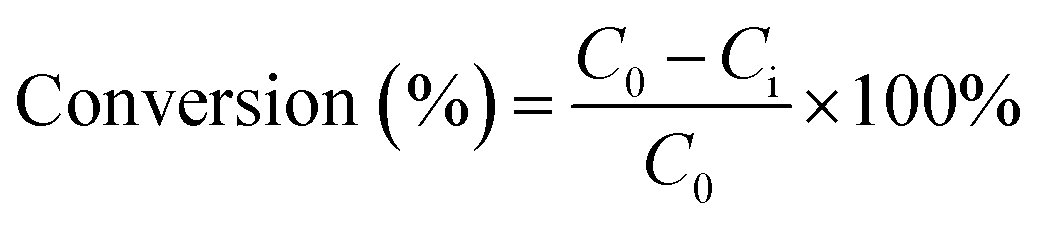 | (4) |
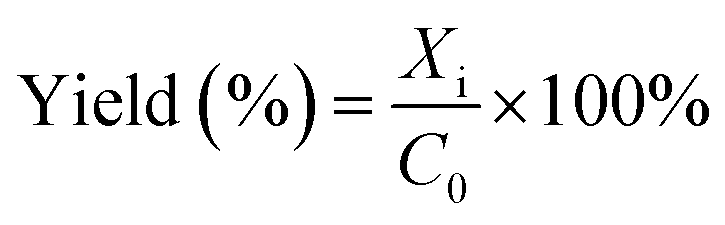 | (5) |
3 Results and discussion
3.1 Kinetics study
Fig. 2 shows the results for the investigations of the reaction orders with respect to the catalyst, α-pinene and H2O2. The natural log graph between the initial rates and the catalyst concentrations gives a straight line having a gradient of ∼1, showing that the first order in terms of catalyst concentration (Fig. 2A). Similarly, the gradient obtained from the natural log graph between the initial rates and the α-pinene concentration confirms a first-order reaction (Fig. 2B). Fig. 2C shows that the gradient for the natural log graph between the initial rates and the H2O2 concentration was ∼0.5, which indicates a stepwise process where H2O2 will form the peroxo species with the catalyst precursor, tungstate (WO42−) and phosphate (PO43−). The peroxo species in turn oxidise the double bonds to form epoxides. Therefore, according to eqn (2), increasing the concentration of H2O2 increases the formation of the active peroxo species, but this is limited by the concentration of WO42− and PO43−. Since the concentration of both WO42− and PO43− is constant, increasing the amount of H2O2 does not cause a first-order kinetic reaction.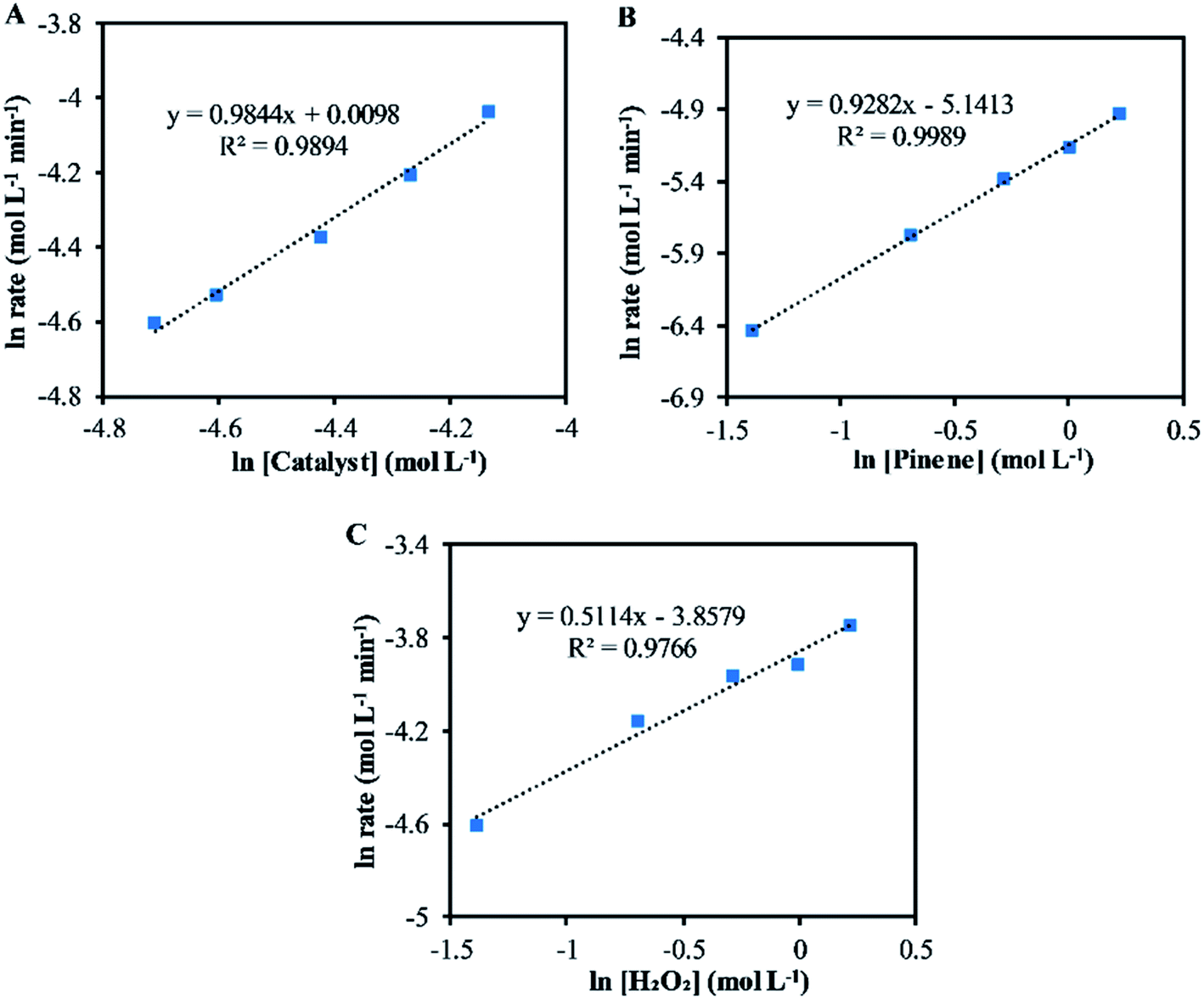 | ||
| Fig. 2 The plot of the natural log between the initial reaction rate and (A) catalyst concentration (B) α-pinene concentration (C) H2O2 concentration. | ||
3.2 Effect of temperature
Fig. 3 shows the effects of reaction on the epoxidation of α-pinene. The rates of conversions of α-pinene increased with temperatures from 30–70 °C, as shown in Fig. 3A. This trend was also followed by the α-pinene oxide yields, except at 70 °C, where a reduction in the α-pinene oxide yield was observed after 60 min reaction time. The reduction in the yield was due to the α-pinene oxide undergoing both hydrolytic decomposition and rearrangement, resulting in the formation of side products: mainly sobrerol (∼3%) and campholenic aldehyde (∼1%). Allylic oxidation becomes significant at temperatures higher than 60 °C, where the yield of verbenol is ∼5% (negligible verbenone). While higher temperatures increase the rate of α-pinene conversion, they reduce selectivity to α-pinene oxide. At temperatures below 50 °C, the process was highly selective to α-pinene oxide, with little to no formation of any side products. Hence this temperature was used in all subsequent experiments, as the highest temperature at which no side products were detected.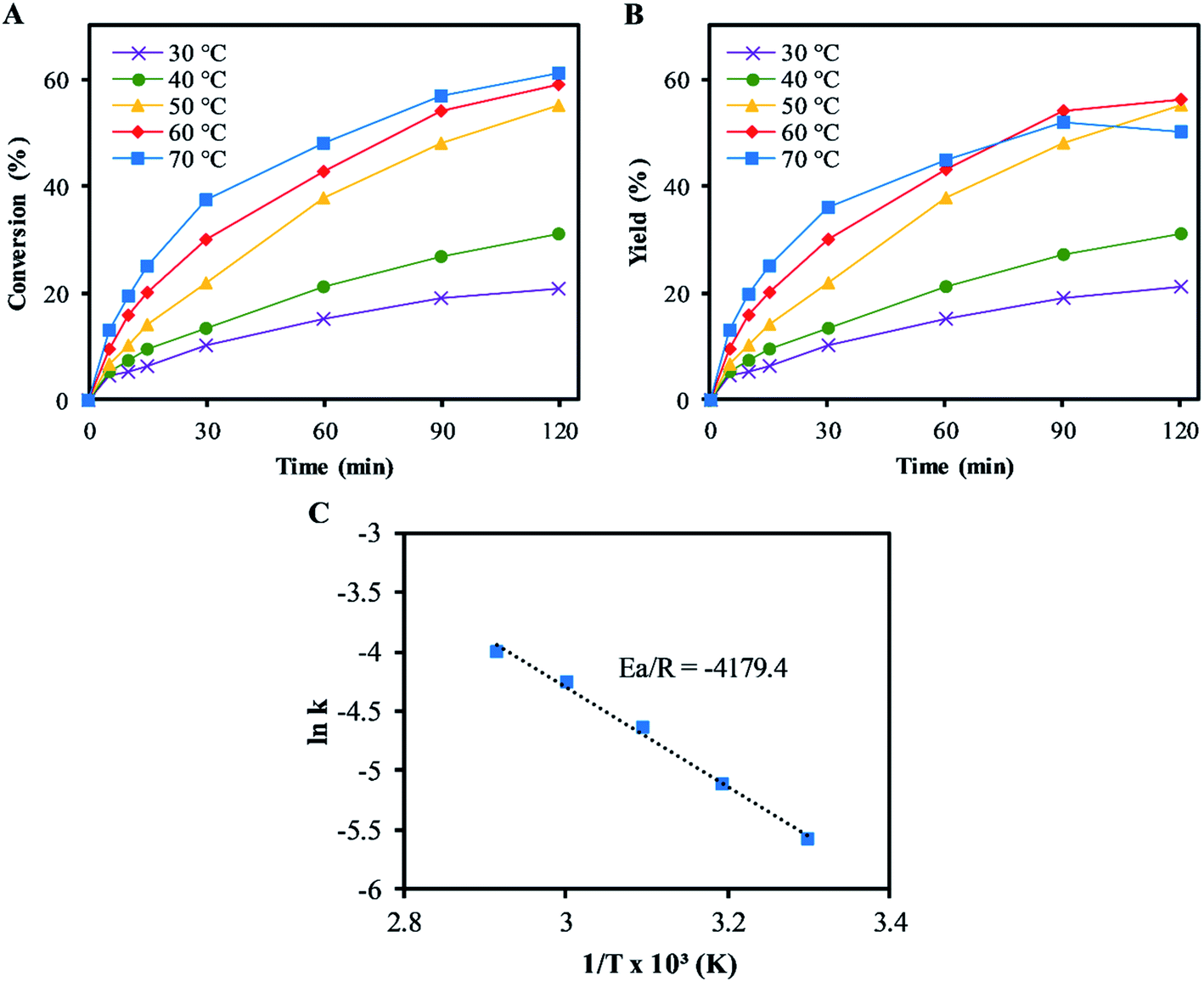 | ||
| Fig. 3 Effect of change in temperature on α-pinene epoxidation: (A) conversion of α-pinene; (B) yield of α-pinene oxide. (Reaction conditions: α-pinene (1.25 M), H2O2 (1.25 M), Na2WO4·2H2O (0.012 M), H2SO4 (0.05 M) and toluene (500 mol%); (C) determination of Arrhenius activation energy for α-pinene epoxidation. The lines represent the trends between data points. | ||
The Arrhenius activation energy (Ea) for the epoxidation was determined to be ∼35 kJ mol−1, using the gradient of the plot of ln(kepox) versus 1/T in Fig. 3C, which is not far from ∼40 kJ mol−1 reported by Becerra et al.13 and lower than that suggested by Cánepa et al. (∼68 kJ mol−1) for the epoxidation of α-pinene.42 The lower activation energy was obtained from this study compared to ∼68 kJ mol−1 in Cánepa et al. when a heterogeneous catalyst was used,42 indicating a faster epoxidation reaction rate in homogeneous catalysis. The rate constant, kepox at 50 °C was determined to be 9.2 × 10−3 L mol−1 s−1.
3.3 Effect of oxidant amount
Results of the H2O2 oxidant amount on the epoxidation of α-pinene at the concentrations of 100–200 mol% are shown in Fig. 4. As depicted in Fig. 4, the selectivity to α-pinene oxide decreased by increasing the oxidant amount. The yields of α-pinene oxide were 55% and 12% for the oxidant amounts of 100 and 200 mol%, respectively. At a greater-than-equimolar amount of oxidant, epoxidation and allylic oxidation occur simultaneously.45 The yield of verbenol increases from 4–13% with an increase in oxidant amount from 125–200 mol%, which subsequently increases the yield of verbenone. These results are in accord with an existing study, which states that an increase in H2O2 concentration generates more radicals and favours the allylic oxidation route, which in turn oxidises verbenol to verbenone.46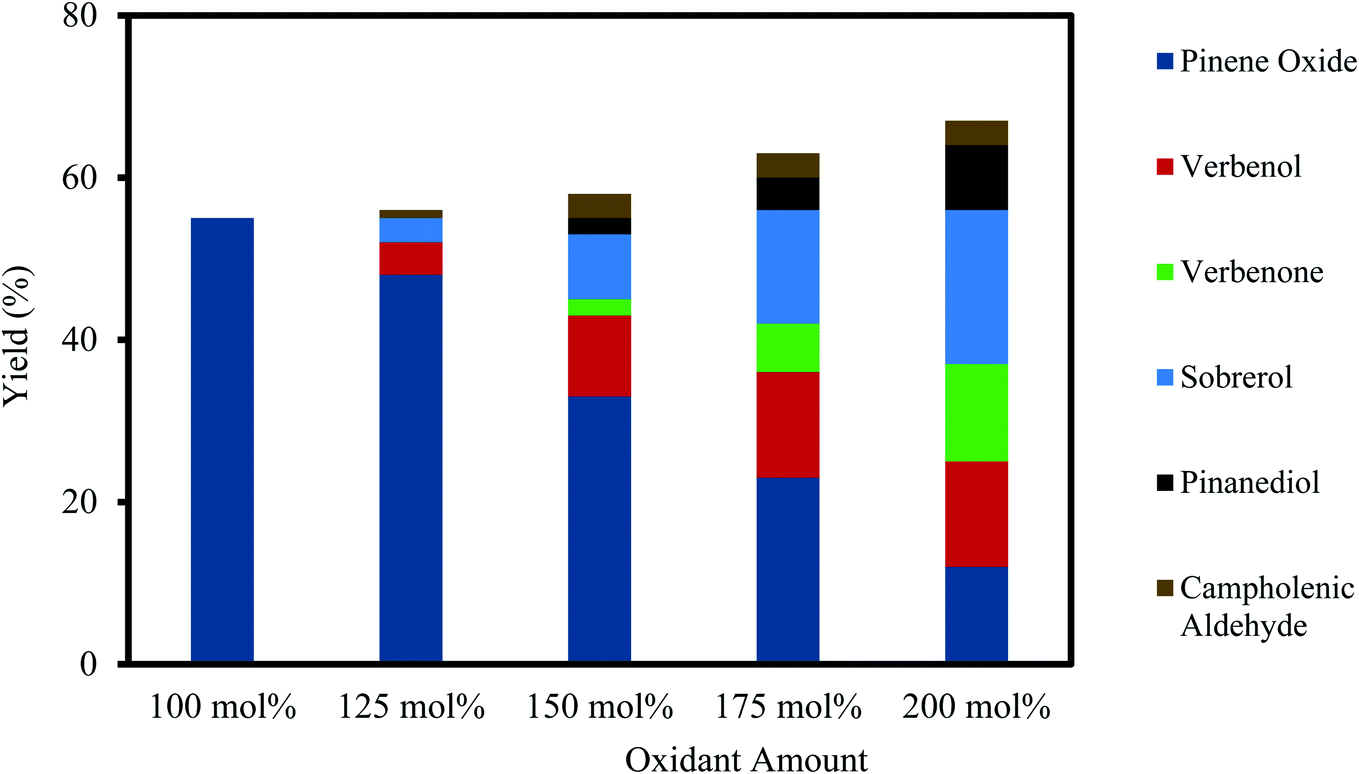 | ||
| Fig. 4 Effect of the oxidant amount on α-pinene epoxidation and the distribution of the products. Reaction conditions: temperature (50 °C), reaction time (120 min), α-pinene (1.25 M), Na2WO4·2H2O (0.012 M), H2SO4 (0.05 M) and toluene (500 mol%). | ||
As shown in Fig. 4, a higher oxidant amount also causes a lower yield of α-pinene oxide, as the rate of hydrolytic decomposition increases, which results in the formation of sobrerol and pinanediol. The increased rate of hydrolytic decomposition is mainly influenced by the increasing concentration of H2O, which is proportionate to the oxidant amount, as 30% (wt/wt) H2O2 solution was used. The yield of sobrerol rose from 3–19% when the oxidant amount is increased from 125–200 mol%. Pinanediol was observed between an oxidant amount of 150 and 200 mol%, with its yield increasing from 2–8%. A small amount of campholenic aldehyde (∼3%) is always present in the reaction mixture at higher oxidant amounts but does not correlate to the increasing amount of oxidant. It appears that increasing amounts of oxidant have little effect on the transformation of α-pinene oxide to campholenic aldehyde. To maintain the highest selectivity towards α-pinene oxide, an equimolar amount of oxidant and α-pinene is recommended.
3.4 Effect of acid concentration
The effect of acid concentration in the reaction media was investigated by varying the H2SO4 concentrations from 0.04–0.09 M, as shown in Fig. 5, for the investigations using H2SO4 concentrations from 0.04–0.09 M. The selectivity to α-pinene oxide decrease dramatically with an increasing acid concentration, for example, the yields were 38% at 0.04 M and 4% at 0.09 M acid concentrations.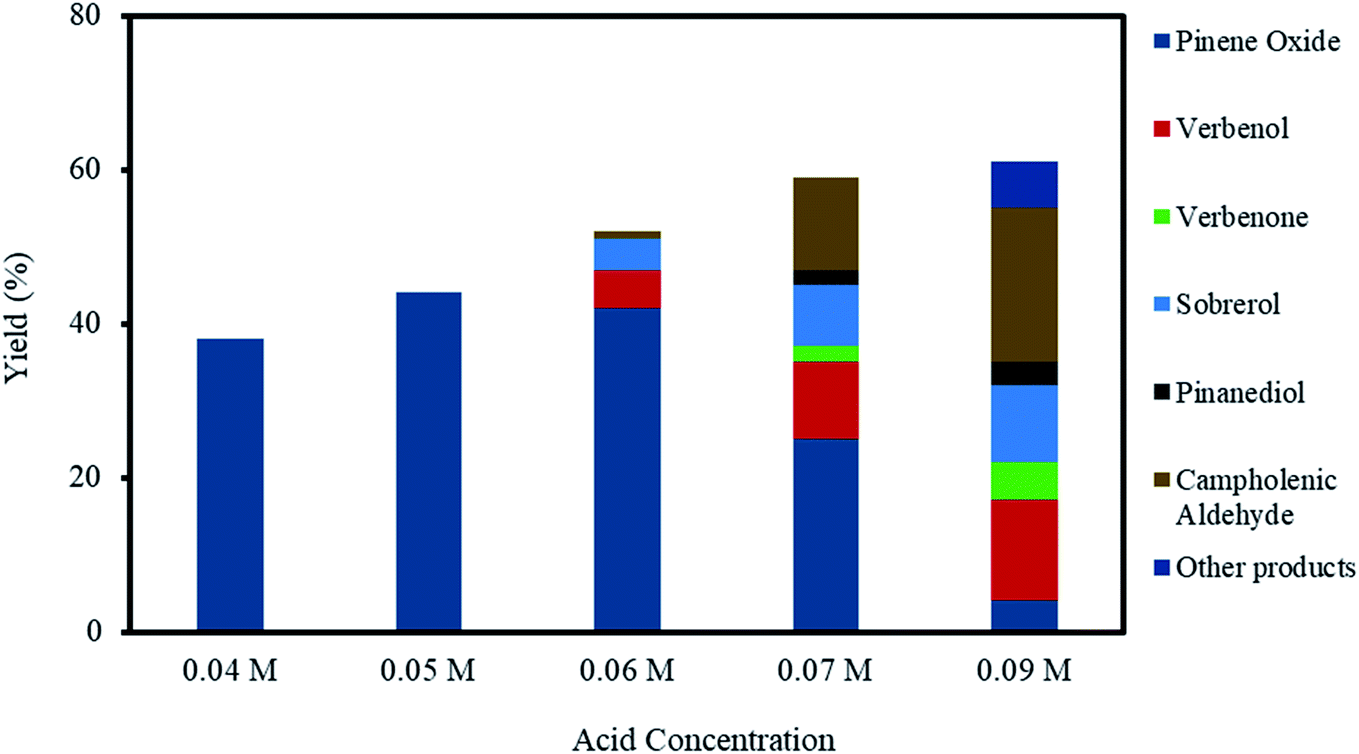 | ||
| Fig. 5 Effect of acid concentration on α-pinene epoxidation and distribution of the products. Reaction conditions: temperature (50 °C), reaction time (120 min), α-pinene (1.25 M), H2O2 (1.25 M), Na2WO4·2H2O (0.012 M) and toluene (500 mol%). | ||
At acid concentrations of 0.06 M and higher, the decrease in the final yield of α-pinene oxide is primarily due to the significant increase in α-pinene oxide rearrangement; α-pinene oxide was progressively consumed to an almost complete conversion, mainly forming campholenic aldehyde. The yield of campholenic aldehyde rises from ∼1 to ∼20% with increasing acid concentrations. Furthermore, at an acid concentration of 0.09 M, other products consist of mainly aldehyde and alcohol were detected by the analytical methods with a total calculated yield of 6%. These observations are largely influenced by the increase in H+ concentration, which acts as a Lewis acid. The H+ reacts with the oxygen atoms in α-pinene oxide, causing the C–C bonds to split and form aldehyde and alcohol.12,45
The decrease in α-pinene oxide yield is also due to the increase in the hydrolytic decomposition of the oxide. The yield of sobrerol increases from 4–10% with the increasing acid concentration. On the other hand, the yield of pinanediol was only 2 and 3% at acid concentrations of 0.07 and 0.09 M, respectively. The hydrolytic decomposition of α-pinene oxide becomes less prevalent with increasing acid concentrations than with increases in the oxidant amount, as discussed in Section 3.3. This confirms the need for high H2O concentrations to degrade α-pinene oxide through hydrolysis, while increasing acid (H+) concentrations favour the rearrangement process.
The competing allylic oxidation is apparent when acid concentration is higher than 0.06 M, with the yield of verbenol increasing from 5–13%. Verbenone was only observed at acid concentrations of 0.07 and 0.09 M, with yields of ∼2 and 5%, respectively. From these results, it can be deduced that the epoxidation route is more kinetically favoured than allylic oxidation, even when the acid concentration is increased, as confirmed by the increasing total yield of α-pinene oxide rearrangement and hydrolysis products as compared to allylic oxidation products.
The epoxidation of α-pinene is only 100% selective to α-pinene oxide when the acid concentration is below 0.05 M. The pH of the aqueous phase increases throughout the reaction time mainly caused by the formation of H2O as H2O2 was consumed. At acid concentrations of 0.06 M and higher, the final pH was less than 2, while acid concentrations lower than 0.05 M resulted in a final pH of about ∼2.5. It is presumed that higher acid concentrations not only destabilise the peroxo species but also reduce H+ concentration, which could affect the selectivity to α-pinene oxide. In this regard, the use of a pH buffer might favour the acid-catalysed formation of side products.
The reaction having a 0.05 M acid concentration at 70 °C (Section 3.2) did not yield as many side products as the reaction with a 0.09 M acid concentration at 50 °C. This highlights the importance of acid concentration to this type of epoxidation, which holds the key to developing a highly selective epoxidation of α-pinene. It is worth mentioning that a control experiment was carried out in the absence of an acid. This raised the pH of the aqueous peroxide solution above 4, which presumably retarded the formation of the active peroxo species. No conversion of α-pinene to any product was observed during the reaction time.
3.5 Effects of organic solvents
The exothermic nature of the α-pinene epoxidation with H2O2 is typically mitigated by the drop-wise addition of H2O2 and by the addition of an organic solvent. Therefore, a selection of solvents, from polar to non-polar, in addition to an organic solvent-free system using α-pinene in a higher molar ratio (>200 mol%) were investigated as shown in Fig. 6.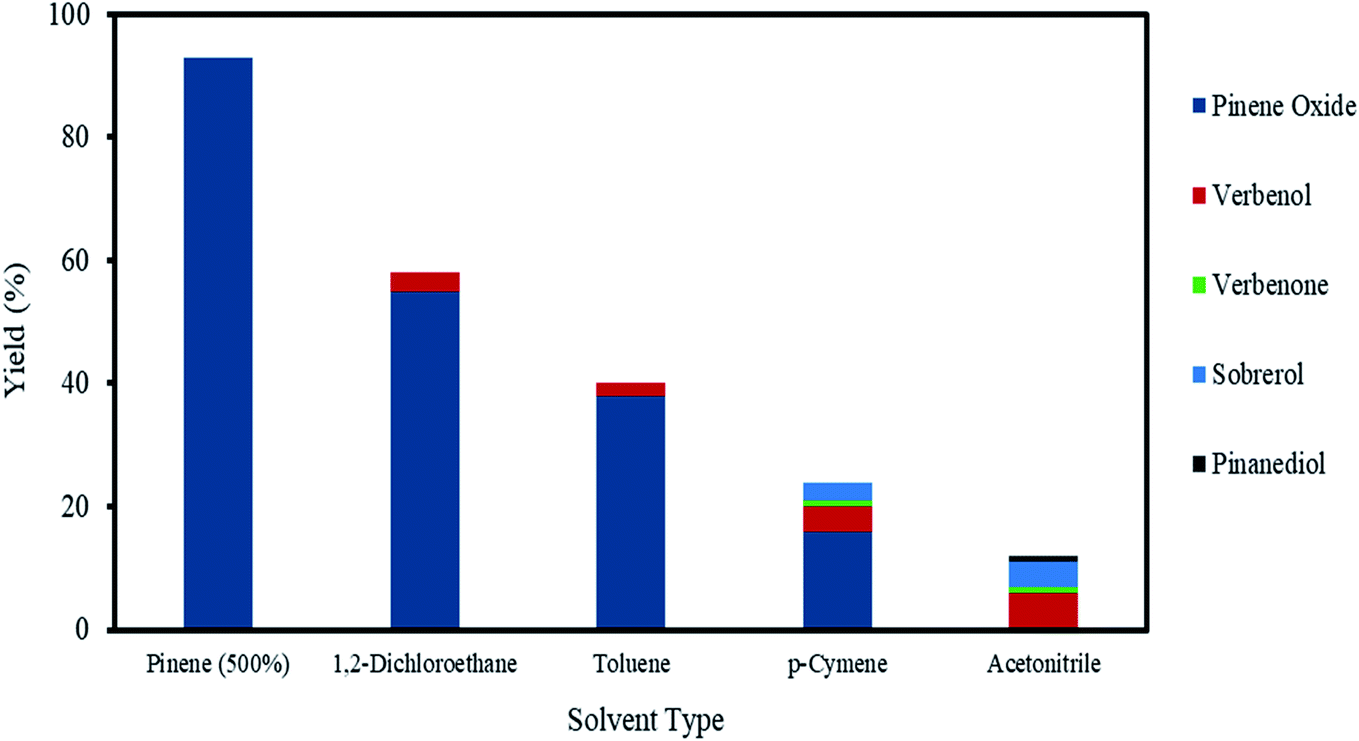 | ||
| Fig. 6 The effects of solvents on α-pinene epoxidation. Reaction conditions: temperature (50 °C), reaction time (120 min), α-pinene (1.25 M), H2O2 (1.25 M), Na2WO4·2H2O (0.012 M), H2SO4 (0.05 M) and solvent amount (500 mol%) for each solvent. | ||
As shown in Fig. 6, in the presence of various solvents, the conversion of α-pinene decreases in the following order: 1,2-dichloroethane (58%) > toluene (40%) > p-cymene (24%) > acetonitrile (12%). Of all the solvents, 1,2-dichloroethane improved the conversion of α-pinene epoxidation most. Under these conditions, the yield of α-pinene oxide is 55%, with only verbenol (3%) being observed as a side product. Toluene afforded yields of α-pinene oxide and verbenol of about 38 and 2%, respectively. The conversion of α-pinene in p-cymene as a solvent after a reaction time of 120 min was about 24%. p-Cymene was less selective to α-pinene oxide than toluene and 1,2-dichloroethane. The conversion of α-pinene is the lowest when acetonitrile is used, with no α-pinene oxide being detected within a reaction time of 120 min. This was due to the complete conversion of α-pinene oxide to its hydrolysis products, sobrerol (4%) and pinanediol (∼1%). Allylic oxidation is more prevalent in acetonitrile, in which the yields of verbenol and verbenone are 6 and ∼1%, respectively. This is consistent with an existing study showing that allylic oxidation is favoured when acetonitrile is used.47 There was no formation of campholenic aldehyde observed with any of the solvents used. This can be attributed to the lower acid concentration (0.05 M), which hampered the rearrangement of α-pinene oxide as explained in Section 3.4.
The highest yield and selectivity to α-pinene oxide was obtained when α-pinene was used without any additional organic solvent in a higher molar ratio than H2O2 (Fig. 7). The calculated conversion and yield were determined on the basis of H2O2 as the limiting reactant. Fig. 7 shows the increasing yield of α-pinene oxide when the amount of α-pinene is increased to 500 mol%. In the absence of an organic solvent and with an amount of α-pinene equimolar to H2O2 (100 mol%), the yield of α-pinene oxide reached its maximum (12%) before completely converting to various side products. Interestingly, the mass balance shows that the conversion of H2O2 does not account for all the products detected by GC. It is presumed that an oligomeric and polymeric compound was formed throughout the reaction time, assisted by the exothermicity. This became evident when a viscous layer of liquid was observed in the organic phase, justifying the need for a solvent in such processes.
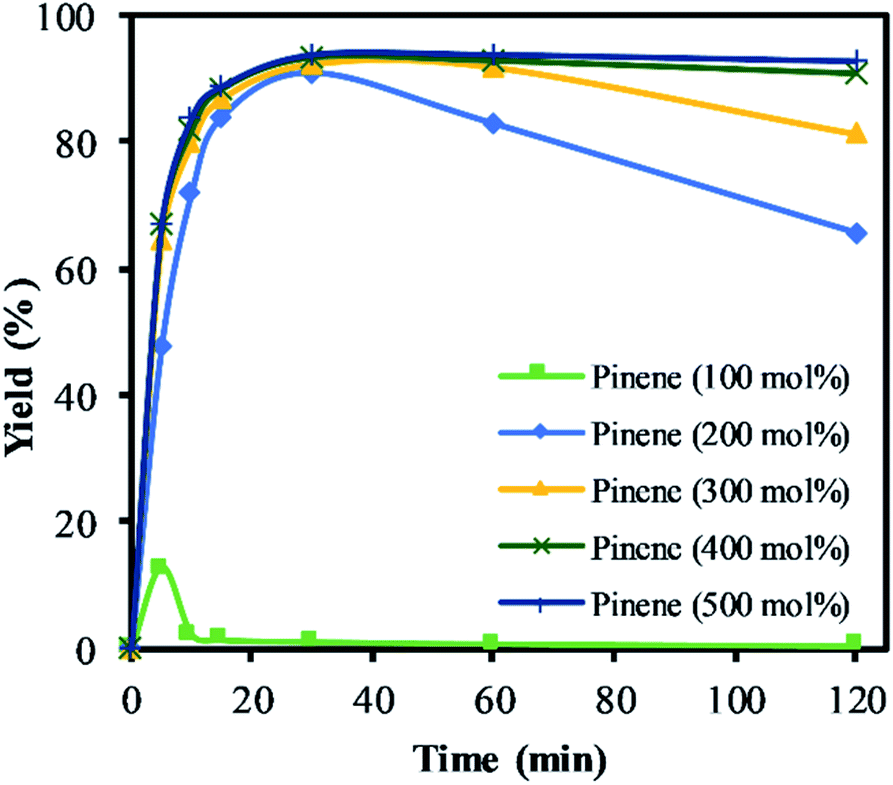 | ||
| Fig. 7 The yield of α-pinene oxide vs. time based on the amount of α-pinene. Reaction conditions: temperature (50 °C), H2O2 (1.25 M), Na2WO4 (0.012 M) and H2SO4 (0.05 M). The lines represent the trends between data points. | ||
The yield of α-pinene oxide improves significantly when α-pinene was used in a higher molar ratio (>200 mol%), to double up as solvent. Maximum yield was reached within 20 min of the reaction time due to a high reaction rate. The yields of α-pinene oxide at 200 and 300 mol% reached a maximum of 91 and 93%, respectively. However, the selectivity to α-pinene oxide decreases gradually over time due to the combination of the hydrolytic decomposition and rearrangement processes. Further increasing the amount of α-pinene up to 500 mol% improves the selectivity of α-pinene oxide and results in isothermal conditions. With 500 mol% of α-pinene, the yield of α-pinene oxide reaches 93%, with 100% selectivity to α-pinene oxide throughout the reaction. This result can be illustrated by the rapid formation of α-pinene oxide, which fully utilised the oxidant in the reaction. This is confirmed by the titration of H2O2. The rapid consumption of active oxygen, coupled with the lower acid concentration, significantly reduces the propagation of radicals. This, in turn, suppresses the allylic oxidation and favours epoxidation as the dominant route; at α-pinene amounts of 200 mol% and higher, little to no formation of allylic products was observed. The large reservoir of α-pinene not only helps to mitigate the exotherm but also to isolate α-pinene oxide in the organic phase, minimising its contact with H+ and H2O presents in the aqueous phase in this biphasic reaction.
The easy separation of the organic phase and the subsequent separation of α-pinene from α-pinene oxide in a vacuum evaporator allows the unreacted α-pinene to be recycled and reused. This results in a rapid process whereby α-pinene is converted to α-pinene oxide with high selectivity in an organic solvent-free environment within a short reaction time (∼20 min). The reduced reaction time is achieved by adding the oxidant with a single-step addition. Catalyst recycling should be considered to further improve the process, making it industrially and environmentally feasible.
4 Conclusions
The epoxidation of α-pinene typically leads to many side products and is usually performed in excess solvent or using drop-wise addition of the oxidant, which reduces the reaction rates and lengthens the reaction time, while the use of solvent causes additional separation step. In this work, 93% conversion of H2O2 with 100% selectivity to α-pinene oxide within 20 min reaction time was achieved using a single-step addition of the oxidant without an organic solvent. It was demonstrated that a low concentration of acid (∼0.05 M) should be used, as this prevented the formation of various side products. The selective process was able to be achieved through the rapid and efficient consumption of oxidants by increasing the substrate molar ratio in the absence of an organic solvent and lowering the concentration of acid to allow a predominantly epoxidation reaction route. This study also found that increasing the amount of oxidant increases the rate of allylic oxidation. Reaction kinetics revealed that the reaction is first order for α-pinene and the catalyst, and a reaction order of 0.5 for the H2O2. The activation energy for the α-pinene epoxidation was determined to be 35 kJ mol−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Engineering and Physical Science Research Council (EPSRC) funding for Sustainable Polymers (Project reference EP/L017393/1) and the Ministry of Higher Education, Malaysia.References
- N. Fdil, A. Romane, S. Allaoud, A. Karim, Y. Castanet and A. Mortreux, Terpenic olefin epoxidation using metals acetylacetonates as catalysts, J. Mol. Catal. A: Chem., 1996, 108(1), 15–21 CrossRef CAS.
- H. Mimoun, Catalytic opportunities in the flavor and fragrance industry, Chimia, 1996, 50(12), 620–625 CAS.
- A. J. Silvestre and A. Gandini, Terpenes: major sources, properties and applications, Monomers, Polymers and Composites from Renewable Resources, Elsevier, 2008, pp. 17–38 Search PubMed.
- A. Calogirou, B. Larsen and D. Kotzias, Gas-phase terpene oxidation products: a review, Atmos. Environ., 1999, 33(9), 1423–1439 CrossRef CAS.
- W. F. Erman, Chemistry of Monoterpenes, Marcel Dekker, New York, 1985 Search PubMed.
- C. Chapuis and D. Jacoby, Catalysis in the preparation of fragrances and flavours, Appl. Catal., A, 2001, 221(1–2), 93–117 CrossRef CAS.
- K. Bauer, D. Garbe and H. Surburg, Common fragrance and flavor materials: preparation, properties and uses, John Wiley & Sons, 2008 Search PubMed.
- M. N. Belgacem and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, Elsevier, The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, UK, 2008 Search PubMed.
- L. A. Paquette, Handbook of Reagents for Organic Synthesis, Chiral Reagents for Asymmetric Synthesis, John Wiley & Sons, 1999 Search PubMed.
- Y. M. Ahmat, S. Madadi, L. Charbonneau and S. Kaliaguine, Epoxidation of terpenes, Catalysts, 2021, 847 CrossRef.
- G. Fráter, J. A. Bajgrowicz and P. Kraft, Fragrance chemistry, Tetrahedron, 1998, 54(27), 7633–7703 CrossRef.
- M. Stekrova, N. Kumar, A. Aho, I. Sinev, W. Grünert, J. Dahl, J. Roine, S. S. Arzumanov, P. Mäki-Arvela and D. Y. Murzin, Isomerization of α-pinene oxide using Fe-supported catalysts: selective synthesis of campholenic aldehyde, Appl. Catal., A, 2014, 470, 162–176 CrossRef CAS.
- J.-A. Becerra, L.-M. González and A.-L. Villa, Kinetic study of α-pinene allylic oxidation over FePcCl16-NH2-SiO2 catalyst, J. Mol. Catal. A: Chem., 2016, 423, 12–21 CrossRef CAS.
- U. Neuenschwander, F. Guignard and I. Hermans, Mechanism of the Aerobic Oxidation of α-Pinene, ChemSusChem, 2010, 3(1), 75–84 CrossRef CAS PubMed.
- U. Neuenschwander, E. Meier and I. Hermans, Peculiarities of β-Pinene Autoxidation, ChemSusChem, 2011, 4(11), 1613–1621 CrossRef CAS PubMed.
- G. Sienel, R. Rieth and K. T. Rowbottom, Ulmann's Encyclopedia of Industrial Chemistry, 6th edn, Verlag Chemie, Weinheim, 2003 Search PubMed.
- K. A. D. Swift, Catalytic transformations of the major terpene feedstocks, Top. Catal., 2004, 27(1–4), 143–155 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chemical Routes for the Transformation of Biomass into Chemicals, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- G. Carr, G. Dosanjh, A. P. Millar and D. Whittaker, Ring opening of α-pinene epoxide, J. Chem. Soc., Perkin Trans. 2, 1994,(7), 1419–1422 RSC.
- J. Van der Waal, M. Rigutto and H. Van Bekkum, Zeolite titanium beta as a selective catalyst in the epoxidation of bulky alkenes, Appl. Catal., A, 1998, 167(2), 331–342 CrossRef CAS.
- F. Skrobot, A. Valente, G. Neves, I. Rosa, J. Rocha and J. Cavaleiro, Monoterpenes oxidation in the presence of Y zeolite-entrapped manganese (III) tetra (4-N-benzylpyridyl) porphyrin, J. Mol. Catal. A: Chem., 2003, 201(1–2), 211–222 CrossRef CAS.
- J. M. d. S. e. Silva, F. a. S. Vinhado, D. Mandelli, U. Schuchardt and R. Rinaldi, The chemical reactivity of some terpenes investigated by alumina catalyzed epoxidation with hydrogen peroxide and by DFT calculations, J. Mol. Catal. A: Chem., 2006, 252, 186–193 CrossRef CAS.
- G. A. Eimer, I. Diaz, E. Sastre, S. G. Casuscelli, M. E. Crivello, E. R. Herrero and J. Perez-Pariente, Mesoporous titanosilicates synthesized from TS-1 precursors with enhanced catalytic activity in the α-pinene selective oxidation, Appl. Catal., A, 2008, 343(1–2), 77–86 CrossRef CAS.
- A. Feliczak-Guzik and I. Nowak, Mesoporous niobosilicates serving as catalysts for synthesis of fragrances, Catal. Today, 2009, 142(3–4), 288–292 CrossRef CAS.
- P. Levecque, H. Poelman, P. Jacobs, D. D. Vosa and B. Sels, Regio- and stereoselective terpene epoxidation using tungstate-exchanged takovites: a study of phase purity, takovite composition and stable catalytic activityw, Phys. Chem. Chem. Phys., 2009, 11, 2964–2975 RSC.
- B. Qi, X.-H. Lu, D. Zhou, Q.-H. Xia, Z.-R. Tang, S.-Y. Fang, T. Pang and Y.-L. Dong, Catalytic epoxidation of alkenes with 30% H2O2 over Mn2+-exchanged zeolites, J. Mol. Catal. A: Chem., 2010, 322(1–2), 73–79 CrossRef CAS.
- A. L. Villa, B. F. Sels, D. E. De Vos and P. A. Jacobs, A heterogeneous tungsten catalyst for epoxidation of terpenes and tungsten-catalyzed synthesis of acid-sensitive terpene epoxides, J. Org. Chem., 1999, 64(19), 7267–7270 CrossRef.
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Hybrid polyoxotungstate/MIL-101 materials: synthesis, characterization, and catalysis of H2O2-based alkene epoxidation, Inorg. Chem., 2010, 49(6), 2920–2930 CrossRef CAS PubMed.
- C. B. Woitiski, Y. N. Kozlov, D. Mandelli, G. V. Nizova, U. Schuchardt and G. B. Shul'pin, Oxidations by the system “hydrogen peroxide–dinuclear manganese (IV) complex–carboxylic acid”: Part 5. Epoxidation of olefins including natural terpenes, J. Mol. Catal. A: Chem., 2004, 222(1–2), 103–119 CAS.
- M. Rüsch gen Klaas and S. Warwel, Chemoenzymatic epoxidation of alkenes by dimethyl carbonate and hydrogen peroxide, Org. Lett., 1999, 1(7), 1025–1026 CrossRef.
- C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, Roy. Soc. Chem., Cambridge, 1999 Search PubMed.
- G. Strukul, Catalytic Oxidation with Hydrogen Peroxide as Oxidant, Kluwer, Dordrecht, 1992 Search PubMed.
- W. Hölderich, J. Röseler, G. Heitmann and A. Liebens, The use of zeolites in the synthesis of fine and intermediate chemicals, Catal. Today, 1997, 37(4), 353–366 CrossRef.
- Y. Kon, H. Hachiya, Y. Ono, T. Matsumoto and K. Sato, An Effective Synthesis of Acid-Sensitive Epoxides via Oxidation of Terpenes and Styrenes Using Hydrogen Peroxide under Organic Solvent-Free Conditions, Synthesis, 2011,(7), 1092–1098 CAS.
- C. Venturello and R. D'Aloisio, Quaternary Ammonium Tetrakis(diperoxotungsto)phosphates(3-) as a New Class of Catalysts for Efficient Alkene Epoxidation with Hydrogen Peroxide, J. Org. Chem., 1988, 53, 1553–1557 CrossRef CAS.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant, Green Chem., 2003, 5(1), 1–7 RSC.
- D. C. Duncan, R. C. Chambers, E. Hecht and C. L. Hill, Mechanism and dynamics in the H3[PW12O40]-catalyzed selective epoxidation of terminal olefins by H2O2. Formation, reactivity, and stability of {PO4[WO(O2)2]4}3, J. Am. Chem. Soc., 1995, 117(2), 681–691 CrossRef CAS.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, Hydrogen-Peroxide Oxidation Catalyzed by Heteropoly Acids Combined with Cetylpyridinium Chloride - Epoxidation of Olefins and Allylic Alcohols, Ketonization of Alcohols and Diols, and Oxidative Cleavage of 1,2-Diols and Olefins, J. Org. Chem., 1988, 53(15), 3587–3593 CrossRef CAS.
- E. Poli, J. M. Clacens, J. Barrault and Y. Pouilloux, Solvent-free selective epoxidation of fatty esters over a tungsten-based catalyst, Catal. Today, 2009, 19–22 CrossRef CAS.
- M. F. M. Gunam Resul, A. M. López Fernández, A. Rehman and A. P. Harvey, Development of a selective, solvent-free epoxidation of limonene using hydrogen peroxide and a tungsten-based catalyst, React. Chem. Eng., 2018, 747–756 RSC.
- H. Hachiya, Y. Kon, Y. Ono, K. Takumi, N. Sasagawa, Y. Ezaki and K. Sato, Unique Salt Effect on Highly Selective Synthesis of Acid-Labile Terpene and Styrene Oxides with a Tungsten/H2O2 Catalytic System under Acidic Aqueous Conditions, Synthesis, 2012, 44(11), 1672–1678 CrossRef CAS.
- A. L. Cánepa, E. R. Herrero, M. E. Crivello, G. A. Eimer and S. G. Casuscelli, H2O2 based α-pinene oxidation over Ti-MCM-41. A kinetic study, J. Mol. Catal. A: Chem., 2011, 347(1–2), 1–7 CrossRef.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, Hydrogen peroxide oxidation catalyzed by heteropoly acids combined with cetylpyridinium chloride. Epoxidation of olefins and allylic alcohols, ketonization of alcohols and diols, and oxidative cleavage of 1,2-diols and olefins, J. Org. Chem., 1988, 53(15), 3587–3593 CrossRef CAS.
- G. Grigoropoulou and J. H. Clark, A catalytic, environmentally benign method for the epoxidation of unsaturated terpenes with hydrogen peroxide, Tetrahedron Lett., 2006, 47(26), 4461–4463 CrossRef CAS.
- A. L. Cánepa, C. M. Chanquía, V. M. Vaschetti, G. A. Eimer and S. G. Casuscelli, Biomass toward fine chemical products: oxidation of α-pinene over sieves nanostructured modified with vanadium, J. Mol. Catal. A: Chem., 2015, 404, 65–73 CrossRef.
- A. Negoi, V. I. Parvulescu and M. Tudorache, Peroxidase-based biocatalysis in a two-phase system for allylic oxidation of α-pinene, Catal. Today, 2018, 306, 199–206 CrossRef CAS.
- S. Shylesh and A. Singh, Synthesis, characterization, and catalytic activity of vanadium-incorporated,-grafted, and-immobilized mesoporous MCM-41 in the oxidation of aromatics, J. Catal., 2004, 228(2), 333–346 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |