
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of silver in several samples using a new ionophore polymer membrane as an optical sensor
Hesham H. El-Feky *,
Abdelrazek M. Askar and
Alaa S. Amin
*,
Abdelrazek M. Askar and
Alaa S. Amin
Department of Chemistry, Faculty of Science, Benha University, Benha, Egypt. E-mail: hesham.elfeky@fsc.bu.edu.eg; Tel: +201060204453
First published on 1st November 2021
Abstract
Growing concerns about the possible toxicity of silver to aquatic organisms, bacteria, and humans have led to newly issued regulations by the United States Environmental Protection Agency (US EPA) and the Food and Drug Administration (FDA) regarding the use of silver. However, the increase in bacterial resistance to antibiotics has led to a resurgence in the use of silver as a biocidal agent in applications ranging from washing machine additives to the drinking water treatment system on the International Space Station (ISS). For Ag+ ion detection, a highly sensitive and reversible optical sensor has been established. The optode relies on a novel Schiff base, namely 2-[(benzo[d]thiazol-2-ylimino)methyl]phenol (BTMP) immobilized within PVC film and also incorporated with tris(2-ethylhexyl)phosphate (TEHP) and Aliquat 336 as an ion carrier. Under optimum conditions (i.e. pH 8.5), the proposed sensor displayed a linear response to Ag+ over 4.8 × 10−9 to 1.0 × 10−5 M (0.8494–1698.7 μg L−1) with limits of detection and quantification of 1.5 × 10−9 and 4.8 × 10−9 M (0.2548 and 0.8494 μg L−1), respectively. The sensor's response time was found to be 8.0 min. The sensor was applied successfully to determine Ag+ ion in some real samples, including food, biological, water, and environmental samples.
Introduction
One of the most difficult analytical tasks is the determination of trace element cations in different environmental samples. Difficulties in analysis are due to the low levels of elements in samples, besides matrix interferences; consequently, their analyses require new methods and highly sensitive procedures of preconcentration. It is crucial to remove trace elements from wastewater streams for better public and environmental health. However, the matrix effect is very strong in some analyses and methods for matrix separation are also used; the detection methods for trace metals have been previously indicated.1–5 Silver ions, as heavy metal ions, in comparison with Cr3+, Cd2+, Hg2+, and Cu2+, are one of the most poisonous types of toxic ions.6 Ag+ can be predigested in the lungs, mucous membranes, gastrointestinal tract, and also the skin. Very low concentrations of silver are in the human body but it does not play any biochemical or physiological role in the body.7 It has been revealed that silver cations may have a high risk as water pollutants because the process of recycling mixed silver is very complicated. Silver ions can be used as an indicator for geochemical prospecting in natural waters because the areas that are rich in silver are likely to be enhanced. The maximum amount of silver ions reported in drinking water for cattle is 2.5 × 10−3 μg L−1.8Given the growing applications of silver in medicine and industry, its entrance into the environment has been increased due to its anti-inflammatory, antibacterial, and antifungal properties. The widespread use of silver in wound care products, medical devices, pharmaceuticals, cosmetics, textiles, food packaging, and drinking-water treatment systems has been observed.7,9 On the other hand, towering levels of silver in the environment may cause critical damage to the terrestrial and aquatic ecosystems, and lead to adverse health influences accumulating in human organs.10,11 Therefore, the development of facile and sensitive methods to detect ultra-trace amounts of silver is required due to its utility as well as toxicity.
Among the available traditional techniques, voltammetry,12 potentiometry, atomic absorption spectroscopy (AAS), inductively coupled plasma atomic emission spectrometry (ICP-OES),13 electrothermal atomic absorption spectrometry (ETAAS), and inductively coupled plasma mass spectroscopy (ICP-MS),14 are the most common methods used for Ag+ determination. Significant advantages such as high sensitivity and good accuracy are demonstrated by these techniques but their application in routine Ag+ determination remains limited due to the disadvantages of needing expensive advanced instruments, elaborate sample preparation, well-trained personnel and time-consuming procedures.15 Significant attention has been directed toward developing new methods that are simple and not expensive. However, these techniques involve many complicated steps and critical interferences from other metal cations. The spectrophotometric method is still simple and does not need expensive or complicated equipment. Thus, there has been a wide range of spectrophotometric methods for the detection of Ag+ (Table 1). Each chromogenic system has advantages and disadvantages according to sensitivity, selectivity, and rapidity.16,17
| Reagent | Bead | Samples | Linear range | Detection limit | RSD (%) | Ref. |
|---|---|---|---|---|---|---|
| N,N′-bis(3-methyl-1-phenyl-4-benzylidine-5-pyrazolone) propylenediamine | PVC | — | 10−6 to 10−1 M | 4.0 × 10−5 | — | 38 |
| Tetrakis(p-chlorophenyl)borate and 1-(dansylamidopropyl)-1-aza-4,7,10-trithiacyclododecane | PVC | Medical radiological film, photographical fixing solution and bleaching solution. | 5.0 × 10−7 to 1.7 × 10−2 M | 1.0 × 10−7 M. | 3.6 | 39 |
| Diphenylthiocarbazone | Transparent triacetyl cellulose (produced from waste photographic film) | Synthetic samples, different kinds of photographic solutions and pharmaceutical | Up to 3.4 × 10−4 | 0.01 μg L−1 (9.2 × 10−8 M) | 1.16 | 40 |
| Hexathia-18-crown-6 and 1,2-benzo-3-octadecanoylimino-7-diethylaminophenoxazine | PVC | Drinking water sample | 5.0 × 10−9–5.0 × 10−5 M | 1.8 × 10−9 M | — | 41 |
| 7-(1H-indol-3-ylmethyl)-5,6,7,8,9,10-hexahydro-2H-1,13,4,7,10-benzodioxatriaza cyclopentadecine 3,11(4H,12H)-dione, | PVC | Water and poly-metallic ore | 2.27 × 10−11 to 1.13 × 10−3 | 9.5 × 10−12 M | 1.4 | 6 |
| BTMP | PVC | Water, medical radiological film, biological, and environmental samples | 0.85–1698.7 μg L−1 (4.8 × 10−9 to 1.0 × 10−5 M) | 0.255 μg L−1 (1.5 × 10−9 M) | 2.25 | This work |
Optical chemical sensors (optodes) have attracted considerable attention in real-time detection because of their simplicity, facility, and naked-eye estimation. The basic strategy for the development of the optodes is to immobilize optical indicators, which can establish a color response towards certain metal cations, on the membrane surface or within the membrane matrixes by either physical (adsorption, sol–gel, etc.) or chemical (covalent bond) methods.18 Porous, membranes, having the ability for separation, are widely used in water treatment. Many optical chemical sensors utilize colored complexes or reducing and oxidizing reagents immobilized on a membrane of a suitable polymer. The most convenient immobilization technique for producing highly stable compositions of the molecules that strictly dissolve the reagent is a covalent attachment with a functionalized support by the direct immobilization of the ligand on the surface of the membrane or by increasing the lipophilicity of the corresponding reagent. Optodes have numerous benefits such as facile construction, good selectivity and sensitivity, and high dynamic concentration range accompanied by a low detection limit.19–21
The detection of silver cations by optical sensors based on colorimetric methods has been discussed. Several optical chemical sensors have utilized colored complexes immobilized in a suitable matrix.22–24 There has been significant interest in PVC-based membranes, especially PVC, as they can easily trap the ionophore-based liquid membrane system. Spectrophotometric methods supply wide characteristics such as simple instrumentation, rapid response times, and facile operation that are required for the design of portable analytical instruments for Ag detection.
In the current work, a highly selective, accurate, and sensitive silver optode was constructed and successfully operated in the analysis of different real environmental samples. It has many advantages such as operation in the range of the visible region, high selectivity, accuracy and sensitivity to Ag+ ions, facile preparation of ionophores, a wide dynamic range of detection, low RSD, high recoveries, and low detection and quantification limits.
Material and methods
Instrumentation
The pH values were adjusted using a pH meter (HANA instruments; model HI 8014) with a glass-bodied combined pH electrode (924005) and calibrated with Merck pH standards of pH 4.00, 7.00, and 10.00. Small volumes of silver salt solution were transferred into the cuvette using a Hamilton syringe (10 μL). The absorption spectra were obtained using a Jasco UV/Vis spectrophotometer (Jasco; model V-670) with a scanning speed of 400 nm min−1, bandwidth 2.0 nm, and equipped with 1.0 cm matched pair quartz cells. The absorbance measurements were obtained by bearing the optical membrane sensors inside the quartz cuvette. The spectral measurements of the optical membrane sensor samples were obtained against air and against a blank optode sample. A Shimadzu model 670 atomic absorption spectrometer with flame atomization was used in a nitrous oxide–acetylene flame. Atomic absorption measurements were carried out in an air-acetylene flame. The following parameters were applied: absorption line: 328.1 nm; slit widths: 0.4 nm; and lamp currents: 5 mA. The ratio of the flow rate of air to that of acetylene was 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8. FT-IR spectra of the product in the range of 4000–400 cm−1 at room temperature were measured on an FT-IR spectrometer (Thermo Scientific; model Nicolet iS10) using the standard KBr pellets. 1H-NMR spectra were obtained on a Bruker Avance Drx 300 MHz spectrometer with TMS as an internal standard in a Micro-analytical center, Cairo University, Cairo, Egypt.
:1.8. FT-IR spectra of the product in the range of 4000–400 cm−1 at room temperature were measured on an FT-IR spectrometer (Thermo Scientific; model Nicolet iS10) using the standard KBr pellets. 1H-NMR spectra were obtained on a Bruker Avance Drx 300 MHz spectrometer with TMS as an internal standard in a Micro-analytical center, Cairo University, Cairo, Egypt.
Materials and reagents
All reagents employed in this study were from Merck and were of analytical reagent grade and were used without further purification. Polyvinyl chloride (MW 200000) PVC (high relative molecular mass), cellulose triacetate (CTA), dioctylphthalate (DOP), sodium tetraphenylborate (NaTPB), tributyl phosphate (TBP), o-nitrophenyl octylether (NPOE), tris(2-ethylhexyl) phosphate (TEHP), tricaprylylmethyl ammonium chloride (Aliquat 336), triton X-100, oleic acid and lauric acid, were purchase from Sigma-Aldrich (Steinheim, Switzerland). Silver nitrate (AgNO3) and tetrahydrofuran (THF) also were from Merck.
Sodium chloride (NaCl) and sodium hydroxide (NaOH) were obtained from Sigma-Aldrich (Steinheim, Switzerland). Different acids used in this work such as sulphuric acid (H2SO4), acetic acid (CH3COOH), nitric acid (HNO3), and dichloromethane (DCM) were obtained from BDH. Ethylenediamine, potassium chloride, EDTA, triethanolamine, phosphoric acid, and boric acid were obtained from Merck.
A standard stock solution of Ag+ (1000 μg L−1) was prepared by dissolving an appropriate amount of dried silver salt in doubly distilled water and diluting to 100 mL in a volumetric flask and was standardized against 0.1 M standard sodium chloride using Mohr's method.25 Acetate, borate, thiel, phosphate, and universal buffer solutions of different pH values 2.5–11 were prepared as described previously.26 Stock solutions of 20000 mg L−1 of interfering ions were prepared by dissolving appropriate amounts of their suitable salts in double-distilled water.
Synthesis of reagent
Salicylaldehyde (1.0 mol) and 2-aminobenzothiazol (1.0 mol) were dissolved in an ethanolic solution (20 mL) with a few drops of concentrated H2SO4 as a catalyst. The mixture was refluxed for about 5.0 h and cooled to room temperature to give a clear yellow precipitate of 2-[(benzo[d]thiazol-2-ylimino)methyl] phenol).Preparation of the membrane sensor
The sensor membranes were established by mixing 62 mg plasticizer (TEHP), 31 mg PVC, 2.0 mg of BTMP, and 2.0 mg Aliquat-336. The previous components were mixed and dissolved in exactly 3.0 mL THF. An appropriate volume from the dissolved cocktail (≈200 μL) was transferred onto the glass plate using a pipette. Before the coating process, all glass plates were washed with pure THF to exclude organic impurities and dust. The membrane was allowed to dry in air for 20 min after spinning for 7.0 min with a rotation frequency of 600 rpm. The membrane thickness was about 5.0–7.0 μm. The reference blank membrane had the same composition except for the reagent.General procedure
The cuvette was filled with 2.5 mL from a universal buffer of pH 8.5,26 then the optical sensor membrane was transferred into it. After the addition of a known amount of 1000 μg L−1 Ag+ solution, the absorption spectrum of the sample was measured (after one min) against a reference blank membrane in the range between 350–650 nm at 1.0 cm interval cell (Fig. 1).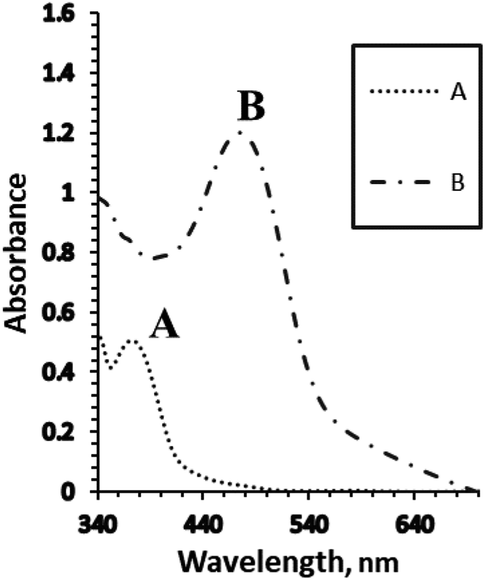 | ||
| Fig. 1 The absorption spectra of the optical sensors using (A) the blank membrane with BTMP, and (B) the blank membrane in the presence of 1.0 × 10−5 M of Ag+ ions under the optimum conditions. | ||
Sample preparation and analysis
Results and discussion
The IR spectrum of the prepared Schiff base showed that characteristic bands appeared at 3449 cm−1 for υ(O–H), 1609 cm−1 for υ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1568 cm−1 for υ(CN) (thiazole ring),1283 cm−1 for υ(C–O), and 755 cm−1 for υ(C–S–C) (Fig. 2). The formation of the Schiff base was indicated by the absence of CO and NH2 peaks from the spectrum of the ligand.30 1H-NMR (DMSO-d6, ppm): 10.28 and 10.47 (s, OH; 2H); 6.80–8.75 (m, 16H).
N), 1568 cm−1 for υ(CN) (thiazole ring),1283 cm−1 for υ(C–O), and 755 cm−1 for υ(C–S–C) (Fig. 2). The formation of the Schiff base was indicated by the absence of CO and NH2 peaks from the spectrum of the ligand.30 1H-NMR (DMSO-d6, ppm): 10.28 and 10.47 (s, OH; 2H); 6.80–8.75 (m, 16H).
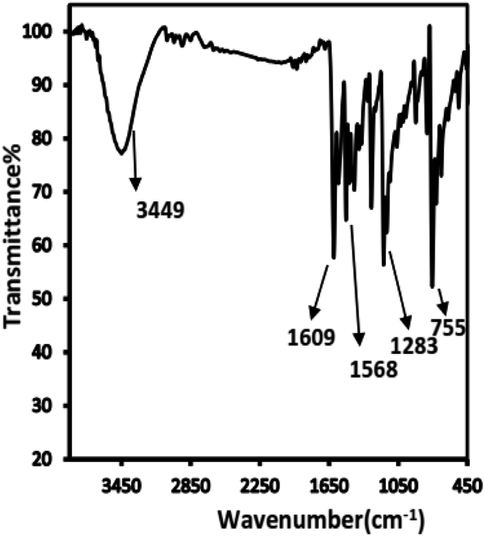 | ||
| Fig. 2 The FT-IR spectra of BTMP. | ||
Silver ions react with BTMP in basic media at pH 8.5 to form a 1:1 (Ag:BTMP) complex in aqueous media containing methanol (40%) (V V−1). Consequently, the addition of suitable amounts of Ag+ to a methanol solution of BTMP led to a rapid change in the absorption spectrum of the solution, changing the color from yellow to reddish-orange, and showed an absorption band at 386 nm for BTMP, whereas for its complex with 1.0 × 10−3 M of Ag+, λmax was at 520 nm (Scheme1).
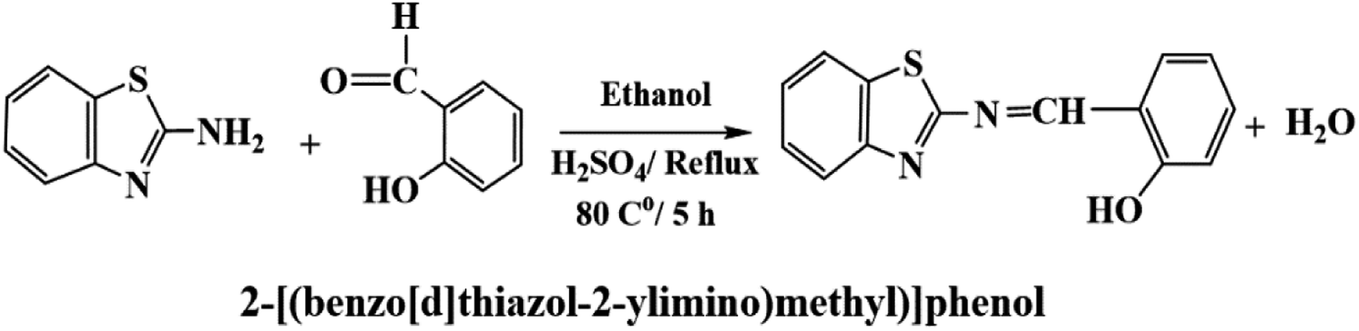 | ||
| Scheme 1 The preparation of the Schiff base BTMP (ionophore). | ||
In the present work, Ag+ reacted with BTMP in the sensor membrane, with λmax of the membrane located at 481 nm. This wavelength was selected for further studies due to the high selectivity and sensitivity at this λmax. In aqueous solutions at pH 8.5, the BTMP membrane showed an absorption band at 375 nm, whereas, for its complex with 1.0 × 10−5 M of Ag+, there was an absorption band at 481 nm, due to the extraction and complexation of Ag+ ions in the membrane.
Optimization of the optode sensor preparation
It is very important to illustrate the influence of all factors that could affect the preparation of the proposed sensor. The different studied parameters were composition, the concentration of the component, type of buffer solution, pH of the buffer, response time, and thickness of the optical membrane sensor.The effect of the membrane composition
It is well recognized that the selectivity and sensitivity achieved for a given ionophore depend significantly on the nature of the solvent mediator and additives and also on the membrane composition.24 The types of plasticizer, polymer, ion carrier, and the lipophilic ionic additive were taken into consideration in order to illustrate their effect on the membrane composition. Consequently, membranes with different compositions were prepared. The response of each of the membranes was examined in Ag+ solutions.First, the sensor matrix should be chosen. Two types of matrix PVC and CTA were tested. PVC is more suitable than CTA as the membrane base, due to numerous indications like appropriate transmittance, reliable permeability to Ag+ ions, good mechanical stability, and the reagent BTMP is also immobilized well without any drainage.31 The plasticizer (solvent mediators) must be compatible physically with the used polymer to achieve a homogenous organic phase, in the membrane preparation.
In the present work, TBP, DOP, TEHP, and NOPE were tested as plasticizers with different degrees of polarities. Using NOPE as a plasticizer for the proposed membrane causes incongruous physical properties. In the case of TBP, and also DOP they gave similar sensitivities as plasticizers, whereas the addition of TEHP gave better characteristic sensitivity of the proposed sensor membrane. The spectral measurements for the effects of the different used plasticizers for the optical sensor membrane were made in 1.0 × 10−5 M of Ag+ against a reference blank membrane.
The existence of an ion carrier is necessary in order to help in completely transferring all Ag+ ions into the sensor membrane and it also decreases the response time. These ion carriers include Aliquat-336, oleic acid, Triton X-100 and lauric acid. The ion carriers also facilitate the ion-exchange equilibrium. From the obtained results, Aliquat-336 gave the best sensor response.
Anionic additives like NaTPB may increase the response of Ag+ ions in the sensor membrane and also decrease the response time; thus, the effect of its presence was studied. From the results, the addition of NaTPB did not have any significant effect on its response.
The effect of the concentration of the membrane composition
The effect of BTMP weight was investigated in the range of 0.25–3.50 mg at fixed silver ions concentration (1.0 × 10−5 M). With the standard addition of the reagent up to 2.0 mg, the concentration of Ag+ ions that migrated to the sensor membrane increased significantly and the complex formation was significantly increased. An increase in the amount of the reagent did not have any significant effect on the sensitivity and the response of the sensor. Thus, 2.0 mg of BTMP was considered the best amount used from the reagent.To optimize the amounts of the plasticizer and PVC in the membrane, the absorbance with different ratios of TEHP/PVC (weight ratios) was investigated. The obtained results indicated that the selectivity and sensitivity of the proposed sensor improved with 2.0/1.0 weight ratio of TEHP/PVC, Moreover, the addition of 31 mg of PVC, and 62 mg of TEHP are the optimum amounts to achieve the best results.
The effect of the amount of Aliquat-336 on the sensor membrane response was investigated. The amount of Aliquat-336 that gave the best sensor response was 3.0 mg, leading to an improvement in the response of the optode (Fig. 3). A greater amount of Aliquat-336 did not have any significant effect on the sensor response and consequently, 3.0 mg of Aliquat-336 was optimized for the next study.
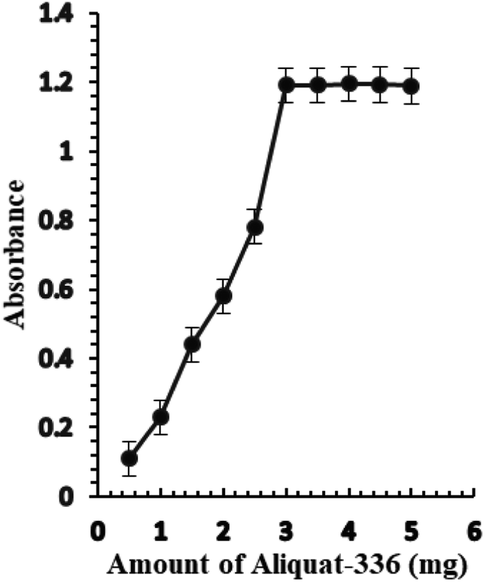 | ||
| Fig. 3 The effect of the amount of Aliquat-336 (mg) on the optical sensor response of BTMP with 1.0 × 10−5 M of Ag+. | ||
The effects of different buffer media
Taking into consideration the structure of Aliquat-336, it contains nitrogen atoms that readily react with Ag+ ions with high pH dependency for complexation; consequently, the response of the sensor based on the incorporation of the BTMP ionophore is significantly influenced by the pH of the buffer. Different types of buffer solution such as acetate, borate, thiel, phosphate, triethanolamine, and universal buffer solutions of different pH values 2.5–11 were examined. Triethanol amine, borate, and universal buffer solutions showed that the optode sensor of Ag+ ions had high absorbance and response in the basic media. Furthermore, the universal buffer with high pH values gave higher absorbance and response results. The effect of pH on the response of the sensor membrane was studied in the range of pH 2.5–11 (Fig. 4). The maximum sensor response was obtained at pH 8.5. At a high concentration of protons (lower pH), the reactive sites are protonated and protons may compete with Ag+ ions. Thus, the sensitivity significantly decreases at lower pH. In another way, at higher pH, the Ag+ ion is hydrolyzed and the carriers may be deprotonated, and charged species may also be formed in the membrane, with possible bleeding from the membrane into the aqueous solution, thus decreasing the sensitivity. On the other hand, at high pH, probably due to the formation of negative charges of the ionophores, their bleeding into the bulk solution is significantly increased and leads to a decrease in the response and sensitivities of optode.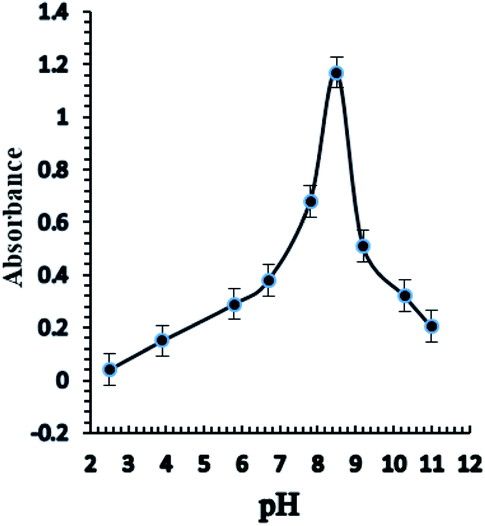 | ||
| Fig. 4 The effect of pH on the response of the optical sensor of BTMP with 1.0 × 10−5 M of Ag+ at optimum conditions. | ||
Thickness of the optode cocktail
The effect of the thickness of the cocktail volume that spread on the surface of the glass plate was studied by measuring the sensor response to 1 × 10−5 M Ag+ ions, at pH 8.5 using 2.0 mL for different membranes in the mixture cocktail range from (50–400 μL). The formed optode gave the best response to the silver cations when 250 μL of the cocktail was spread on the surface of the glass plate, which decreased the concentration of the silver cations in the sample solution.Response time
The time needed to spread the analyte from the bulk solution to the optode interface and to associate with the reagent is called the response time, which is a significant analytical indicator for all the optodes. On the other hand, the response time of the proposed sensor is the required time for the analyte diffusion from the bulk solution into the optical membrane interface, followed by the association of BTMP with 1.0 × 10−5 M of Ag+ ions. The response time of the optical sensor was determined from absorption data at 481 nm, the detected response time of the optode sensor was found to be 5.0–10 min. Response time is affected by different parameters including the ionophore loading technique as well as membrane thickness and Ag+ ion concentration that controls its diffusion into the membrane.32Membrane properties
The properties of the optical sensor membrane were measured at 481 nm from individual solutions 1.0 × 10−5 M of Ag+ ions. These properties include lifetime and stability, repeatability, reproducibility, regeneration, and selectivity.Lifetime
The lifetime of the optode was measured by adding a universal buffer solution of pH 8.5 to a cuvette containing the sensor membrane. For 12 h, at λmax of 481 nm, the absorbance was measured. There was no bleeding of the BMTP into the bulk solution from the sensor membrane; consequently, no significant loss happened during this time. The absorbance of the membrane remained unchanged, which may be attributed to the fixed composition of the membrane. The stability of the proposed sensor membrane was detected by submerging the membrane under water for hours. The sensor membrane stability, as shown by the result, did not change during this period.Repeatability and reproducibility
The repeatability of the sensor membrane response was tested at λmax 481 nm using only a single sensor membrane by recording the absorbance of different replicate measurements for 1.0 × 10−5 M of Ag+. The relative standard deviation (RSD) for the sensor membrane response for Ag+ (1.0 × 10−5 M) was found to be ≤1.85%. The reproducibility of the proposed sensor using several similar sensor membranes was tested by performing measurements. The relative standard deviation (RSD) for these determinations was found to be ≤ 2.20.Regeneration
The regeneration of the membrane response was tested by multiple usages of each sensor for Ag+ ion detection in test solutions of 1.0 × 10−5 M Ag+. The sensor membrane was regenerated by using different regenerating reagents after each absorbance measurement. EDTA, KCl, HCl, H2SO4, HNO3, ethylenediamine, and NaOH solutions (0.1 M) were used as regenerating reagents. The best results were obtained after immersing of the optode in 0.1 M HNO3 for 30 seconds, rinsing with deionized water, and steeping in a universal buffer solution of pH 8.5 for a few mins (Scheme 2). High regeneration was achieved for Ag+ ions of 1.0 × 10−5 M and the corresponding RSD value was ± 1.22%. The membrane could be used ten times after regeneration.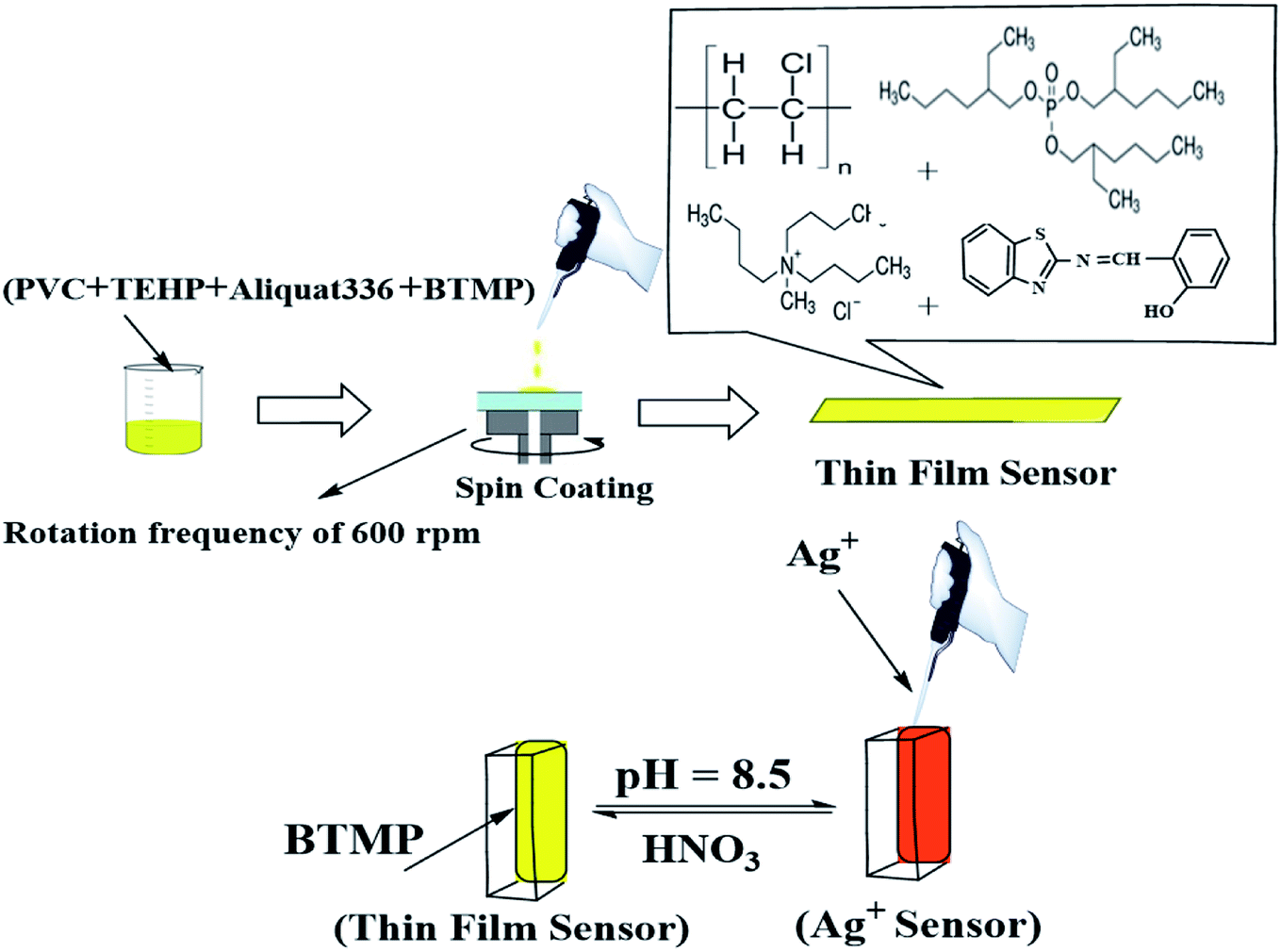 | ||
| Scheme 2 Fabrication, detection, and regeneration of the optical sensor. | ||
Sensor selectivity
The effects of interfering ions in real water samples on the recovery of silver cations were also studied. The tolerance limit for each interfering ion was detected as that which caused a change in the absorbance by ±5.0% of its original value. A selectivity coefficient higher than 15000 was obtained for Na+, K+, Li+, Tl+, NH4+, Mg2+, Ca2+, F−, Cl−, Br−, I−, ClO4−, NO3−, SO42−, PO43−, and thiourea; and higher than 12500 for Fe2+, Ni2+, Mn2+, La3+, Y3+, Sc3+, Cd2+, Al3+, Cr3+, Cr6+, citrate and tartrate; and higher than 10000 for oxalate, Fe3+, Cu2+, Sm3+, Yb3+, Co2+, La3+, Lu3+, V5+, and Zn2+.
In most biological samples, the most abundant elements include Na, Ca, K, O, N, Mg, Fe, O, C, P, and the most common microelements include Cu, Mn, Ni, Zn, Co, Al, and Cr, with other elements being present in only trace amounts.33 The tolerance limits for these elements were in excess of 1500 μg, especially K, Ca, Na, Fe, and Mg. This property can be used to obtain a desirable procedure for detecting trace amounts of Ag+ ions in plants, and seeds without any further separation. Because the entire procedure has excellent selectivity, Ultra trace concentrations of Ag+ were easily analyzed in biological samples by increasing the concentration of the sample.
Dynamic range
The dynamic range for the determination of Ag+ ions concentration was linear under the optimum conditions from 4.8 × 10−9 to 1.0 × 10−5 M (0.8494–1698.7 μg L−1) (Table 1, Fig. 5), and the calculated detection (LOD) and quantification limits were found to be 1.5 × 10−9 and 4.8 × 10−9 M (0.2548 and 0.8494 μg L−1), respectively. The obtained LOD34 was found to be much lower than the previously reported methods6,35–50 (Tables 1 and 2). In this case, it was established that the extraction of Ag+ ions from the aqueous phase increased using PVC, and consequently, the sensor sensitivity was enhanced. The proposed sensor also exhibited better sensitivity as compared with other methods reported for Ag+ ions.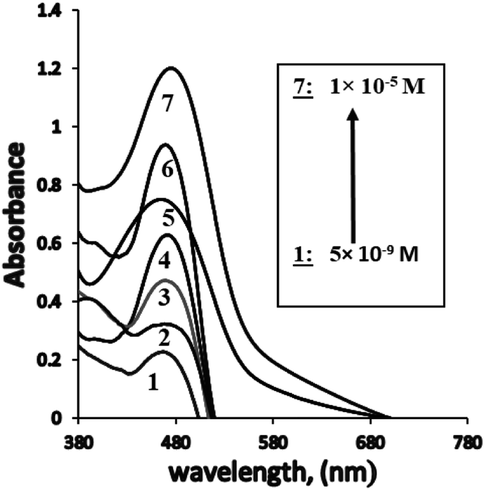 | ||
| Fig. 5 The absorption spectra obtained for BTMP with different bulk concentrations of Ag+[ 5.0 × 10−9 M: 1.0 × 10−5 M] at optimum conditions. | ||
| Method | Linear range, M | Detection limit, M | Ref. |
|---|---|---|---|
| Ion selective electrode | 5.0 × 10−8 to 1.0 × 10−1 | 1.0 × 10−8 | 42 |
| Ion selective electrode | 1.0 × 10−7 to 1.5 × 10−2 | 8.6 × 10−8 | 43 |
| Ion selective electrode | 1.0 × 1 0−2 to 1.0 × 10−7 | 4.0 × 10−8 | 44 |
| Differential pulse anodic stripping voltammetry | 5.09 × 10−8 to 2.0 × 10−6 | 4.8 × 10−8 | 45 |
| Inductively coupled plasma Mass spectrometry | 5.9 × 10−7 to 1.69 × 10−3 | 3.8 × 10−7 | 46 |
| Electrospray-ionization tandem mass spectrometry | 1.77 × 10−7 to 28 × 10−2 | 2.7 × 10−9 | 47 |
| Dispersive liquid–liquid microextraction | 1.0 × 10−6 to 2.0 × 10−2 | 3.0 × 10−8 | 48 |
| Ionic liquid in simultaneous microextraction -ET-AAS | 9.2 × 10−9 to 1.1 × 10−6 | 2.7 × 10−9 | 49 |
| Liquid–liquid microextraction based on ionic liquid | 4.9 × 10−8 to 1.18 × 10−4 | 3.2 × 10−8 | 50 |
| Optical chemical sensor | 10−6 to 10−1 | 4.0 × 10−5 | 38 |
| Optical chemical sensor | 5.0 × 10−9 to 5.0 × 10−5 | 1.8 × 10−9 | 41 |
| Optical chemical sensor | 2.27 × 10−11 to 1.13 × 10−3 | 9.5 × 10−12 | 6 |
| Optical chemical sensor | 5.0 × 10−9 to 1.0 × 10−5 | 1.5 × 10−9 | This work |
Analytical applications
For evidence of the useful advantages of the proposed optode, different natural water samples were analyzed. The system was investigated under the optimized conditions mentioned in (Table 3). The accuracy was evaluated by comparing the obtained results with those achieved using FAAS. By applying the F-test and t-test,51 there was no significant difference at the 95% confidence level. The standard addition technique was applied for the determination of silver in terms of validation.27| Sample | Added, μg L−1 | Founda (μg L−1) | Recovery (%) | t-test | F-value | |
|---|---|---|---|---|---|---|
| Proposed | FAAS | |||||
| a Mean of five extractions. ND: not detected.b From the drinking water system of Beha, Egypt.c From the Benha river water (Nile river).d Mediterranean Sea water.e Collected at Benha City, Egypt (Dec. 2020).f From Sewa mineral water.g From the rinse water in photography. | ||||||
| Tap waterb | — | ND | — | |||
| 75 | 74.0 ± 0.2 | 73.9 ± 0.9 | 98.7 | 1.23 | 2.69 | |
| 150 | 152.3 ± 0.4 | 151.6 ± 1.0 | 101.5 | 1.56 | 2.94 | |
| River waterc | — | ND | — | |||
| 40 | 40.9 ± 0.2 | 39.0 ± 0.8 | 102.3 | 1.52 | 2.57 | |
| 80 | 78.6 ± 0.5 | 81.5 ± 0.9 | 98.3 | 1.42 | 2.79 | |
| Sea waterd | — | ND | — | |||
| 90 | 89.0 ± 0.3 | 91.8 ± 1.1 | 98.9 | 1.91 | 3.69 | |
| 180 | 184.0 ± 0.5 | 176.4 ± 1.3 | 102.2 | 1.71 | 3.06 | |
| Rain watere | — | ND | — | |||
| 120 | 122.6 ± 0.5 | 118.7 ± 1.0 | 102.2 | 1.37 | 2.76 | |
| 240 | 236.0 ± 0.2 | 246.5 ± 0.7 | 98.3 | 1.34 | 2.69 | |
| Mineral waterf | — | NDd | — | |||
| 100 | 102 5 ± 0.4 | 98.7 ± 0.8 | 102.5 | 1.16 | 2.37 | |
| 200 | 197.0 ± 0.2 | 65.7 ± 0.9 | 98.5 | 1.73 | 3.13 | |
| Waste waterg | — | 63.0 ± 1.8 | 62.5 ± 1.6 | — | ||
| 50 | 111.0 ± 1.6 | 113.9 ± 1.7 | 98.3 | 1.55 | 3.02 | |
| 100 | 161.2 ± 1.4 | 165.5 ± 1.8 | 98.9 | 1.77 | 3.56 | |
The reliability of the proposed procedure was verified by applying it in the detection of trace quantities of Ag+ ions in some water (Table 3), food, and biological real samples (Table 4). The accuracy of the proposed procedure was established by carrying out some recovery experiments. Different quantities of Ag+ ions were transferred into the different samples before any pretreatment. The obtained results were recorded in Tables 3 and 4. The proposed method achieved recoveries between 98.2% and 102.5%, which proved the accuracy of the procedure. It was also successfully applied for determining Ag+ ions in sulphadiazine cream and radiological film samples. The determination of Ag+ in both samples was done using both the proposed sensor and FAAS methods by applying the standard addition method, and the results are listed in (Table 5). The obtained results from both methods were in good agreement.
| Sample | Added | Founda | Recovery (%) | ||
|---|---|---|---|---|---|
| Proposed | FAAS | Proposed | FAAS | ||
| a Mean ± SD (n = 6). | |||||
| Corn (μg g−1) | — | — | — | ||
| 35 | 34.2 ± 0.5 | 35.8 ± 0.6 | 98.5 | 102.2 | |
| 70 | 70.8 ± 0.6 | 68.9 ± 0.6 | 101.1 | 98.4 | |
| 140 | 143.5 ± 0.8 | 137.2 ± 0.6 | 102.5 | 98.0 | |
| Lentils (μg g−1) | — | — | — | ||
| 75 | 74.4 ± 0.3 | 75.7 ± 0.6 | 99.2 | 100.9 | |
| 150 | 148.4 ± 0.4 | 151.9 ± 0.6 | 98.9 | 101.2 | |
| 225 | 221.6 ± 0.5 | 228.7 ± 0.6 | 98.4 | 101.6 | |
| Green tea (μg g−1) | — | 0.00 | 0.00 | — | — |
| 25 | 25.5 ± 0.3 | 24.7 ± 0.5 | 102.0 | 98.8 | |
| 50 | 48.1 ± 0.4 | 51.6 ± 0.6 | 98.2 | 103.2 | |
| 100 | 99.0 ± 0.2 | 102.3 ± 0.7 | 99.0 | 102.3 | |
| Rice (μg g−1) | — | — | — | ||
| 60 | 60.9 ± 0.5 | 59.0 ± 0.6 | 101.5 | 98.3 | |
| 120 | 118.5 ± 0.7 | 121.6 ± 0.6 | 98.7 | 101.3 | |
| 180 | 182.6 ± 0.3 | 177.8 ± 0.6 | 101.4 | 98.7 | |
| Spices (μg g−1) | — | — | — | ||
| 45 | 44.4 ± 0.3 | 45.8 ± 0.6 | 98.6 | 101.7 | |
| 90 | 91.0 ± 0.5 | 89.0 ± 0.6 | 101.1 | 98.8 | |
| 180 | 177.8 ± 0.8 | 183.1 ± 0.6 | 98.7 | 101.7 | |
| Wheat (μg g−1) | — | — | — | ||
| 30 | 30.6 ± 0.5 | 29.4 ± 0.6 | 102.0 | 98.0 | |
| 60 | 59.0 ± 0.6 | 61.0 ± 0.6 | 98.3 | 101.6 | |
| 120 | 118.3 ± 0.7 | 122.2 ± 0.6 | 98.5 | 101.8 | |
| Human blood (μg L−1) | — | — | — | ||
| 50 | 50.8 ± 0.2 | 49.2 ± 0.5 | 101.6 | 98.4 | |
| 100 | 98.2 ± 0.1 | 102.6 ± 0.4 | 98.2 | 102.6 | |
| 200 | 203.5 ± 0.3 | 195.7 ± 0.7 | 101.7 | 97.8 | |
| Human urine (μg L−1) | — | 0.16 | — | — | |
| 40 | 40.6 ± 0.2 | 39.2 ± 0.7 | 101.5 | 98.0 | |
| 80 | 79.2 ± 0.5 | 80.9 ± 0.3 | 99.0 | 101.1 | |
| 160 | 157.5 ± 0.1 | 161.6 ± 0.4 | 98.4 | 101.0 | |
| Sample | Added μg L−1 | Founda (μg L−1) | Recovery% | t-testb | F-valueb | |
|---|---|---|---|---|---|---|
| Proposed | FAAS | |||||
| a Results average of six consecutive measurements.b Theoretical values for t and F at the 95% confidence limit are 2.57 and 5.05, respectively. | ||||||
| Silver sulphadiazine | 0.00 | 7.3 ± 0.3 | 7.6 ± 0.6 | — | ||
| 25.0 | 32.7 ± 0.2 | 31.9 ± 0.7 | 101.2 | 1.26 | 2.59 | |
| 50.0 | 56.9 ± 0.4 | 58.6 ± 0.9 | 98.7 | 1.18 | 2.56 | |
| 75.0 | 81.9 ± 0.5 | 82.8 ± 0.9 | 99.1 | 1.31 | 2.74 | |
| Radiological film | 0.00 | 12.5 ± 0.5 | 12.7 ± 0.9 | — | ||
| 75 | 87.2 ± 0.3 | 88.8 ± 0.8 | 99.6 | 1.67 | 3.22 | |
| 150 | 161.4 ± 0.2 | 164.8 ± 0.7 | 99.3 | 1.43 | 2.78 | |
| 225 | 240.3 ± 0.2 | 234.5 ± 0.8 | 101.1 | 1.69 | 3.37 | |
| Photographic plate | 0.00 | 45.5 ± 0.6 | 45.3 ± 1.3 | — | 1.52 | 3.03 |
| 50 | 96.2 ± 0.4 | 93.4 ± 1.1 | 100.7 | 1.46 | 2.95 | |
| 100 | 144.1 ± 0.3 | 147.6 ± 0.8 | 99.0 | 1.83 | 3.87 | |
| 150 | 196.9 ± 0.3 | 192.7 ± 0.9 | 100.7 | 1.37 | 2.98 | |
In order to validate the advantage of the proposed system, silver solutions with concentrations between 0.04 μg L−1 and 0.2 μg L−1 were spiked to prepare samples by using 100 mL of the water sample. Table 3, represents the calculated results for the spiked silver samples. The RSD was less than 2.15%. The recovery values calculated for the water ranged between 98.25% and 102.50%. The obtained results were compared with the data obtained from FAAS in order to evaluate the accuracy of the proposed method. No considerable difference at 95% confidence level was observed after applying both t-test and F-value.51
The accuracy of the proposed procedure was confirmed by applying the method to determine Ag+ in several food and plant leaf samples obtained from supermarkets in Benha, Egypt. The proposed procedure was applied to different sets of samples including, rice, lentils, wheat products, spices, corn, and green tea. From the results given in (Table 4), it is clear that the Ag+ ion content varies from 0.025 to 0.225 μg g−1. Otherwise, the values of recovery obtained by using the standard addition method confirmed that the investigated procedure is not affected by the matrix interferences. The investigated procedure can be also applied for natural food and plant leaves analysis with good agreement. The reliability of the proposed method was evaluated by spiking the sample and/or comparing the results with the data detected by FAAS. From the obtained data presented, the recovery by spiking the sample is good, with satisfactory agreement between the obtained results and data achieved from the analysis of FAAS, confirming that the proposed method is reliable and applicable for the samples examined.
Conclusion
The proposed sensor membrane based on the BTMP Schiff base showed high selectivity for Ag+ ions as compared to other interfering metal ions. The proposed optode is adequate for the direct detection of Ag+ ions in different categories of samples such as natural water, food, environmental and biological samples since the membrane possesses high sensitivity with limits of detection and quantification of 1.5 × 10−9 and 4.8 × 10−9 M (0.2548 and 0.8494 μg L−1), respectively. The establishment of the ascribed sensor membrane is very simple and the formation of a covalent bond between the membrane and BTMP gives optodes with long lifetimes and short response times without any proof of reagent bleeding. The ascribed membrane was easily regenerated by 0.1 M HNO3 and the performance was reproducible. A comparison was made between the results obtained in the present study and other recent studies using different techniques for Ag+ monitoring.Ethics approval and consent to participate
All biological studies were carried out in strict accordance with the animal welfare guidelines of the World Organization for Animal Health. All biological experiments were performed using protocols approved by the Laboratory Animal Ethics Committee of “Egypt”.Conflicts of interest
There are no conflicts to declare.References
- C. Göde, M. L. Yola, A. Yılmaz, N. Atar and S. Wang, J. Colloid Interface Sci., 2017, 508, 525–531 CrossRef PubMed.
- V. K. Gupta, M. L. Yola, N. Atar, A. O. Solak, L. Uzun and Z. Üstündağ, Electrochim. Acta, 2013, 105, 149–156 CrossRef CAS.
- V. K. Gupta, M. L. Yola, N. Atar, Z. Ustundağ and A. O. Solak, Electrochim. Acta, 2013, 112, 541–548 CrossRef CAS.
- M. L. Yola, N. Atar, M. S. Qureshi, Z. Üstündağ and A. O. Solak, Sens. Actuators, B, 2012, 171, 1207–1215 CrossRef.
- M. L. Yola, T. Eren, H. İlkimen, N. Atar and C. Yenikaya, J. Mol. Liq., 2014, 197, 58–64 CrossRef CAS.
- A. R. Firooz, A. A. Ensafi, N. Kazemifard and R. Khalifeh, Sens. Actuators, B, 2013, 176, 598–604 CrossRef CAS.
- A. B. G. Lansdown, Adv. Pharm. Sci., 2010, 2010, 1–16 CrossRef PubMed.
- P. Biparva and M. R. Hadjmohammadi, Clean–Soil, Air, Water, 2011, 39, 1081–1086 CrossRef CAS.
- R. Behra, L. Sigg, M. J. Clift, F. Herzog, M. Minghetti, B. Johnston, A. Petri-Fink and B. Rothen-Rutishauser, J. Royal Soc., Inter., 2013, 10, 20130396 CrossRef PubMed.
- M. Korani, E. Ghazizadeh, S. Korani, Z. Hami and A. Mohammadi-Bardbori, Eur. J. Nanomed., 2015, 7, 51–62 CAS.
- M. L. Soriano, C. Ruiz-Palomero and M. Valcárcel, Anal. Bioanal. Chem., 2019, 411, 5023–5031 CrossRef CAS PubMed.
- A. Mohadesi and M. A. Taher, Talanta, 2007, 71, 615–619 CrossRef CAS PubMed.
- T. Wang, X. Jia and J. Wu, J. Pharm. Biomed. Anal., 2003, 33, 639–646 CrossRef CAS PubMed.
- D. M. Mitrano, A. Barber, A. Bednar, P. Westerhoff, C. P. Higgins and J. F. Ranville, J. Anal. At. Spectrom., 2012, 27, 1131–1142 RSC.
- P. Nagaraja, M. S. H. Kumar and H. S. Yathirajan, Anal. Sci., 2002, 18, 815–817 CrossRef CAS PubMed.
- G. Fang, S. Meng, G. Zhang and J. Pan, Talanta, 2001, 54, 585–589 CrossRef CAS PubMed.
- M. Ghaedi, Spectrochim. Acta, Part A, 2007, 66, 295–301 CrossRef PubMed.
- W. M. Mukhtar, R. M. Halim, K. A. Dasuki, A. R. Abdul Rashid and N. A. M. Taib, Mal. J. Fund. Appl. Sci., 2017, 13, 619–622 CrossRef.
- W. E. Morf, K. Seiler, B. Lehmann, C. Behringer, K. Hartman and W. Simon, Pure Appl. Chem., 1989, 61, 1613–1618 CAS.
- M. Phichi, A. Imyim, T. Tuntulani and W. Aeungmaitrepirom, Anal. Chim. Acta, 2020, 1104, 147–155 CrossRef CAS PubMed.
- X. Wang, Y. Zhou, V. Decker, M. Meyerhoff, M. Sun and Y. Cui, Anal. Methods, 2020, 12, 2547–2550 RSC.
- Z. Al-Mallah and A. S. Amin, J. Ind. Eng. Chem., 2018, 63, 281–287 CrossRef CAS.
- A. Amin, Eurasian J. Anal. Chem., 2018, 13, 5–13 Search PubMed.
- H. H. El-Feky, S. M. El-Bahy, A. M. Hassan and A. S. Amin, Inter. J. Env. Anal. Chem., 2021, 1–18 Search PubMed.
- Y. K. Reddy, S. Rao and N. Raju, Talanta, 1975, 22, 545–547 CrossRef CAS PubMed.
- H. T. S. Britton, Hydrogen Ions, Chapman and Hall, London, 4th edn, 1952 Search PubMed.
- M. K. Amini, M. Ghaedi, A. Rafi, I. Mohamadpoor-Baltork and K. Niknam, Sens. Actuators, B, 2003, 96, 669–676 CrossRef CAS.
- H. H. Nadiki, M. A. Taher, H. Ashkenani and I. Sheikhshoai, Analyst, 2012, 137, 2431–2436 RSC.
- M. Ghaedi and A. Shokrollahi, Fresenius Envir. Bull., 2006, 15, 1373–1381 CAS.
- H. Li, J. Li, H. Chen, Y. Zhang and D. Huang, J. Chem. Crystallogr., 2011, 41, 1844–1849 CrossRef CAS.
- O. Yavuz, M. Sezen, Y. Alcay, M. S. Yildirim, K. Kaya, Y. Ozkilic, N. Ş. Tuzun and I. Yilmaz, Sens. Actuators, B, 2021, 329, 129002 CrossRef CAS.
- C. De Stefano, L. Ferrigno, F. Fontanella, L. Gerevini and A. S. di Freca, Pattern Recognit. Lett., 2020, 135, 375–381 CrossRef.
- K. Caballero-Gallardo, M. Alcala-Orozco, D. Barraza-Quiroz, J. De la Rosa and J. Olivero-Verbel, Chemosphere, 2020, 242, 125173 CrossRef CAS PubMed.
- IUPAC, Spectrochim. Acta, Part B, 1978, 33, 241–245 CrossRef.
- S. Chaiyo, W. Siangproh, A. Apilux and O. Chailapakul, Anal. Chim. Acta, 2015, 866, 75–83 CrossRef CAS PubMed.
- N. Lamaiphan, C. Sakaew, P. Sricharoen, P. Nuengmatcha, S. Chanthai and N. Limchoowong, J. Korean Ceram. Soc., 2021, 58, 314–329 CrossRef CAS.
- S. Motia, B. Bouchikhi, E. Llobet and N. El Bari, Talanta, 2020, 216, 120953 CrossRef CAS PubMed.
- M. Hassouna, S. Elsuccary and J. Graham, Sens. Actuators, B, 2010, 146, 79–90 CrossRef CAS.
- M. Shamsipur, K. Alizadeh, M. Hosseini, C. Caltagirone and V. Lippolis, Sens. Actuators, B, 2006, 113, 892–899 CrossRef CAS.
- M. Noroozifar, M. K. Motlagh, A. Taheri and R. Z. Dorabei, Turk. J. Chem., 2008, 32, 249–257 CAS.
- M. Shamsipur, S. Rouhani, A. Mohajeri and M. R. Ganjali, Anal. Bioanal. Chem., 2003, 375, 692–697 CrossRef CAS PubMed.
- J. Q. Lu, D. W. Pang, X. S. Zeng and X. W. He, J. Electroanal. Chem., 2004, 568, 37–43 CrossRef CAS.
- M. H. Mashhadizadeh, A. Shockravi, Z. Khoubi and D. Heidarian, Electroanalysis, 2009, 21, 1041–1047 CrossRef CAS.
- E. Jeong, M. S. Ahmed, H.-S. Jeong, E.-H. Lee and S. W. Jeon, Bull. Kor. Chem. Soc., 2011, 32, 800–804 CrossRef CAS.
- J. B. Raoof, R. Ojani, A. Alinezhad and S. Z. Rezaie, Monatsh. Chem., 2010, 141, 279–284 CrossRef CAS.
- Q. B. Lin, B. Li, H. Song and H. J. Wu, Food Addit. Contam., Part A, 2011, 28, 1123–1128 CrossRef CAS PubMed.
- K. Minakata, I. Yamagishi, H. Nozawa, K. Gonmori, K. Hasegawa, M. Suzuki, F. Horio, K. Watanabe and O. Suzuki, Forensic Toxicol., 2012, 30, 149–155 CrossRef CAS.
- L. Kocúrová, I. S. Balogh, L. Nagy, F. Billes, A. Simon and V. Andruch, Microchem. J., 2011, 99, 514–522 CrossRef.
- H. Ashkenani and M. A. Taher, Microchem. J., 2012, 103, 185–190 CrossRef CAS.
- M. Vaezzadeh, F. Shemirani and B. Majidi, J. Anal. Chem., 2012, 67, 28–34 CrossRef CAS.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Prentice-Hall, London, 5th edn, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |