
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Na4PMo11VO40-catalyzed one-pot oxidative esterification of benzaldehyde with hydrogen peroxide†
Castelo Bandane Vilanculo *a and
Márcio José da Silvab
*a and
Márcio José da Silvab
aChemistry Department, Pedagogic University of Mozambique, FCNM, Campus of Lhanguene, Av. De Moçambique, Km 1, Maputo, 4040, Mozambique. E-mail: castelovilanculo@gmail.com; Tel: +258 825573337
bChemistry Department, Federal University of Viçosa, Minas Gerais State 36590-000, Brazil
First published on 28th October 2021
Abstract
The activity of the sodium salts of vanadium-doped phosphomolybdic acid was assessed in the oxidative esterification reaction of benzaldehyde with hydrogen peroxide in alkyl alcohol solutions. The effect of main reaction parameters, such as temperature, catalyst load, vanadium doping level, and reactant stoichiometry, on the conversion and reaction selectivity was investigated. Among the tested heteropoly salts, Na4PMo11VO40 was the most active and selective catalyst, achieving almost complete conversion of benzaldehyde and high ester selectivity regardless of the alcohol investigated. The efficiency of the catalyst was correlated with its vanadium content. The size of the carbon chain of alcohol and the steric hindrance on the hydroxyl group played a key role in the reaction selectivity. While methyl and ethyl alcohols selectively provided the ester as the main product (ca. 90–95%) and benzoic acid as a subproduct, the other alcohols also afforded acetal, a condensation product, and benzaldehyde peroxide, an oxidation reaction intermediate, as secondary products. The use of an inexpensive, environmentally benign, and atom-efficient oxidant, mild conditions, and short reaction times were the positive aspects of this one-pot process.
1. Introduction
The synthesis of aromatic esters has gained attention due to the wide utility of these compounds as raw materials in the industrial production of resins, perfumes, cosmetics, fibers, plasticizers, and dyes.1 However, the traditional routes of ester production involve hazardous and environmentally unfriendly reagents, which leads to large generation of stoichiometric quantities of residues and effluents.2To circumvent these drawbacks, alternative routes to the traditional stoichiometric oxidation processes, such as the direct transformation in one-pot reactions of aldehydes into esters, have been developed.3 Indeed, the oxidative esterification of aldehydes has been raised as a sustainable and efficient alternative to classical synthesis.4,5 Several Lewis acid metal-catalyzed reactions using tertbutyl peroxide or hydrogen peroxide as an oxidant have been described.6–9
In this sense, hydrogen peroxide is an easy-handling liquid reactant that is inexpensive, atom-efficient, and non-flammable; it is also a green oxidant that generates only water as a by-product.10,11 The choice of an adequate solvent avoids the use of a phase transfer agent as well as the addition of pH controllers.12–14 Nonetheless, hydrogen peroxide requires an activation step, which is generally performed by a metal catalyst such as an oxide, salt, or organometallic compound.15–18
Solid-supported catalysts have been demonstrated to be active in the oxidation of aldehydes with hydrogen peroxide.19–23 Thakur et al. investigated oxidation with hydrogen peroxide of aromatic and aliphatic aldehydes to methyl esters over a VO(acac)2/TiO2 catalyst.24 In this sense, vanadium catalysts have been highlighted as effective catalysts in several oxidation reactions, mainly when used in Keggin heteropoly compounds. Keggin heteropolyacids (HPAs) are polyoxometalates that are widely used as catalysts due to their acidic and redox properties; thus, they are potentially active in oxidative esterification reactions.25,26 They have been widely used as catalysts in oxidation reactions with hydrogen peroxide.27–30
Keggin HPAs are well-defined metal–oxygen clusters in which oxygen atoms link tungsten or molybdenum atoms, resulting in octahedral units that are tetrahedrally arranged around a heteroatom (i.e., phosphorus or silicon atom).31,32 Because Keggin HPAs are soluble in polar solvents, they have been used as solid-supported catalysts.33,34 Alternatively, Keggin HPAs can be converted to solid salts, exchanging their protons by larger radium cations as cesium or potassium.35,36
Two other interesting approaches may improve the performance of Keggin HPAs in catalytic oxidation reactions: the removal of an MO unit (i.e., M = W or Mo), generating lacunar catalysts,37,38 and the exchange of an addenda atom (i.e., tungsten or molybdenum) by vanadium atom.39,40 Lacunar Keggin HPA catalysts were successfully used in the oxidation of aldehydes with hydrogen peroxide.41,42
On the other hand, the simple exchange of Mo or W by V atoms in the primary structure of a heteropolyanion can accelerate the steps of oxidation–reduction, enhancing the activity and selectivity of oxidation reactions.43–45 Particularly, molybdenum-based HPAs have been found to be better catalysts for oxidation reactions than their tungsten counterparts.46,47 Moreover, vanadium-doped phosphomolybdic catalysts have been generally used as acids, an aspect that can compromise the selectivity of oxidation reactions.
In this work, for the first time as far as we know, sodium phosphomolybdate salts were doped with different vanadium loads and used as catalysts in oxidative esterification reactions of benzaldehyde with hydrogen peroxide in alcoholic solutions. The focus was to assess the impacts of the vanadium load on the conversion and selectivity of the reactions. To accomplish this, sodium salts of phosphomolybdic acid, containing Keggin-type heteropolyanions with the general formula PMo12−nVnO40(3+n)− (n = 0, 1, 2, or 3), were synthesized and evaluated. The effects of the main reaction parameters of the reaction, such as the oxidant load, type, and concentration of the metal catalyst, temperature, and nature of the alcohol, were investigated.
2. Experimental
2.1. Materials and methods
All chemicals were purchased from commercial sources. Benzaldehyde and alkyl alcohols (i.e., methyl, ethyl, propyl, butyl, sec-propyl, sec-butyl) were all obtained from Sigma-Aldrich (99 wt%). Hydrogen peroxide was obtained from Moderna (34 wt%). Hydrated heteropolyacid H3PMo12O40 (99 wt%) was acquired from Sigma-Aldrich. V2O5 (99.6 wt%), MoO3 (99.5 wt%), H3PO4 (85 wt%), NaVO3 (98 wt%), Na2MoO4 (≥98 wt%), and CH3CN (99 wt%) were also purchased from Sigma-Aldrich. Aqueous hydrogen peroxide (35 wt%) was acquired from Alphatec, and H2SO4 (95–98 wt%) was obtained from Dinâmica.2.2. Synthesis of the Na4PMo11VO40, Na5PMo10V2O40, and Na6PMo9V3O40 catalysts
The Na4PMo11VO40 salt was synthesized according to the literature as depicted in Scheme 1.48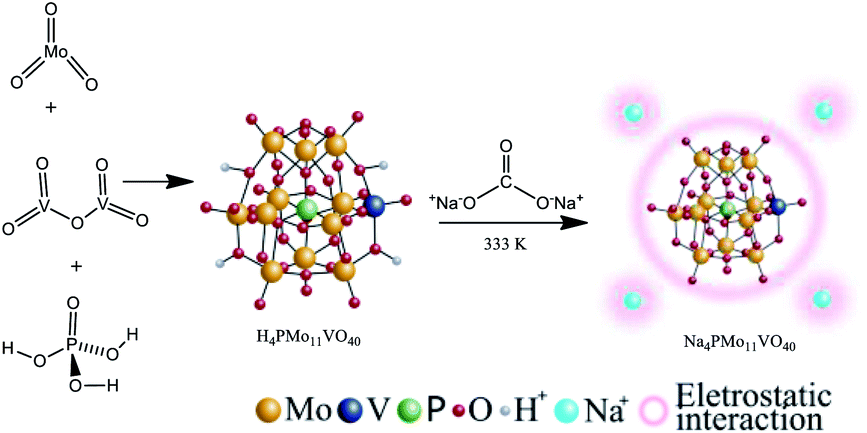 | ||
| Scheme 1 Route of synthesis of Na4PMo11VO40. | ||
Typically, stoichiometric amounts of MoO3 and V2O5 were dissolved in deionized water and heated to the boiling point. Then, phosphoric acid was added, and the resulting mixture was refluxed for 6 h. Cooling to room temperature afforded a clean solution. Evaporation of the solvent resulted in the solid acid (i.e., H4PMo11VO40), which was recrystallized. This solid was dissolved in aqueous, and an aqueous solution of sodium carbonate was added; the mixture was stirred and heated at 333 K for 3 hours. Finally, evaporation of the solvent led to the Na4PMo11VO40 salt, which was recrystallized from water and then dried at 373 K/5 h.
2.3. Synthesis of the Na5PMo10V2O40 and Na6PMo10V3O40 salts
These catalysts were synthesized according to the original and modified procedures.49,50 An aqueous solution of sodium diphosphate was added to a hot aqueous solution of sodium meta-vanadate in an adequate stoichiometric ratio (Scheme 2).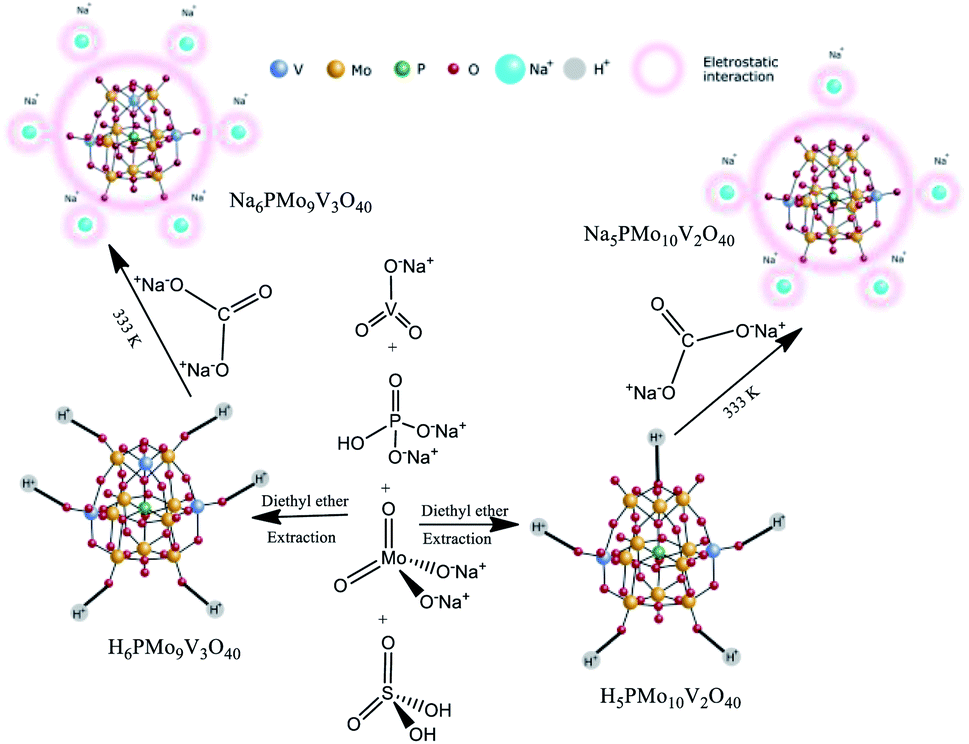 | ||
| Scheme 2 Route of synthesis of Na5PMo11V2O40 and Na6PMo11V3O40. | ||
After the mixture was cooled to room temperature, concentrated sulfuric acid (ca. 5 mL) was slowly added, and the solution developed a red color. Subsequently, a solution of sodium molybdate was added under vigorous stirring. Sulfuric acid (ca. 85 mL) was slowly added, and the solution was cooled to room temperature. The etherate extract was vapored under airflow, affording the solid acid H5PMo10V2O40. The resulting solid was dried at 343 K and later dried at 373 K/5 h. The solid acid was solved in water, and a Na2CO3 solution was added, mixed, and heated at 333 K for 3 hours. Evaporation of the water afforded a Na5PMo10V2O40 salt, which after recrystallization was dried at 373 K/5 h.
A similar procedure was used to synthesize H6PMo9V3O40 and the Na6PMo9V3O40 salt, except by taking the required amounts of sodium metavanadate and sodium molybdate. In this case, the resulting solution became cherry red.
2.4. Catalyst characterization
Infrared spectroscopy analyses were recorded on a Varian 660-IR spectrometer at 400 to 1300 cm−1 wavenumbers, which is the fingerprint region of the typical absorption bands of Keggin anions. UV-visible spectroscopy analyses were obtained in a AJX-6100 PC double beam Micronal spectrometer fitted with tungsten and deuterium lamps. The spectra were recorded from CH3CN solutions with concentrations of 0.002 mol L−1, the concentration used in the catalytic reactions.Powder X-ray diffraction patterns of the vanadium heteropoly salts were obtained using an X-ray diffraction system (model D8-Discover, Bruker) using Ni-filtered Cu-kα radiation (λ = 1.5418 Å), working at 40 kV and 40 mA, with a counting time of 1.0 s in an angle (2θ) range from 5 to 80 degrees.
The porosimetry of the catalysts was analyzed by N2 adsorption/desorption using a NOVA 1200e High Speed Automated Surface Area and Pore Size Analyzer (Quantachrome Instruments). The samples were previously degassed for 1 h. The surface areas of the salts were calculated by applying the Brunauer–Emmett–Teller equation (BET) to the desorption/adsorption isotherms. Thin sections of salts were selected to characterize their surfaces, which were metalized with carbon and analyzed through scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) using a JEOL JSM 6010LA SEM.
The catalyst acidity was estimated by potentiometric titration, as described by Pizzio et al.51 The electrode potential variation was measured with a potentiometer (i.e., Bel, model W3B). Typically, 50 mg of vanadium salts were dissolved in CH3CN and then titrated with n-butylamine toluene solution (ca. 0.05 mol L−1).
2.5. Catalytic runs
Catalytic tests were carried out in a glass reactor (ca. 25 mL), fitted with a reflux condenser and sampling septum, in a glycerine bath. Typically, benzaldehyde (ca. 2.75 mmol) and the vanadium heteropoly salt catalyst (ca. 1.77 mol%) were dissolved in C2H5OH (ca. 10 mL solution) at reaction temperature (ca. 333 K). The solution was magnetically stirred, and aqueous H2O2 solution (ca. 8.25 mmol) was slowly added, starting the reaction.The reactions were followed by GC analysis of regularly collected samples (GC 2010 Shimadzu, capillary column, FID). The reaction products were identified by GC-MS analysis (GC-MS 2010 ultra mass, i.e., 70 eV) and co-injection in GC equipment with analytical patterns (i.e., benzoic acid), or by comparison with authentic samples that were previously synthesized (i.e., acetal and esters). The mass balance of the reaction was checked by comparing the GC peak area of the substrate consumed with the sum of the corrected GC peak area of the main products.
The reaction conversions were calculated through eqn (1), comparing the GC peak area of benzaldehyde in each reaction (Ai) with the initial area (A0).
| % conversion = (A0 − Ai)/A0 × 100 | (1) |
The selectivity was calculated from eqn (2), where the corrected area of the GC peak of each product (Ap) was compared to the initial area of the GC peak of benzaldehyde (A0) – eqn (2)
| % selectivity = (Ap/A0) × 100 | (2) |
The selectivity of benzaldehyde peroxide, which is an undetected product by GC analysis, was calculated using eqn (3).
| % benzaldehyde peroxide = (consumed GC peak area of benzaldehyde − ΣGC peak corrected area of products)/consumed GC peak area of substrate × 100 | (3) |
3. Results and discussion
3.1. Catalyst characterization
The characterization of the vanadium-doped sodium phosphomolybdate catalysts was previously discussed in other work, where they were used in the epoxidation of terpene alcohols with hydrogen peroxide.52 Notwithstanding, all the important data obtained in the characterization (i.e., infrared spectra, powder XRD patterns, EDS analyses, and measurements of the strength of the acidic sites) are shown in the supplemental material (Fig. 1SM–3SM†). The most important characterization data and the respective assumptions are summarized as follows:• The integrity of the primary structures of the vanadium-doped heteropolyanions was confirmed by infrared spectroscopy analysis (Fig. 1SM†). The fingerprint region of the infrared spectra presents the typical absorption bands of a Keggin anion.53 A comparison of the infrared spectra of the vanadium salts and undoped sodium phosphomolybdate salt clearly showed that the primary structure of the catalyst (i.e., Keggin heteropolyanion) remained almost untouched after the synthesis of the salts (Fig. 1SM†). A more detailed characterization and better discussion of the infrared spectra of these catalysts was previously presented when they were used in oxidation reactions of terpenic alcohols.52
• The analysis of the powder XRD patterns allows us to verify if any changes occurred in the secondary structure of Keggin HPAs when their protons were exchanged by sodium cations and when vanadium ions were introduced into the heteropolyanion. X-Ray diffractograms of the sodium phosphomolybdate salts evidenced that vanadium doping increased the level of crystallinity, preserving the main diffraction peaks between the 5 and 40° 2θ angles. This suggests that both the primary (i.e., Keggin anion) and secondary structures were preserved. New diffraction signals were noticed in the low angle region (ca. 2θ 10°) of all the XRD diffractograms of the salts. Moreover, new diffraction peaks at 2θ angles greater than 40° (ca. 47° and 50° angles) appeared in the diffractogram of the monosubstituted salt (Fig. 2SM†). These changes are attributed to the difference between the ionic radius of the hydrate protons (i.e., H3O+, H2O5+) and Na+ ions, which may affect the packaging of the heteropolyanions on the secondary structure as well as the different hydration levels of the salts.54 A comparison of the XRD patterns of the sodium salts with their respective acids allow us to conclude that the sodium salts presented a body-centered cubic structure, which remained almost intact after the inclusion of one vanadium atom.55,56
• The strength of the acidic sites of the phosphomolybdic catalysts was estimated by measuring the initial electrode potential (i.e., Ei) of their acetonitrile solutions (Fig. 3SM†). While the phosphomolybdic acid displayed a value of Ei = 680 mV,52 its unsubstituted and vanadium-monosubstituted sodium salts presented Ei values equal to 400 and 370 mV, respectively. However, an increase in vanadium load drastically reduced the acidity strength of the sodium phosphomolybdate salts; the values of Ei measured in the solutions of Na5PMo10V2O40 and Na6PMo9V3O40 salts were equal to 65 and −100 mV, respectively (Fig. 3SM†). According to the literature, the two first salts have very strong acidic sites (Ei > 100 mV) and Na5PMo10V2O40 has strong acidic sites (0 < Ei < 100 mV), while the Na6PMo9V3O40 salt has weak acidic sites (−100 < Ei < 0 mV).51
• The MEV images obtained from the vanadium-doped phosphomolybdate sodium salts revealed that the particles of Na4PMo11VO40 are smaller than those of the undoped Na3PMo12O40. Therefore, the vanadium doping increased their surface area, as demonstrated by BET analysis.52 In the elemental analysis, the percentual elemental compositions of the sodium phosphomolybdate salts were confirmed by EDS analysis.52
• The hydration levels of the vanadium-doped phosphomolybdate sodium salts were determined by TG/DTG analyses.52,57 It was verified that upon increasing the number of vanadium atoms doped into the heteropolyanion, the number of water molecules increased (ca. 7, 10, and 13 water moles when 1, 2, or 3 vanadium atoms were doped into the anion, respectively). Some of the changes observed in the XRD patterns of the sodium salts (Fig. 2SM†) can be assigned to the distinct hydration levels.52 Finally, the DSC analyses of the phosphomolybdate salts also confirmed that the vanadium doping increased the thermal stability.52
3.2. Catalytic tests
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of oxidant to substrate.
:1 molar ratio of oxidant to substrate.Comparing the performance of the sodium phosphomolybdate catalysts with and without vanadium, we can realize that the doping triggered a noticeable improvement in the activity of the heteropoly catalyst. However, when the content of vanadium was increased from V1 to V2 or V3, a decrease in the conversion of the reactions was noticed (Fig. 1).
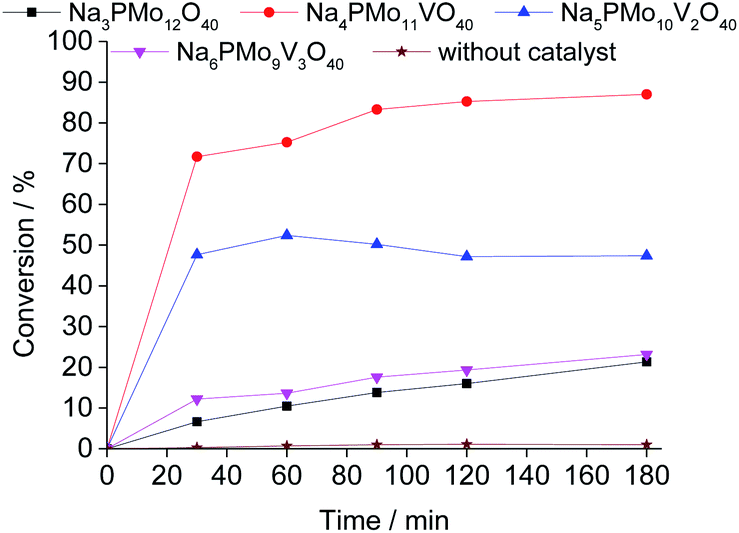 | ||
| Fig. 1 Kinetic curves of oxidative esterification reactions of benzaldehyde with H2O2 in C2H5OH solutions in the presence of undoped and banadium-doped sodium phosphomolybdate catalysts. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), catalyst (1.77 mol%), toluene (0.1 mL), C2H5OH (10.0 mL), 333 K. | ||
In terms of selectivity, it was clear that the Na4PMo11VO40 salt was the most efficient catalyst (Fig. 2). In general, 4 products were formed in all the reactions (Scheme 3). Benzaldehyde peroxide and benzoic acid were oxidation products and ethyl benzoate was the product of oxidation, followed by esterification; also, benzaldehyde acetal was formed through the condensation reaction of benzaldehyde and ethyl alcohol.
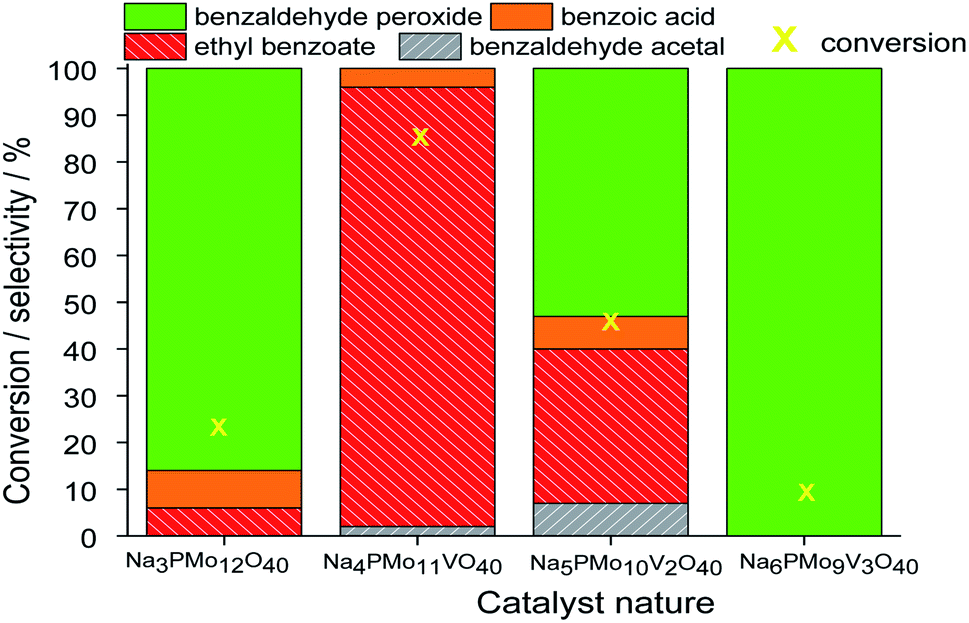 | ||
| Fig. 2 Effect of the catalyst nature on the conversion and selectivity of oxidative esterification reactions of benzaldehyde with H2O2 in C2H5OH in the presence of undoped and vanadium-doped sodium phosphomolybdate catalysts. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), catalyst (1.77 mol%), toluene (0.1 mL), C2H5OH (10.0 mL), 333 K. | ||
 | ||
| Scheme 3 Products of the sodium phosphomolybdate-catalyzed oxidative esterification of benzaldehyde with hydrogen peroxide in ethyl alcohol solutions. | ||
While the reaction rate was almost unaffected, the vanadium content had a remarkable impact on the product distribution (Fig. 3). Without vanadium, the reaction does not provide oxidation products in a significant amount. Conversely, the Na4PMo11VO40-catalyzed reaction achieves the maximum conversion (ca. 90%) and highest ester selectivity within the first reaction hour (Fig. 3). From this vanadium content, as the load increased, the conversion and the ester selectivity decreased. Noticeably, the efficiency of Na6PMo9V3O40 was lower than that of Na3PMo12O40. This effect deserves to be investigated in future work.
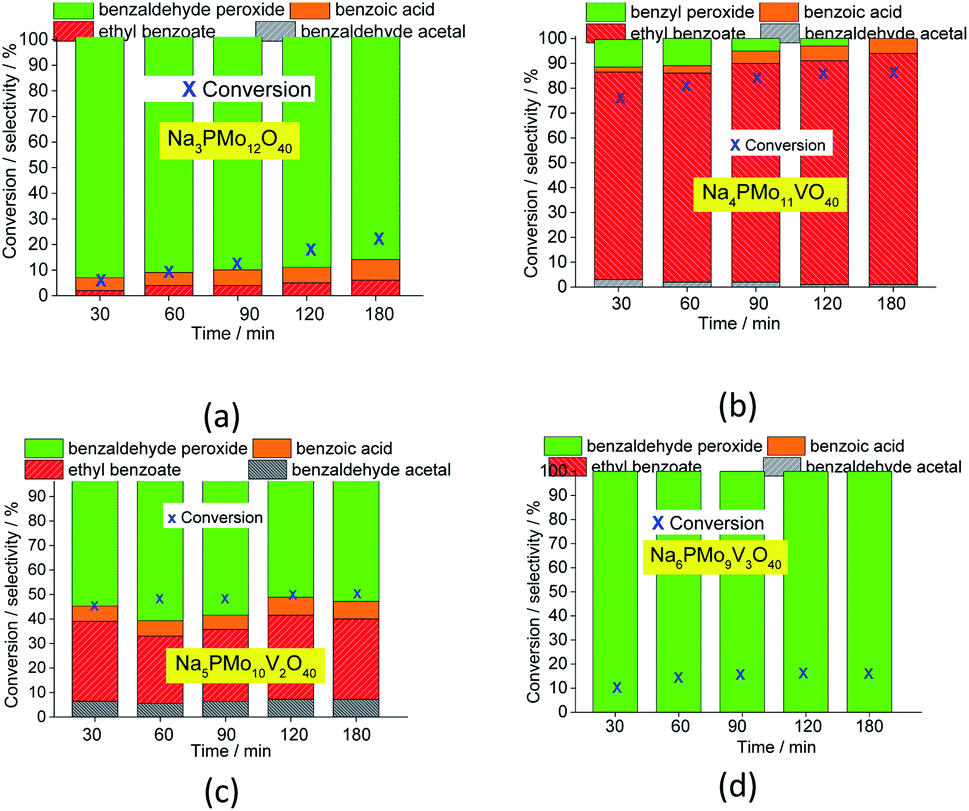 | ||
| Fig. 3 Variations of the conversion and selectivity of oxidative esterification reactions of benzaldehyde with H2O2 in C2H5OH solutions in the presence of pristine (a) and vanadium-doped (b–d) sodium phosphomolybdate catalysts. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), catalyst (1.77 mol%), toluene (0.1 mL), C2H5OH (10.0 mL), 333 K. | ||
In all the other reactions, in addition to the lower conversions, benzaldehyde peroxide, which is an intermediate of the oxidation reaction, was the main product. Conversely, in the Na4PMo11VO40-catalyzed reaction, methyl benzoate, a product of one-pot oxidative esterification, was more selectively formed. It is probably obtained through consecutive steps of oxidation to benzoic acid and esterification with methyl alcohol reactions. The same effect was observed when benzaldehyde was oxidatively esterified over TiO2-supported vanadium-doped cesium phosphomolybdate (i.e., Cs3+nPMo12−nVnO40/TiO2, n = 0–3) catalysts.58 Indeed, despite the different oxidants used by those authors (i.e., molecular oxygen), they verified that while the monosubstituted supported catalyst (i.e., Cs4PMo11VO40/TiO2) achieved the highest conversion, a greater vanadium content led to a lower conversion.
Fig. 3 presents the reaction selectivity variation with time for the 4 sodium phosphomolybdate catalysts. It is possible to observe that among the vanadium salts, only Na4PMo11V1O40 was an efficient catalyst. Indeed, when vanadium was included in the phosphomolybdic anion, there was a significant gain in catalytic performance, either in conversion or oxidation selectivity (Fig. 3a and b). However, an increase in vanadium doping drastically reduced the activity and selectivity of the catalyst.
Recently, we have found that an increase in vanadium doping led to a decline in conversion reached in oxidation reactions of terpenic alcohol.52 This effect was attributed to the higher vanadium load, which increases the energy barrier between the HOMO and LUMO orbitals and hampers the reducibility of the di-or tri-substituted heteropolyanions.59 As shown in Fig. 2, the same effect occurred herein.
In general, in the presence of the most active catalyst (i.e., Na4PMo11VO40), benzaldehyde was quickly oxidized to benzoic acid and quickly esterified to ethyl benzoate; these two reactions were performed in a one-pot process, in the presence of aqueous H2O2 and ethyl alcohol solutions containing catalytic amounts of Na4PMo11VO40. Being the most active catalyst, it was selected to study the effects of the main reaction parameters in the next sections.
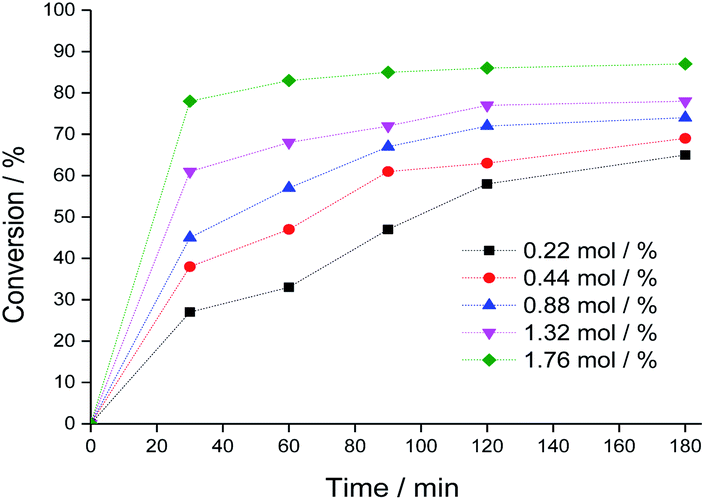 | ||
| Fig. 4 Impacts of Na4PMo11VO40 catalyst load on the kinetic curves of benzaldehyde oxidative esterification reactions with H2O2. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), toluene (internal stander), temperature (333 K), C2H5OH (10 mL). | ||
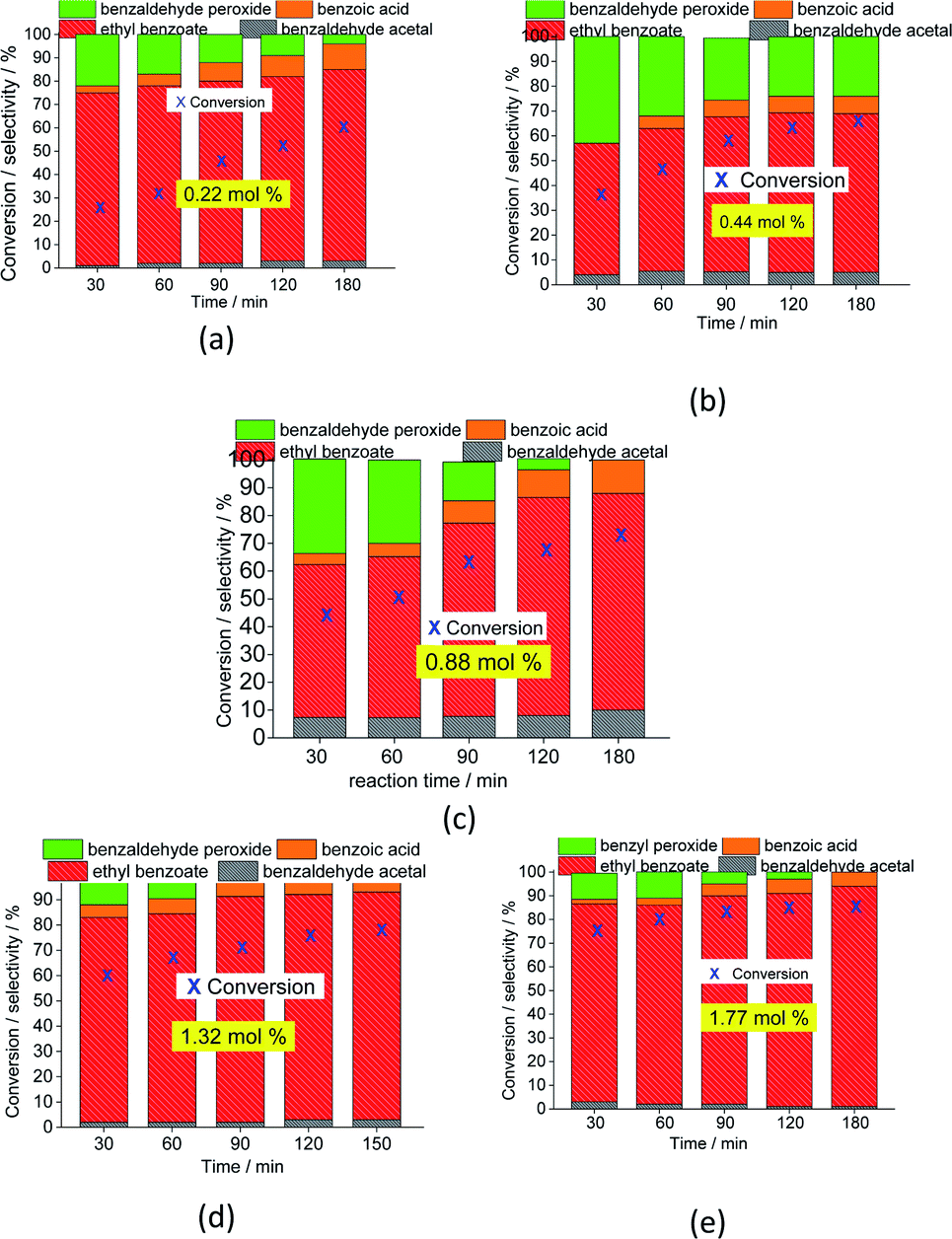 | ||
| Fig. 5 Impacts of the Na4PMo11VO40 catalyst load (a–e) on the conversion and product selectivity after 3 h oxidative esterification reactions of benzaldehyde with H2O2. | ||
The selectivity of the reactions was also impacted by the catalyst load. It is possible to verify that an increase in catalyst load favored the conversion of benzaldehyde peroxide to oxidation products (i.e., benzoic acid and their ethyl ester). Additionally, as the catalyst load increased, the benzoic acid was more efficiently esterified with ethyl alcohol (Fig. 5).
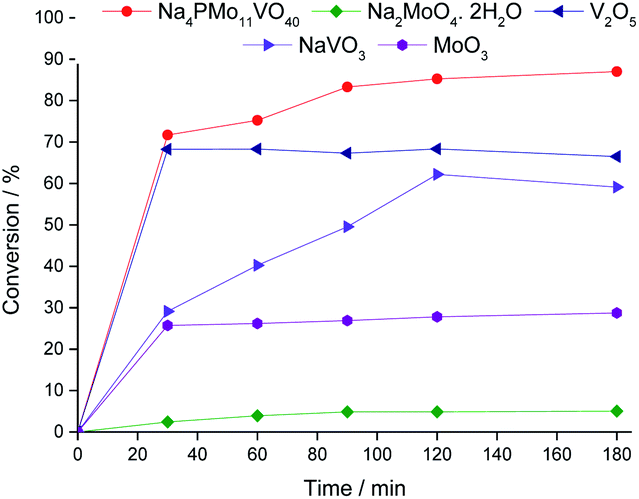 | ||
| Fig. 6 Kinetic curves of oxidative esterification reactions of benzaldehyde with H2O2 in C2H5OH solutions in the presence of the Na4PMo11VO40 catalyst and their synthesis precursors. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), catalyst (1.77 mol%), toluene (0.1 mL), C2H5OH (10.0 mL), 333 K. | ||
In Fig. 7, it is possible to see that while sodium molybdate was almost inactive as a catalyst, the sodium vanadate-catalyzed reaction achieved a reasonable conversion (ca. 59%). Nonetheless, the condensation product (benzaldehyde acetal) was significantly formed. This catalyst was poorly effective in converting benzaldehyde peroxide to the acid or ester.
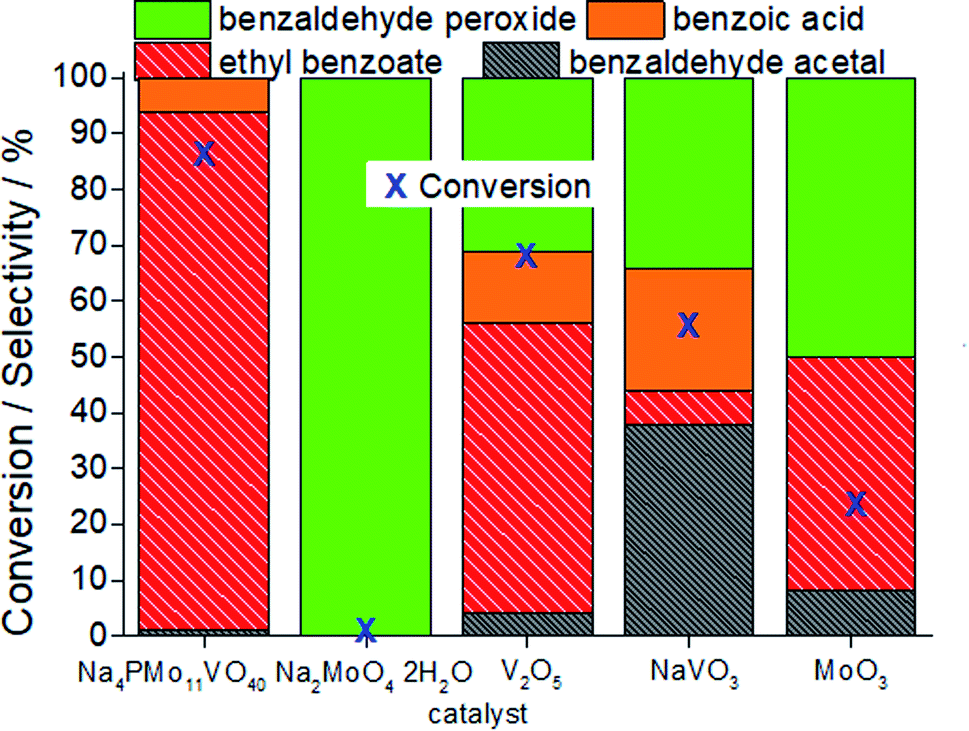 | ||
| Fig. 7 Conversion and selectivity after 3 h of oxidative esterification of benzaldehyde with H2O2 in C2H5OH solutions in the presence of the Na4PMo11VO40 catalyst or its synthesis precursors. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), catalyst (1.77 mol%), toluene (0.1 mL), C2H5OH (10.0 mL), 333 K. | ||
A comparison of the reactions in the presence of metal oxides allows us to conclude that the molybdenum and vanadium oxides were efficient to oxidatively esterify benzaldehyde, converting it to ethyl benzoate. However, the conversion of the V2O5-catalyzed reaction was much higher than that in the presence of the MoO3 catalyst.
Remarkably, when we compared the catalytic performance of the Na4PMo11VO40 salt to its precursors of synthesis, we can conclude that the vanadium atom plays a key role in the activity of the catalyst. In addition, this effect is even greater when vanadium is entrapped into the Keggin anion. Therefore, there is a synergism between the two species. Notably, as depicted in Fig. 1, if more than one vanadium atom was present in the Keggin anion, this effect was compromised.
On the other hand, the literature describes that ethyl benzoate can also be obtained from benzaldehyde acetal in the presence of Lewis acid metal catalysts.60 Herein, we excluded this hypothesis by carrying out the reaction with the Na4PMoVO40 catalyst without hydrogen peroxide; although benzaldehyde acetal was selectively formed, no significant amount of ester was detected.
When used as catalysts in oxidation with hydrogen peroxide of alcohols or olefins, Keggin heteropolyacids can undergo a peroxidation step and generate peroxide intermediates, which are the most probable active species in these reactions.61,62
Recently, Patel et al. assessed the benzaldehyde oxidative esterification over Ni-exchanged supported phosphotungstic acid and proposed a reaction mechanism, which we suppose probably also operates herein.61 Therefore, as the basis of the literature and our experimental results, we propose that the oxidative esterification of the benzaldehyde can be described as depicted in Scheme 4.
 | ||
| Scheme 4 Reaction pathway of Na4PMo11VO40-catalyzed oxidative esterification of benzaldehyde with H2O2 in C2H5OH solution. | ||
We think that the addition of hydrogen peroxide to the solution containing the Na4PMoVO40 catalyst promotes its peroxidation, generating an intermediate that reacts with benzaldehyde and generates another intermediate, where the transfer of oxygen atom from the oxidant to the substrate is more favorable (Scheme 4).
This intermediate is decomposed, releasing the benzoic acid and the vanadium-doped phosphomolybdate catalyst. The catalyst can promote the interaction between ethyl alcohol and benzoic acid, probably through an intermediate (i.e., omitted by simplification), in which nucleophilic attack on the carbonyl group of benzoic acid by the hydroxyl group of ethyl alcohol occurs, releasing the ester and water and regenerating the catalyst. It must be highlighted that both these steps take place in situ.63 Herein, it is probable that in the beginning, the formation of a peroxided active intermediate involves molybdenum or vanadium atoms.52 However, as previously reported, details of the mechanism of the oxidative esterification of aldehydes involving these POM catalysts are not clear yet.60,61
Two additional experiments were carried out to support this proposal. In the first experiment, the Na4PMoVO40 catalyst was evaluated in the oxidation reaction of benzaldehyde in acetonitrile. A conversion of 40% was achieved, with a selectivity of 60% toward benzoic acid. Secondly, the activity of the Na4PMoVO40 catalyst was evaluated in the esterification of benzoic acid with ethyl alcohol. A high ester selectivity (ca. 90%) toward ethyl benzoate was reached, at a rate of 45% conversion. These two tests confirm that this catalyst can efficiently promote both reactions.
:1 to 1:4.
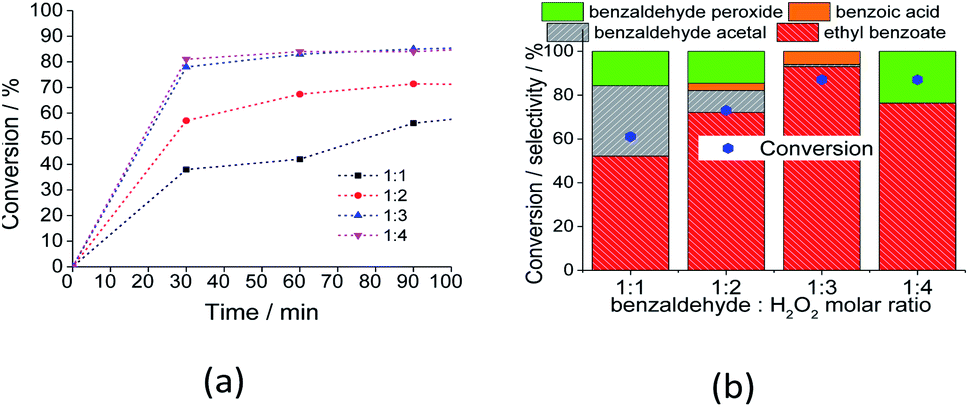 | ||
| Fig. 8 Impacts of the molar ratio of substrate to oxidant on the conversion (a) and selectivity (b) after 3 h of Na4PMo11VO40 catalyzed-oxidative esterification of benzaldehyde with H2O2 in C2H5OH solutions. Reaction conditions: benzaldehyde (2.75 mmol), catalyst (1.77 mol%), toluene (0.1 mL), C2H5OH (10.0 mL), 333 K. | ||
An excess of oxidant increased both the initial rate and reaction conversion of the reactions. Similarly, the ester and acid selectivity were favored. This can be assigned to the reversible character of the esterification reaction, which is favored by higher amounts of reactants. However, at proportions greater than 1:3, the selectivity of the reaction was compromised; when a higher amount of water was present, the Lewis acidity of the catalyst was compromised (i.e., V5+ and or Mo6+), and the conversion of benzaldehyde peroxide to acid and or ester became less favorable.
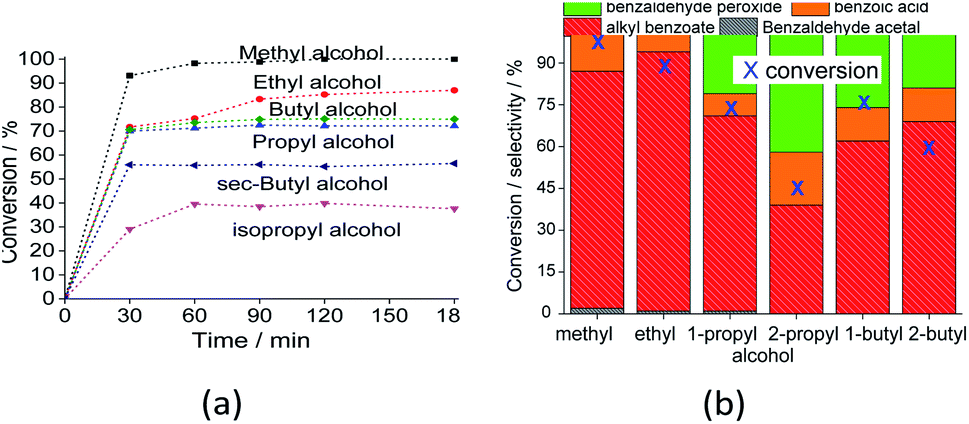 | ||
| Fig. 9 Impacts of alcohol on the conversion (a) and selectivity (b) (i.e., after 3 h) of the Na4PMo11VO40-catalyzed-oxidative esterification reaction of benzaldehyde with H2O2. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), catalyst (1.77 mol%), toluene (0.1 mL), alcohol (10.0 mL), 333 K. | ||
The reaction selectivity was also affected by the increase in the size of the carbon chain and the steric hindrance on the hydroxyl group. Although benzoic ester was the major product in all the runs (Scheme 3), benzoic acid was also obtained.
In runs with less reactive alcohols (i.e., with secondary hydroxyl groups or longer carbon chains), benzaldehyde peroxide, an intermediate product of oxidation, was the secondary product. The more hindered the hydroxyl group of the alcohol, the more difficult the attack on the carbonylic carbon of benzaldehyde, and consequently, the lower the ester selectivity (Scheme 5).
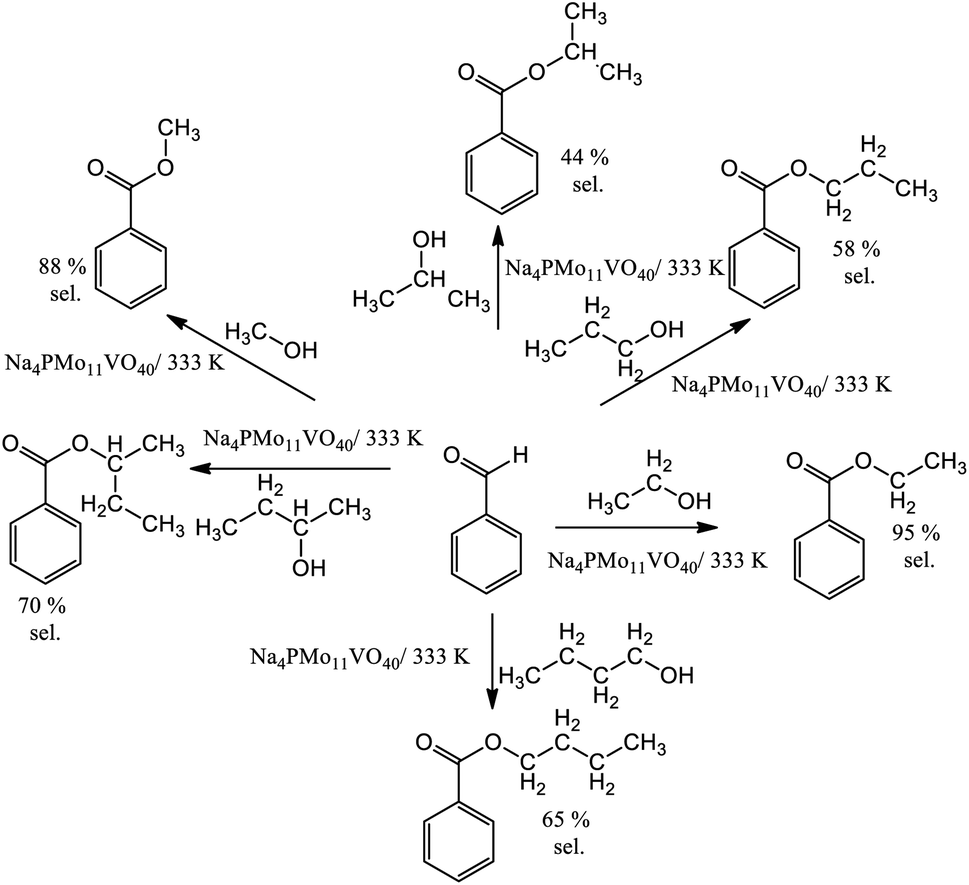 | ||
| Scheme 5 Ester selectivity of the Na4PMo11VO40-catalyzed oxidative esterification reactions of benzaldehyde with hydrogen peroxide in alcoholic medium. Reaction conditions: benzaldehyde (2.75 mmol); H2O2 (8.25 mmol), catalyst (1.77 mol%); reaction volume (10 mL). | ||
Likewise, when alcohols have a greater carbon chain size, the approach to the carbonylic carbon by the hydroxyl group is more difficult. The following trend was observed in terms of conversion of alcohols: CH3OH > C2H5OH > C3H7OH ≈ C4H9OH > sec-C4H9OH > sec-C3H7OH.
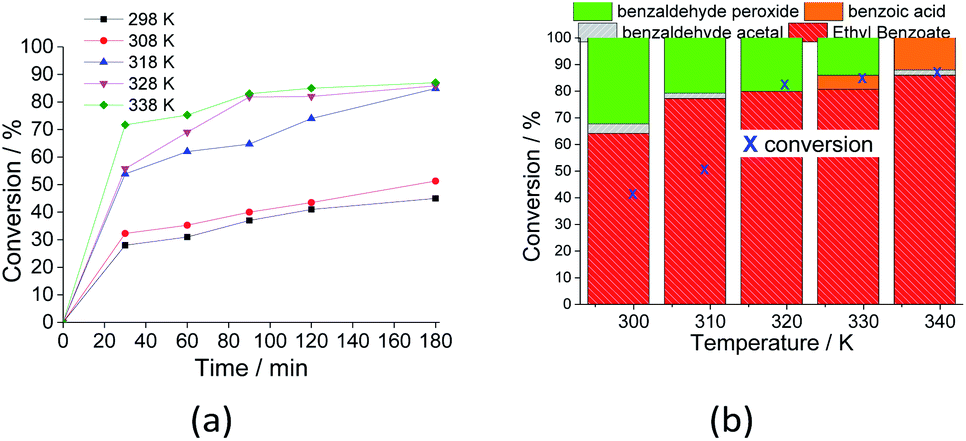 | ||
| Fig. 10 Effects of temperature on the kinetic curves (a) and conversion and selectivity (b) of the Na4PMo11VO40-catalyzed oxidative esterification of benzaldehyde by H2O2. Reaction conditions: benzaldehyde (2.75 mmol), H2O2 (8.25 mmol), (catalyst 1.77 mol%), temperature (variable); ethyl alcohol volume (10 mL). | ||
When the reaction was carried out at room temperature, 45% conversion was achieved, and the ester selectivity was 65%. Although not shown herein, in the absence of catalyst at this temperature, almost no conversion was detected, regardless of the excess of peroxide (ca. 1:4).
An increase in reaction temperature resulted in a higher conversion of benzaldehyde to ethyl benzoate as well as a drastic reduction in the amount of benzaldehyde peroxide at the end of the reaction, which was almost completely converted to the ester or benzoic acid (Fig. 10b). Under these reaction conditions, no significant difference was verified in the reactions carried out at 318 and 333 K.
4. Conclusions
A new route to oxidatively esterify benzaldehyde in a one-pot reaction with an environmentally friendly oxidant (i.e., aqueous H2O2) using a vanadium-doped catalyst in alcoholic solutions was developed. Among the vanadium-doped salts assessed, Na4PMo11VO40 was the most active catalyst. The main aspects that drive the reaction selectivity were studied. Benzaldehyde was efficiently converted to esters (ca. 85–93% selectivity) in the presence of methyl and ethyl alcohols in 3 h of reaction at 333 K with a low oxidant excess (ca. 1:3). Benzaldehyde acetal and benzoic acid were the minor products. Benzaldehyde peroxide was also obtained, mainly when the metal catalyst was less efficient. Secondary alcohols and those with a chain carbon size greater than that of ethyl alcohol provided benzoate esters as the main products; benzoic acid was the minor product, and no traces of acetal were observed. Notably, it was demonstrated that vanadium-doping has a beneficial effect only when one vanadium atom is used. The activity of the Na4PMo11VO40 catalyst was compared to its synthesis precursors, revealing that the Mo and V atoms have a synergic effect which efficiently promotes the oxidative esterification of benzaldehyde.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Federal University of Viçosa, the PPGMQ-MG, and the development agencies CNPq and FAPEMIG. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.References
- C. Yu, B. Ozkaya and F. W. Patureau, Chem. –Eur. J., 2021, 27, 3682 CrossRef CAS PubMed
.
- A. Liang, S. Han, L. Wang, J. Li, D. Zou, Y. Wu and Y. Wu, Adv. Synth. Catal., 2015, 357, 3104 CrossRef CAS
- M. J. da Silva and A. A. Rodrigues, Mol. Catal., 2020, 493, 111104 CrossRef CAS
- S. Gaspa, A. Porcheddu and L. De Luca, Tetrahedron Lett., 2016, 57, 3433 CrossRef CAS
- F. Rajabi, R. A. D. Arancon and R. Luque, Catal. Commun., 2015, 59, 101 CrossRef CAS
- Y. Li, R. Wang, R. Yan, J. Han and S. Zhang, Catal. Sci. Technol., 2015, 5, 3682 RSC
- B. Manoj, A. K. Rathi, J. Tucek, K. Safarova, N. Bundaleski, M. N. D. Orlando, L. Kvitek, R. S. Varma and R. Zboril, Green Chem., 2014, 16, 4137 RSC
- S. Dey, S. K. Gadak and A. Sudalai, Org. Biomol. Chem., 2015, 13, 10631 RSC
- X. F. Wu, Tetrahedron Lett., 2012, 53, 3397 CrossRef CAS
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374 CrossRef CAS PubMed
- M. J. da Silva and J. A. Villarreal, Catal. Lett., 2017, 147, 1646 CrossRef CAS
- D. C. Batalha, S. O. Ferreira, R. C. da Silva and M. J. da Silva, Chem. Sel., 2020, 5, 1976 CAS
- M. J. da Silva, P. H. da S. Andrade, S. O. Ferreira, C. B. Vilanculo and C. M. Oliveira, Catal. Lett., 2018, 148, 2516 CrossRef CAS
- M. Kim, J. Jeon, J. Baek, J. Choi, E. J. Park, J. Song, H. Bang, K. H. Suh, Y. H. Kim, J. Kim, D. Kim, K. H. Min and K. O. Lee, Bull. Korean Chem. Soc., 2014, 35, 345 CrossRef CAS
- R. Xie, X. Wang, J. Wang, J. Ye, M. Zhou and S. Zang, J. Saudi Chem. Soc., 2017, 21, 817 CrossRef CAS
- R. Kashyap, D. J. TAlukdar and S. Pratihar, New J. Chem., 2015, 39, 1430 RSC
- A. F. P. Biajoli, F. Peringer and A. L. Monteiro, Catal. Commun., 2017, 89, 48 CrossRef CAS
- M. J. da Silva and C. M. Oliveira, New J. Chem., 2021, 45, 3683 RSC
- J. L. C. Sousa, I. C. M. S. Santos, M. M. Q. Simões, J. A. S. Cavaleiro, H. I. S. Nogueira and A. M. V. Cavaleiro, Catal. Commun., 2011, 12, 459 CrossRef CAS
- M. R. Farsani and B. Yadollahi, J. Mol. Catal. A: Chem., 2014, 392, 8 CrossRef CAS
- L. Zhou, B. Dong, S. Tang, H. Ma, C. Chen, X. Yang and J. Xu, J. Energy Chem., 2013, 22, 659 CrossRef CAS
- A. Patel, S. Pathan and P. Prakashan, RSC Adv., 2016, 6, 51394 RSC
- M.-L. Jia, Y.-P. Zhang, Y.-S. Bao, J. Wang and A.-J. Xu, Green Chem. Lett. Rev., 2018, 11(3), 230 CrossRef CAS
- D. Talukdar, K. Sharma, S. K. Bharadwaj and A. J. Thakur, Synlett, 2013, 24, 963 CrossRef CAS
- S. Pathan and A. Patel, Appl. Catal. A., 2013, 459, 59 CrossRef CAS
- F. Cavani, N. Ballarini and S. Luciani, Top. Catal., 2009, 52, 935 CrossRef CAS
- M. J. da Silva, L. C. de Andrade Leles, S. O. Ferreira, R. C. da Silva, K. V. Viveiros, D. M. Chaves and P. F. Pinheiro, ChemSelect, 2019, 4, 7665 CAS
- M. J. Da Silva, P. H. Da Silva Andrade and V. F. C. Sampaio, Catal. Lett., 2020, 92 Search PubMed
- J. B. Feng, J. L. Gong, Q. Li and X. F. Wu, Tetrahedron Lett., 2014, 55, 1657 CrossRef CAS
- C. B. Vilanculo, M. J. da Silva, M. G. Teixeira and J. A. Villarreal, RSC Adv., 2020, 10, 7691 RSC
- M. J da Silva and N. A. Liberto, Curr. Org. Chem., 2016, 20, 1 CrossRef
- N. C. Coronel and M. J da Silva, J. Cluster Sci., 2018, 29, 195 CrossRef CAS
- S. Magar, G. T. Mohanraj, S. K. Jana and C. V. Rode, Inorg. Nano-Met. Chem., 2020, 50(11), 1157 CrossRef CAS
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed
- D. C. Batalha, S. O. Ferreira, R. C. da Silva and M. J. da Silva, ChemSelect, 2020, 5, 1976 CAS
- M. J. da Silva, P. H. S. Andrade, S. O. Ferreira, C. B. Vilanculo and C. M. Oliveira, Catal. Lett., 2018, 148, 2516 CrossRef CAS
- C. B. Vilanculo and M. J da Silva, New J. Chem., 2020, 44, 2813 RSC
- C. B. Vilanculo, M. J. da Silva, S. O. Ferreira and M. G. Teixeira, Mol. Catal., 2019, 478, 110589 CrossRef CAS
- N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358 CrossRef CAS
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128 CrossRef CAS PubMed
- N. C. Coronel, M. J. da Silva, S. O. Ferreira, R. C. da Silva and R. Natalino, ChemSelect, 2019, 4, 302 CAS
- S. S. Balula, I. C. M. S. Santos, L. Cunha-Silva, A. P. Carvalho, J. Pires, C. Freire, J. A. S. Cavaleiro, B. de Castro and A. M. V. Cavaleiro, Catal. Today, 2013, 203, 95 CrossRef CAS
- K. P. Barteau, J. E. Lyons, I. K. Song and M. A. Barteau, Top. Catal., 2006, 41(1–4), 55 CrossRef CAS
- J. K. Lee, J. Melsheimer, S. Berndt a, G. Mestl, R. Schlögl and K. Köhle, Appl. Catal. A, 2001, 214, 125 CrossRef CAS
- I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 1997, 117, 151 CrossRef CAS
- S. Paul, W. Chu, M. Sultan and E. Bordes-Richard, Sci. China: Chem., 2010, 53, 2039 CrossRef CAS
- R. J. J. Jansen, H. M. V. Veldhuizen, M. A. Schwegler and H. van Bekkum, Recl. Trav. Chim. Pays-Bas, 1994, 113, 115 CrossRef CAS
- F. Jing, B. Katryniok, F. Dumeignil, E. Bordes-Richard and S. Paul, J. Catal., 2014, 309, 121 CrossRef CAS
- C. Y. Chen, H. X. Li and M. E. Davis, Microporous Mater., 1993, 2(1), 17 CrossRef CAS
- G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437 CrossRef CAS
- L. R. Pizzio, P. G. Vázquez, C. V. Cáceres and M. N. Blanco, Appl. Catal., A, 2003, 256(1–2), 125 CrossRef CAS
- C. B. Vilanculo, M. J. da Silva, A. A. Rodrigues, S. O. Ferreira and R. C. da Silva, RSC Adv., 2021, 11, 24072 RSC
- K. T. Venkateswara Rao, P. S. N. Rao, P. Nagaraju, P. S. Sai Prasad and N. Lingaiah, J. Mol. Catal. A: Chem., 2009, 303, 84 CrossRef
- Z. H. Zhang, Z. P. Liao and G. H. Zhang, RSC Adv., 2015, 5(77), 63104 RSC
- Z. Shen, F. Long, T. Ma, H. Li, A. Li, Q. Feng, J. Liu and Y. Sun, J. Mater. Chem., 2020, 13, 6016 CAS
- Z. Q. He, X. H. Gao, P. M. Zhang and S. X. Xiao, J. Chem. Res. Appl., 2001, 13, 253 CAS
- P. S. N. Rao, G. Parameswaran, A. V. P. Rao and N. Lingaiah, J. Mol. Catal. A, 2015, 399, 62 CrossRef CAS
- C. Y. Chen, H. X. Li and M. E. Davis, Microporous Mater., 1993, 2(1), 17 CrossRef CAS
- M. Kanno, T. Yasukawa, W. Ninomiya, K. Ooyachi and Y. Kamiya, J. Catal., 2010, 273, 1 CrossRef CAS
- W.-J. Yoo and C.-J. Li, Tetrahedron Lett., 2007, 48, 1033 CrossRef CAS
- H. Mimoun, I. S. de Roch and L. Sajus, Tetrahedron, 1970, 23, 37 CrossRef
- Z. Weng, J. Wang and X. Jian, Catal. Commun., 2008, 9, 1688 CrossRef CAS
- A. Patel and A. Patel, RSC Adv., 2019, 9, 1460 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06718d |
| This journal is © The Royal Society of Chemistry 2021 |