
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of O-acylhydroxamates via reaction of oxime chlorides with carboxylic acids†
Kai-Kai Wang ab,
Yan-Li Lic,
Ying-Chao Zhaoa,
Shan-Shan Zhanga,
Rongxiang Chen*a and
Aili Sun*ab
ab,
Yan-Li Lic,
Ying-Chao Zhaoa,
Shan-Shan Zhanga,
Rongxiang Chen*a and
Aili Sun*ab
aSchool of Pharmacy, Xinxiang University, Xinxiang 453000, P. R. China. E-mail: chenhlmei@163.com; sunailifly@126.com; Fax: +86-373-3682674
bKey Laboratory of Nano-carbon Modified Film Technology Engineering of Henan Province, Xinxiang 453000, P. R. China
cMedical College, Xinxiang University, Xinxiang 453000, P. R. China
First published on 20th December 2021
Abstract
A simple and efficient method for the synthesis of O-acylhydroxamate derivatives from oxime chlorides and carboxylic acids was developed. The reaction affords clean and facile access to diverse O-acylhydroxamates in high yields (up to 85%). The chemical structure of a typical product was confirmed using single-crystal X-ray structure analysis.
Over recent decades, transition metal-catalyzed C–H activation reactions have emerged as a powerful tool in organic synthesis.1 The functionalization of C–H bonds has a wide range of applications in, for example, the synthesis of drug molecules, natural products and the intermediates of many biologically active compounds.2
Among the many known C–H activation reaction substrates, O-acylhydroxamates are highly effective and play an important role in C–H activation reactions enabling the production of highly regioselective products (Fig. 1). In 2011, the Guimond group developed a mild rhodium(III)-catalyzed synthesis of isoquinolone and 3,4-dihydroisoquinolone via a C–H bond functionalization reaction between O-acylhydroxamates and either alkynes or alkenes.3 Subsequently, in 2015, bulky phosphine ligands were synthesized by Li and Wang through RhIII-catalyzed C–H activation and annulation reactions using 1-alkynylphosphine sulfides and O-acylhydroxamates.4 In 2012, Glorius reported an annulative coupling between O-acylhydroxamates and allenes to synthesise 3,4-dihydroisoquinolin-1(2H)-ones via a rhodium(III)-catalyzed C–H activation reaction.5 Subsequently, the Shi group demonstrated a controllable rhodium(III)-catalyzed C–H functionalization of O-acylhydroxamates with vinylidenecyclopropanes. Product selectivity was obtained through changing the directing group from C(O)NH–OPiv to C(O)NH–OBoc to afford two different major products.6 In addition to cyclization reactions, O-acylhydroxamates are useful compounds that can be converted to anilines and symmetrical ureas in the presence of palladium(II) acetate or triethylamine.7 Because of the importance of O-acylhydroxamates, a distinct strategy or a concise process to synthesize O-acylhydroxamates with diverse structural requirements is still desirable.
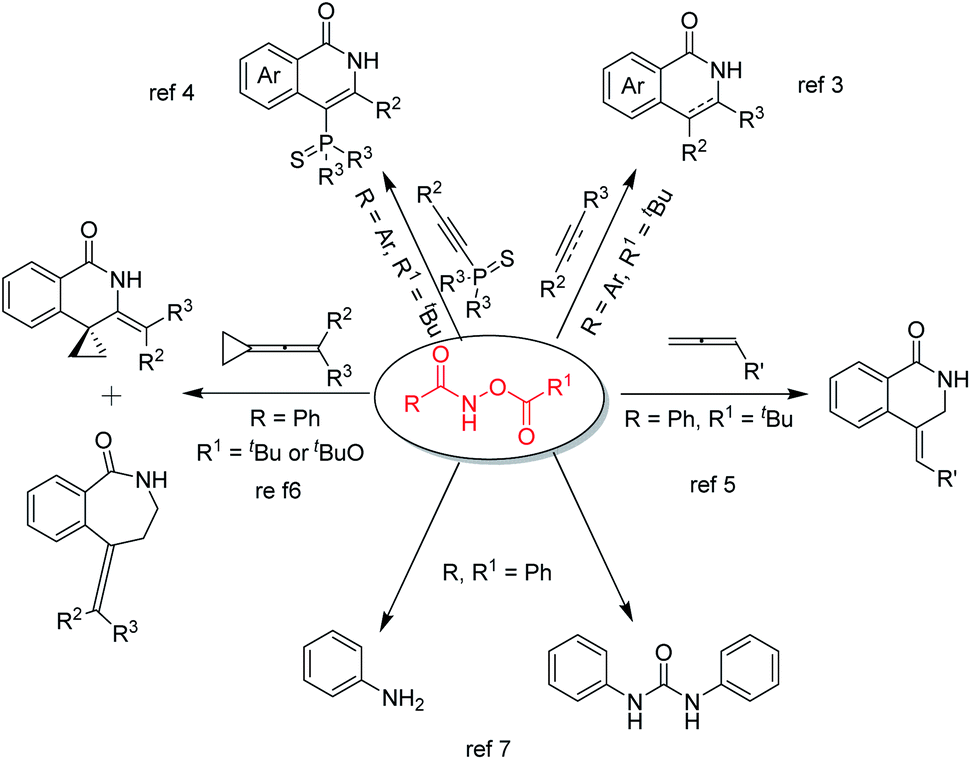 | ||
| Fig. 1 Representative examples of reactions between O-acylhydroxamate and various reaction partners. | ||
To the best of our knowledge, the classical approach reported for the synthesis of O-acylhydroxamates proceeds through an acylation reaction between hydroxamic acids and acyl chlorides under basic conditions (Scheme 1a).7 However, this acylation reaction requires anhydrous conditions when acyl chlorides are used. On the other hand, stable and structurally diverse carboxylic acid compounds exist in many natural products, non-natural products and materials and are frequently accessible from commercial sources.8 Inspired by these studies, we conceived a direct pathway to access O-acylhydroxamates from carboxylic acids (Scheme 1b). Herein, we describe the reaction of oxime chlorides with carboxylic acids under mild reaction conditions, providing convenient and efficient access to functionalized O-acylhydroxamate derivatives (Scheme 1b). In our protocol, the oxime chlorides9 were conveniently and easily synthesized in high yields from oximes using N-chlorosuccinimide (NCS) under mild conditions without any catalysts. In addition, the oximes could be obtained in high yields from various inexpensive aldehydes and hydroxylamine hydrochloride.
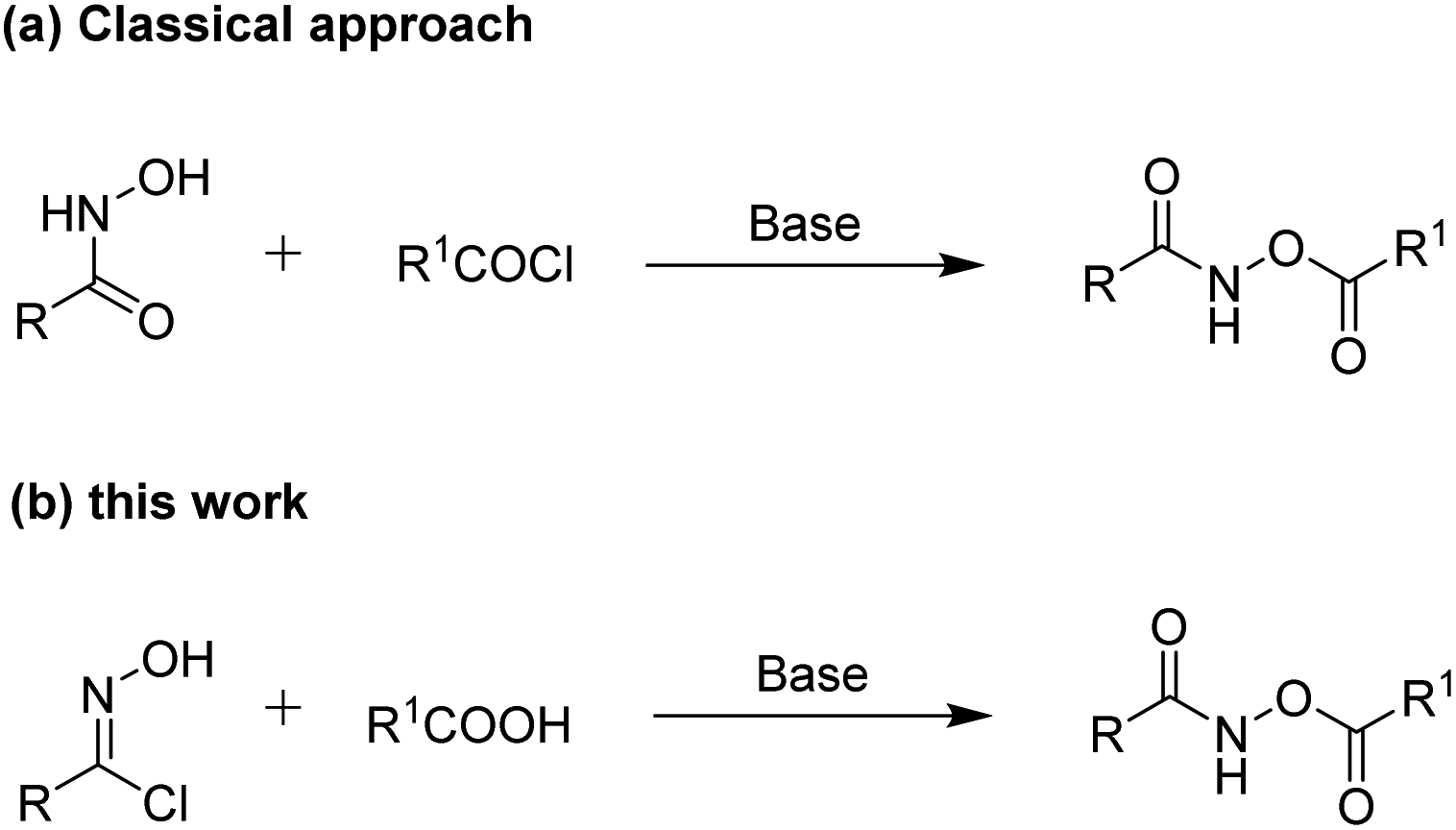 | ||
| Scheme 1 (a) Classical approach and (b) our protocol for the synthesis of O-acylhydroxamates. | ||
In our initial investigation, we chose phenylhydroximoyl chloride 1a with benzoic acid 2a as the model substrates for the optimization of the reaction conditions. The phenylhydroximoyl chloride 1a, which can, in the presence of a base, generate nitrile oxides in situ, has found extensive utility in 1,3-dipolar cycloadditions for the synthesis of a wide variety of important heterocyclic compounds.10 To our delight, the model reaction proceeded smoothly in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) in dioxane at room temperature for 12 h and furnished the corresponding product 3a in an 80% yield (Table 1, entry 1). The chemical structure of product 3a was confirmed using single-crystal X-ray structure analysis (CCDC 1974489).11 Subsequently, various types of base were screened, such as organic bases including TEA and DBU, and inorganic bases including Na2CO3 and NaOH. This revealed that DABCO was the most suitable base for this reaction (entries 2–5). To further improve the product yield, a series of different solvents were screened (entries 6–13). The results showed that product 3a was provided in a moderate yield in Et2O, toluene, DCE and CHCl3 (entries 6, 9, 11 and 13). Whereas the product was obtained in good yields when the reaction was performed in THF or EtOAc (entries 7 and 8). The reaction can be carried out in acetone but with poorer yields than in other solvents. Furthermore, increasing the reaction temperature resulted in a slightly lower yield (entry 14). Moreover, the yield was not obviously improved when the reaction time was prolonged (entry 15). When 2 equiv. of DABCO was used for 6 h product 3a was obtained in a moderate yield (entry 16).
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yield of 3ab (%) |
| a Unless noted otherwise, reactions were performed with oxime chloride 1a (0.22 mmol, 1.1 equiv.) and benzoic acid 2a (0.2 mmol, 1 equiv.), base (0.22 mmol, 1.1 equiv.) in solvent (1.0 mL) at r.t.b Yield of the isolated product.c Reaction was carried out at 60 °C.d The reaction was performed at room temperature for 24 h.e 2 equiv. DABCO were used and the reaction was performed for 6 h. | |||
| 1 | DABCO | Dioxane | 80 |
| 2 | TEA | Dioxane | 71 |
| 3 | DBU | Dioxane | 60 |
| 4 | Na2CO3 | Dioxane | 31 |
| 5 | NaOH | Dioxane | 20 |
| 6 | DABCO | Et2O | 50 |
| 7 | DABCO | THF | 70 |
| 8 | DABCO | EtOAc | 76 |
| 9 | DABCO | Toluene | 53 |
| 10 | DABCO | CH3CN | 30 |
| 11 | DABCO | DCE | 60 |
| 12 | DABCO | Acetone | 30 |
| 13 | DABCO | CHCl3 | 45 |
| 14c | DABCO | Dioxane | 62 |
| 15d | DABCO | Dioxane | 81 |
| 16e | DABCO | Dioxane | 68 |
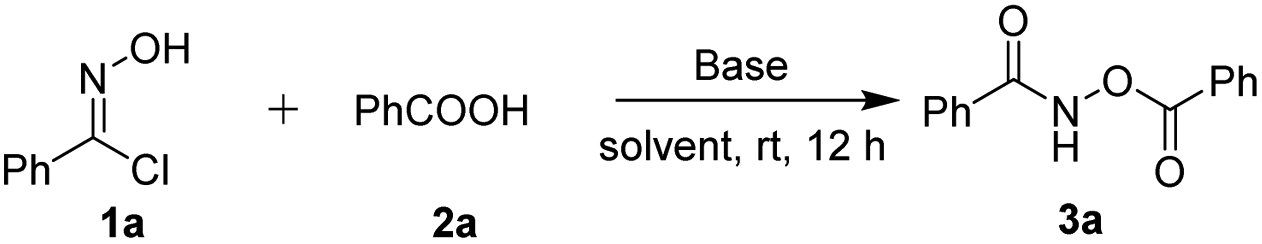
With our optimized reaction conditions in hand, the suitability of these conditions for reactions between various oxime chlorides and carboxylic acids was investigated. The results are shown in Table 2. Firstly, various substituted oxime chlorides 1 were reacted with benzoic acid 2a. Pleasingly, regardless of the electronic properties of (electron-donating or electron-withdrawing) or the heterocyclic substituents on the R group of the oxime chloride moiety, the reaction proceeded successfully under the optimized reaction conditions, affording the corresponding products 3a–3e (entries 1–5) in good yields (74–80%). Additionally, when an oxime chloride 1 bearing an alkyl group was reacted with benzoic acid 2a, the corresponding product 3f was obtained in a 69% yield. Subsequently, a number of aromatic carboxylic acids with either electron-denoting or electron-withdrawing substituents on the phenyl group reacted smoothly with phenylhydroximoyl chloride 1a at ambient temperature under the optimized conditions. The corresponding O-acylhydroxamates 3g–3p (entries 7–16) were obtained in good yields (62–74%). In these reactions, both a benzoic acid with a large sterically hindered group and a bis-substituted benzoic acid afforded the desired products 3i and 3j in 64% and 67% yields, respectively (entries 9 and 10). Additionally, the position of the substituent on the phenyl ring of benzoic acid 2 seemed to have a slight effect on the outcome of this reaction. For example, a para-bromo substrate led to a higher yield (3o, 70%) than the corresponding ortho- and meta-bromo substrates (3m, 67%; 3n, 67%, respectively) (entries 13–15). Some heterocyclic carboxylic acids such as thiophene-2-carboxylic acid and 2-naphthoic acid could also afford the desired products in good yields. It is worth noting that an aliphatic carboxylic acid and cinnamic acid were compatible in this reaction, giving the desired products 3s in a 70% yield (entry 19) and 3t in a 69% yield (entry 20), respectively. This demonstrates that the reaction is not limited to only aromatic carboxylic acids. Especially, an indole-derived carboxylic acid also worked well in the reaction to furnish the corresponding O-acylhydroxamate 3u in a 64% yield (entry 21). O-Acylhydroxamate 3u could not be obtained through a one-step acylation reaction between a hydroxamic acid and an acyl chloride via the classical approach (scheme 1a).
|
|||
|---|---|---|---|
| Entry | R | R1 | Yield of 3b (%) |
| a Reaction conditions: oxime chloride 1 (0.22 mmol), carboxylic acid 2 (0.2 mmol), DABCO (0.22 mmol), dioxane (1 mL), at room temperature for 12 h.b Isolated yield. | |||
| 1 | Ph | Ph | 3a, 80 |
| 2 | 4-MeC6H4 | Ph | 3b, 74 |
| 3 | 4-BrC6H4 | Ph | 3c, 76 |
| 4 | 2-Furyl | Ph | 3d, 70 |
| 5 | 2-Naphthyl | Ph | 3e, 76 |
| 6 | Cy | Ph | 3f, 69 |
| 7 | Ph | 4-MeC6H4 | 3g, 68 |
| 8 | Ph | 4-MeOC6H4 | 3h, 66 |
| 9 | Ph | 4-tBuC6H4 | 3i, 64 |
| 10 | Ph | 3,5-Di-Me-C6H3 | 3j, 67 |
| 11 | Ph | 4-FC6H4 | 3k, 74 |
| 12 | Ph | 4-ClC6H4 | 3l, 70 |
| 13 | Ph | 2-BrC6H4 | 3m, 67 |
| 14 | Ph | 3-BrC6H4 | 3n, 67 |
| 15 | Ph | 4-BrC6H4 | 3o, 70 |
| 16 | Ph | 4-CF3C6H4 | 3p, 62 |
| 17 | Ph | 2-Thienyl | 3q, 74 |
| 18 | Ph | 2-Naphthyl | 3r, 71 |
| 19 | Ph | 2-Styryl | 3s, 70 |
| 20 | Ph | Cy | 3t, 69 |
| 21 | Ph | 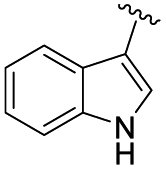 |
3u, 64 |
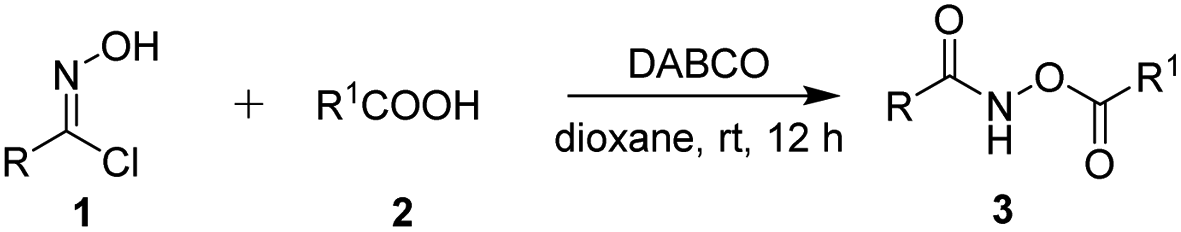
To further highlight the utility of our method to obtain O-acylhydroxamates under the standard conditions, the reaction was carried out on a gram-scale between 5.5 mmol of phenylhydroximoyl chloride 1a and 5 mmol benzoic acid 2a. The reaction proceeded smoothly to afford the desired product 3a without a significant loss of efficiency (0.964 g, in an 80% yield, Scheme 2), showing the reaction to be a practical tool for the synthesis of O-acylhydroxamates.
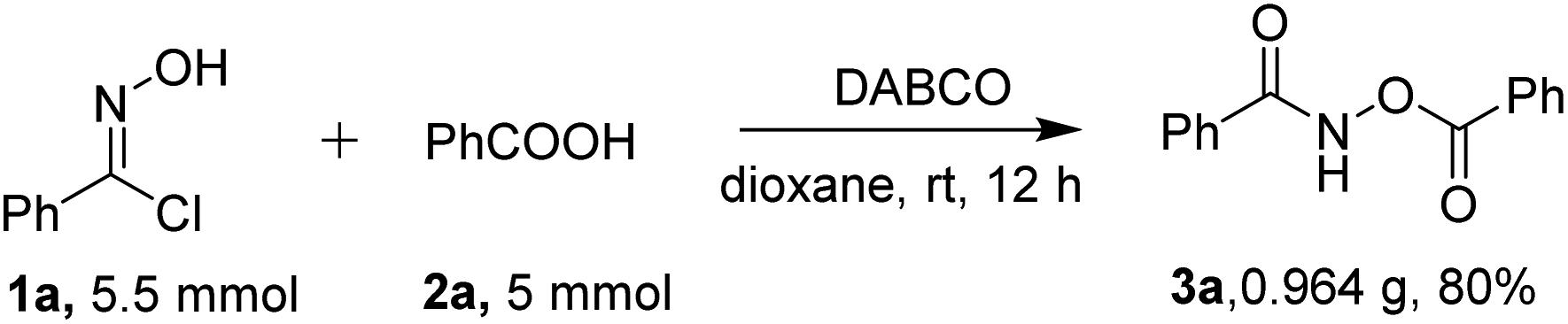 | ||
| Scheme 2 Scaled-up synthesis of O-acylhydroxamate 3. | ||
On the other hand, mesityl nitrile oxide 1a′ was stable enough to be isolated from the corresponding oxime chloride.12 The same reaction conditions tolerated stable nitrile oxide 1a′ and it reacted with benzoic acid 2a delivering the desired product 3a′ in an 85% yield (Scheme 3).
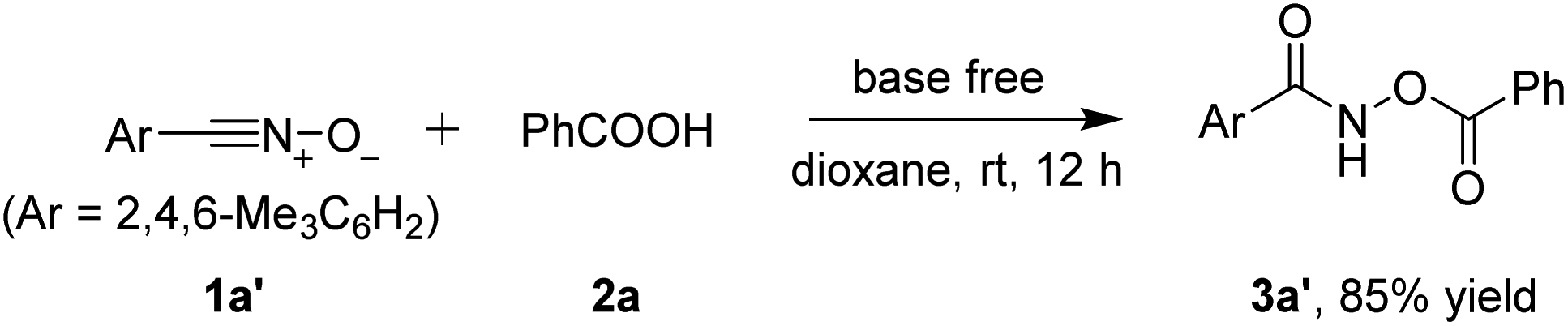 | ||
| Scheme 3 Reaction of isolable 2,4,6-trimethylbenzonitrile oxide (1a′) with benzoic acid 2a. | ||
As shown in Scheme 4, a plausible mechanism is proposed to explain the reaction process. Firstly, the highly active nitrile oxide A is generated from an oxime halide under basic conditions. Then, this nitrile oxide intermediate A reacts with carboxylic acid 2 to afford intermediate E. Finally, the intermediate E would transform into the desired product 3 via keto–enol tautomerization.13
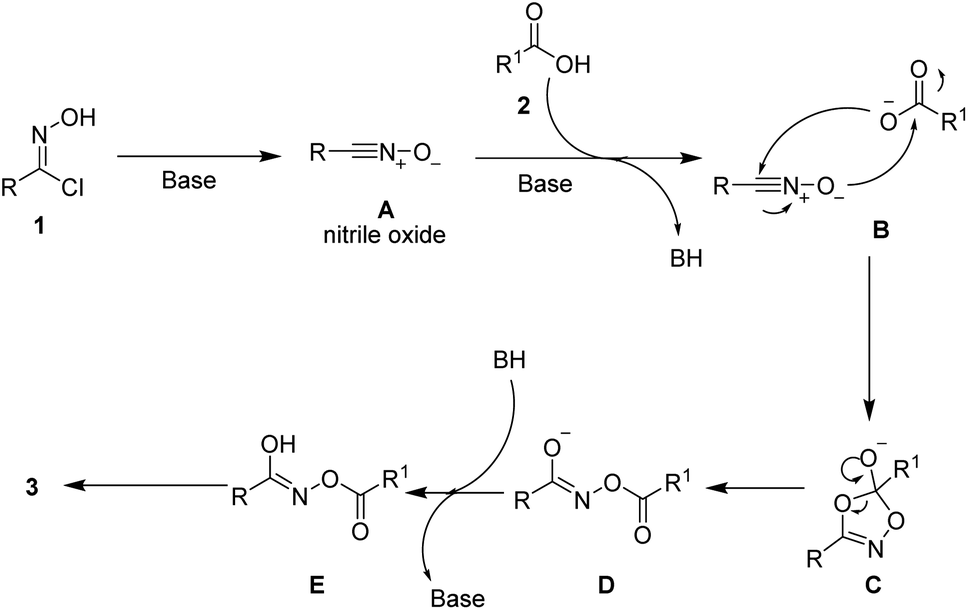 | ||
| Scheme 4 Proposed mechanism. | ||
In conclusion, we have developed a mild, practical and efficient protocol to prepare a broad range of functionalized O-acylhydroxamates in high yields (up to 85%) from oxime chlorides and carboxylic acids. In addition, the methodology allows the synthesis of O-acylhydroxamates on a gram scale. Our approach features some advantages over previously published methods, including the easy manufacture of the start materials, being stable in air and water, having a broad tolerance of functional groups, mild reaction conditions, a simple workup and noble metal-free catalysis. Further applications of this method are presently under active investigation in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (No. 21801214), the Key Scientific Research Project of Colleges and Universities in Henan Province of China (No. 18A150014 and 20B150019), the Natural Science Foundation of Henan Province (No. 202300410016), the Program for Youth Backbone Teacher Training in University of Henan Province (2021GGJS163), the Funding of National College Students Innovation and Entrepreneurship Training Program (202111071025 and 202111071021), the Key Scientific and Technological Project of Xinxiang (21ZD010), and the PhD research start-up foundation of Xinxiang University (1366020133).Notes and references
- (a) P. Chaudhary, J. Kandasamy, A. P. G. Macabeo, R. J. I. Tamargo and Y. R. Lee, Adv. Synth. Catal., 2021, 363, 2037 CrossRef CAS; (b) M. Oliva, G. A. Coppola, E. V. Van der Eycken and U. K. Sharma, Adv. Synth. Catal., 2021, 363, 1810 CrossRef CAS; (c) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767 CrossRef CAS PubMed; (d) W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735 RSC.
- (a) W. J. Kerr, G. J. Knox, M. Reid and T. Tuttle, Chem. Sci., 2021, 12, 6747 RSC; (b) S. D. Nale, D. Maiti and Y. R. Lee, Org. Lett., 2021, 23, 2465 CrossRef PubMed; (c) H. Li, J. Zhao, S. Yi, K. Hu and P. Feng, Organometallics, 2021, 40, 880 CrossRef CAS.
- N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed.
- B. Li, J. Yang, H. Xu, H. Song and B. Wang, J. Org. Chem., 2015, 80, 12397 CrossRef CAS PubMed.
- H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed.
- C. Ji, Q. Xu and M. Shi, Adv. Synth. Catal., 2017, 359, 974 CrossRef CAS.
- E.-S. M. N. AbdelHafez, O. M. Aly, G. E.-D. A. A. Abuo-Rahma and S. B. King, Adv. Synth. Catal., 2014, 356, 3456 CrossRef CAS.
- (a) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 2013, 7453 CrossRef CAS; (b) P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349 CrossRef CAS PubMed; (c) X. Zhang, F. Jordan and M. Szostak, Org. Chem. Front., 2018, 5, 2515 RSC.
- (a) R. J. B. Schäfer, M. R. Monaco, M. Li, A. Tirla, P. Rivera-Fuentes and H. Wennemers, J. Am. Chem. Soc., 2019, 141, 18644 CrossRef PubMed; (b) M. J. H. Ong and R. J. Hewitt, ChemistrySelect, 2019, 4, 10532 CrossRef CAS; (c) Q. V. Vo, C. Trenerry, S. Rochfort, J. Wadeson, C. Leyton and A. B. Hughes, Bioorg. Med. Chem., 2013, 21, 5945 CrossRef CAS PubMed.
- (a) J. S. Oakdale, R. K. Sit and V. V. Fokin, Chem.–Eur. J., 2014, 20, 11101 CrossRef CAS PubMed; (b) Q.-Y. Fang, H.-S. Jin, R.-B. Wang and L.-M. Zhao, Chem. Commun., 2019, 55, 10587 RSC; (c) L.-E. Carloni, S. Mohnani and D. Bonifazi, Eur. J. Org. Chem., 2019, 44, 7322 CrossRef; (d) Y. You, Y.-Z. Chen, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Tetrahedron Lett., 2018, 59, 2622 CrossRef CAS; (e) X. Shang, K. Liu, Z. Zhang, X. Xu, P. Li and W. Li, Org. Biomol. Chem., 2018, 16, 895 RSC; (f) N. N. Korgavkar and S. D. Samant, Synth. Commun., 2018, 48, 387 CrossRef CAS; (g) K.-M. Jiang, J.-Q. Zhang, Y. Jin and J. Lin, Asian J. Org. Chem., 2017, 6, 1620 CrossRef CAS; (h) S. L. Bartlett, Y. Sohtome, D. Hashizume, P. S. White, M. Sawamura, J. S. Johnson and M. Sodeoka, J. Am. Chem. Soc., 2017, 139, 8661 CrossRef CAS PubMed; (i) X. Zhou, X. Xu, Z. Shi, K. Liu, H. Gao and W. Li, Org. Biomol. Chem., 2016, 14, 5246 RSC; (j) W. Li, X. Zhou, Z. Shi, Y. Liu, Z. Liu and H. Gao, Org. Biomol. Chem., 2016, 14, 9985 RSC; (k) X. Lian, S. Guo, G. Wang, L. Lin, X. Liu and X. Feng, J. Org. Chem., 2014, 79, 7703 CrossRef CAS PubMed; (l) K.-K. Wang, Y.-L. Li, W. Zhang, S.-S. Zhang, T.-T. Qiu and X. Ma, Tetrahedron Lett., 2020, 61, 151943 CrossRef CAS.
- CCDC 1974489 contains the supplementary crystallographic data for this paper relating to 3a.
- (a) O. Altintas, M. Glassner, C. Rodriguez-Emmenegger, A. Welle, V. Trouillet and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 5777 CrossRef CAS PubMed; (b) G. Zhao, L. Liang, C. H. E. Wen and R. Tong, Org. Lett., 2019, 21, 315 CrossRef CAS PubMed; (c) W. Sun, F. Jiang, H. Liu, X. Gao, H. Jia, C. Zhang and H. Guo, Chin. Chem. Lett., 2019, 30, 363 CrossRef CAS.
- M. Ruaysap, S. R. Kennedy, C. M. Mayhan, S. P. Kelley, H. Kumari, C. A. Deakyne and J. L. Atwood, Chem. Commun., 2020, 56, 12985 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, structural proofs, CIF file for 3a. CCDC 1974489. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06860a |
| This journal is © The Royal Society of Chemistry 2021 |