
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective unidirectional benzylic sp3 C–H oxidation of dodecahydrotriphenylene with RuCl3–NaIO4: formation of benzylic ketones†‡
Gaurang J. Bhatt a,
Pradeep T. Deota*a,
Deepak Upadhyayb and
Prafulla K. Jhac
a,
Pradeep T. Deota*a,
Deepak Upadhyayb and
Prafulla K. Jhac
aApplied Chemistry Department, Faculty of Technology & Engineering, The Maharaja Sayajirao University of Baroda, Vadodara-390 001, Gujarat, India. E-mail: ptdeota-appchem@msubaroda.ac.in
bDepartment of Applied Physics, Faculty of Technology & Engineering, The Maharaja Sayajirao University of Baroda, Vadodara-390 001, Gujarat, India
cDepartment of Physics, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara-390 001, Gujarat, India
First published on 20th October 2021
Abstract
Dodecahydrotriphenylene, a higher homologue of trindane chemoselectively undergoes unidirectional benzylic sp3 C–H oxidation and the central benzene ring remains intact unlike that in trindane under similar reaction conditions. RuO4 which generally attacks sp2 C–H to form oxidative products is found to give benzylic ketones via sp3 C–H oxidation. Density functional theory (DFT) calculations have also been performed to analyse the potential energy, energy barrier and HOMO–LUMO energy gap of the products.
Introduction
Selective transformation of benzocyclotrimers (BCTs) into their keto derivatives is highly desirable due to the diverse utility of the keto functionality in various synthetic elaborations towards graphene and buckminsterfullerene (Fig. 1). Hence, selective benzylic oxo-functionalization of alkylarenes is an important protocol in organic synthesis.1 Moreover, benzylic oxidation of alkylarenes provides valuable synthons that can lead to many natural products, agrochemicals and pharmaceuticals.2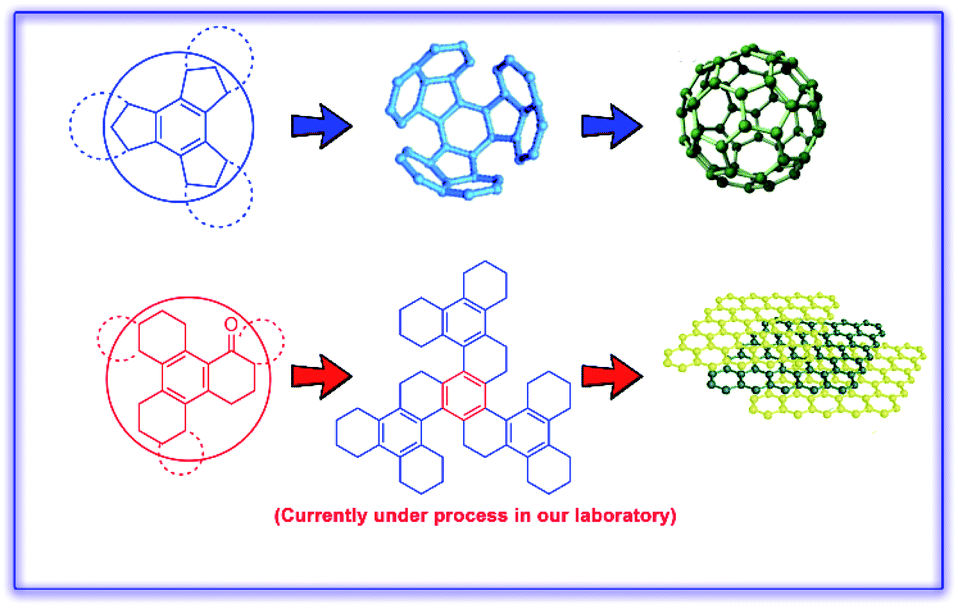 | ||
| Fig. 1 Benzocyclotrimer precursors towards fullerene and graphene. | ||
There are numerous reagents reported in the literature for selective benzylic oxo-functionalization such as NaClO/TEMPO/Co(OAc)2, o-iodoxybenzoic acid, KMnO4/MnO2, bismuth-picolinic acid, t-BuONa, and ascorbic acid.3 Ruthenium complexes have been commonly employed in various oxidative transformations to furnish a variety of oxo-functionalities.4–6
In general, Ru-compounds have been shown to attack alkenes via sp2 C–H activation to form aryl ketones.7 However, RuCl3–NaIO4 has been reported to exhibit poor reactivity during benzylic oxo-functionalization of alkylarenes via sp3 C–H activation.8a Hence, complexes of ruthenium have been employed to accomplish effective benzylic sp3 C–H oxidation of aromatic hydrocarbons to corresponding aromatic ketones.8b
Interestingly, the oxidation of tricyclopentabenzene (trindane, 1) has been shown to yield a highly functionalized tricyclic system 2 (Scheme 1) upon oxidation with ruthenium tetroxide generated in situ.9 It is remarkable to note that the compound 1 undergoes complete cleavage of the central benzene ring.
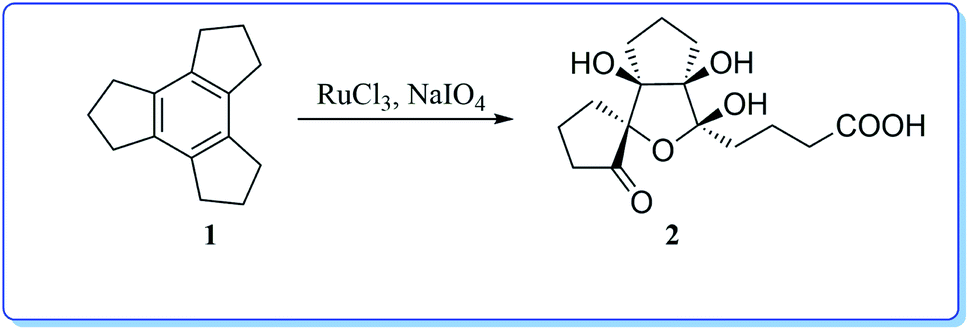 | ||
| Scheme 1 Ru(VIII) mediated oxidation of the benzene ring of trindane 1. | ||
In context with our synthetic studies, we became interested in examining the oxidation of dodecahydrotriphenylene 3 (readily assembled via cyclotrimerization of cyclohexanone), a higher homologue of compound 1. Thus, we treated the compound 3 with RuCl3–NaIO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 in aqueous acetonitrile-carbon tetrachloride system at ambient temperature. The reaction was complete after 30 h (TLC). Usual workup and chromatography furnished the ketone 414 along with hitherto unknown ketones 5 and 6 in a total yield of 37% (Scheme 2).
6 in aqueous acetonitrile-carbon tetrachloride system at ambient temperature. The reaction was complete after 30 h (TLC). Usual workup and chromatography furnished the ketone 414 along with hitherto unknown ketones 5 and 6 in a total yield of 37% (Scheme 2).
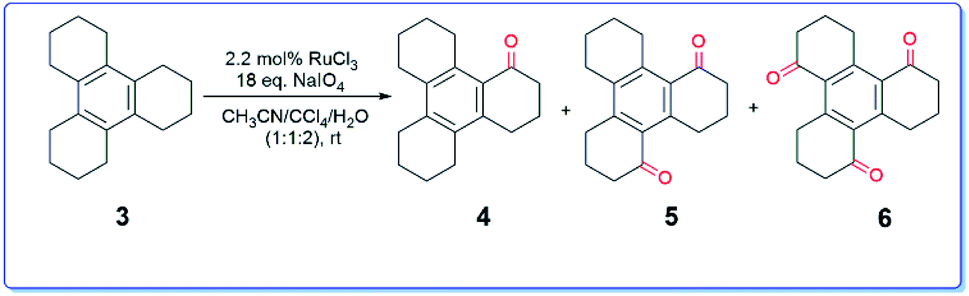 | ||
| Scheme 2 Formation of various benzylic ketones 4–6. | ||
We undertook a time dependent study of the above oxidation by arresting the reaction at various intervals followed by work up and column chromatography of the reaction mixture. Initially, the reaction was done for 4 h during which formation of monoketone 4 was observed after 3 h, after which the formation of dione 5 (6%) was observed (TLC) (Table 1). The highest amounts of compound 5 (16%) was obtained when the reaction was conducted for 20 h. Similarly, the first appearance of trione 6 in trace amount was noticed after 16 h of the reaction which continued to rise and reached to a maximum when the reaction was done for 30 h (Table 1 and Fig. 2). The longer reaction time led to degradation of the products. In case of the entries 1–6, unreacted starting material was recovered.
| Entrya | Reaction time (h) | Yieldb (%) | ||
|---|---|---|---|---|
| Compound 4 | Compound 5 | Compound 6 | ||
| a Compound 3 (5.0 g) for each entry.b Isolated yields obtained after column chromatography.c Starting material 3 completely disappeared.d Complete degradation of reaction products observed.e No reaction (NR) either in presence of RuCl3 alone or NaIO4 alone and unreacted 3 was recovered. | ||||
| 1 | 04 | 22 | 6 | — |
| 2 | 08 | 30 | 9 | — |
| 3 | 12 | 39 | 14 | — |
| 4 | 16 | 33 | 15 | Trace |
| 5 | 20 | 30 | 16 | Trace |
| 6 | 24 | 22 | 13 | 7 |
| 7 | 30c | 19 | 10 | 8 |
| 8 | 72d | — | — | — |
| 9 | 96e | NR | NR | NR |
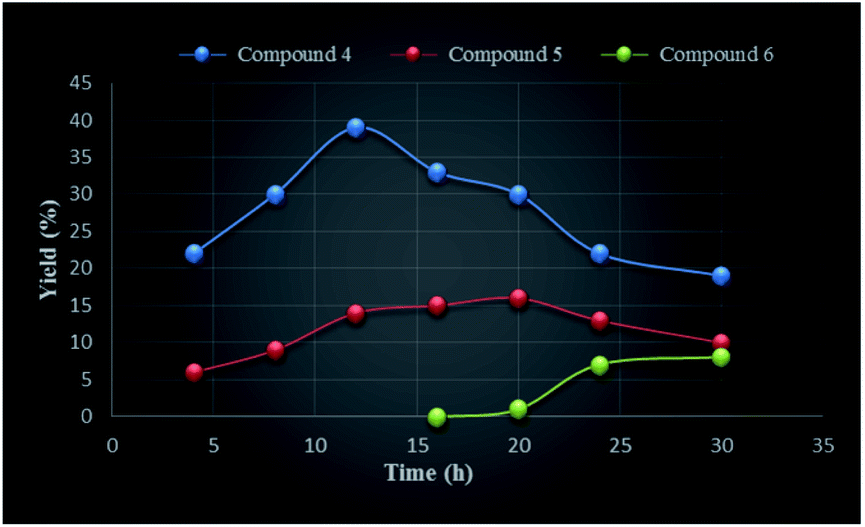 | ||
| Fig. 2 Graphical presentation of the reaction time against yield (%) of compounds 4–6. | ||
The compound 3 does not undergo reaction when treated alone either with RuCl3 and/or NaIO4. It was found that with lesser amounts of NaIO4 or RuCl3 or lesser reaction time, unreacted 3 was isolated. Attempts of benzylic oxidation of 3 using 18 equivalents of NaIO4 alone, in the absence RuCl3 met with no success. Similarly the reaction did not proceed without NaIO4 in the presence of RuCl3 alone indicating involvement of both the reagents in the oxidation cycle.
The structures of the dione 5 and trione 6 were deduced from their spectral features. Thus, the dione 5 exhibited a CO absorption band at 1684 cm−1 in its IR spectrum which clearly indicated the presence of a conjugated carbonyl group. This suggested the presence of aromatic ring in compound 5. The proton NMR spectrum (600 MHz) showed two signals at δ 3.29 (t, J = 6.0 Hz, 2H) and 3.16 (t, J = 6.2 Hz, 2H) due to methylene protons adjacent to carbonyl group. Further, signals were observed at δ 2.83 (q, J = 5.9 Hz, 3H), 2.63 (dt, J = 14.3, 6.1 Hz, 7H), 2.15–2.08 (m, 3H), 2.00–1.94 (m, 2H), 1.81 (dd, J = 11.5, 5.6 Hz, 2H) for other methylenes. 13C NMR (151 MHz, CDCl3) displayed signals at δ 200.98, 200.61 indicating the presence of conjugated carbonyl group. In addition, signals were seen at δ 148.11, 144.95, 134.33, 131.79 and 129.67, 40.71, 40.51, 30.36, 29.40, 29.18, 27.62, 27.47, 22.76, 22.61, 22.43, 22.27, 22.07.
The trione 6 also exhibited a carbonyl absorption band at 1690 cm−1 in its IR spectrum again indicating the presence of a conjugated carbonyl group. The proton NMR spectrum (600 MHz) of 6 showed only three sets of signals at δ 3.33 (t, J = 6.1 Hz, 1H) for methylenes adjacent to carbonyl, 2.68 (t, J = 6.8 Hz, 1H) for benzylic methylenes and 2.05–1.99 (m, 1H) for other methylene protons. 13C NMR (151 MHz, CDCl3) of 6 displayed only one signal at δ 200.12 indicating the presence of conjugated carbonyl groups. Further, only two signals for aromatic carbons were observed at δ 151.60, 131.79 and three signals in the aliphatic region at δ 40.32, 29.81 and 22.44. It is noteworthy to observe only six signals for eighteen carbons indicating C3v symmetry in the molecule.
It is interesting to note that the above reaction furnished mono, di- and tri-keto derivatives in which the aromatic ring remains intact which is contrary to the behaviour of trindane 1 that undergoes complete oxidative cleavage of the aromatic ring.9 Further, the structures of the dione 5 and trione 6 appear to have a unidirectional character in which the keto groups are present in either clockwise or anticlockwise direction. There are six benzylic positions available in compound 3 out of which only three positions are chemoselectively oxidized. None of the C2v symmetric diones (7–9) were observed (Fig. 3). Consequently the unsymmetrical triones 10 and 11 which could result from further oxidation of 7–9 were also not obtained.
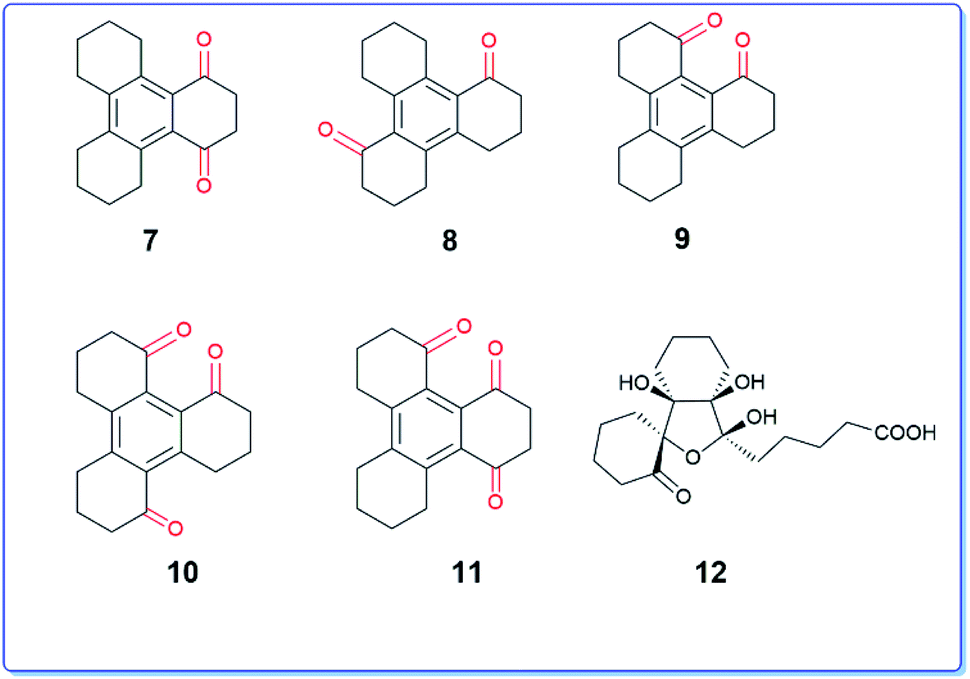 | ||
| Fig. 3 Other possible benzylic diones, triones and the ring-opening product. | ||
It was also interesting to note that the oxidation did not give any ring-opened product of type 12 as expected from the report of S. Ranganathan et al.9 It is known that ruthenium tetroxide is generated in situ using oxidants like periodate or hypochlorite.10 The tetroxide thus formed experiences steric hindrance offered by the folded peripheral cyclohexene rings in compound 3, which perhaps shield the attack of the oxidant on the central benzene ring (Fig. 4a and b) leading to consequent attack on the benzylic sp3 C–H positions.
 | ||
| Fig. 4 (a) Folded peripheral cyclohexene rings around the central benzene ring in compound 3. (b) Energy minimized structure of compound 3 by Chem3D. | ||
RuO4 is isoelectronic with OsO4 and due to its less toxicity, ruthenium is preferentially employed than osmium in oxidations.5b,6b A general mechanism for the oxidation is shown in Fig. 5. Initial benzylic hydrogen abstraction by in situ generated RuO4 (7) leads to formation of a corresponding free radical which on reaction with water gives an alcohol of type (2) forming Ru-oxo/hydroxo species (3).8a Further oxidation of (2) via intermediate (4) then leads to formation of a ketone (5) generating monooxoruthenium(IV) species (6) which is oxidized back to RuO4 (7) by the oxidant periodate for the next redox cycle.10 Periodate itself gets reduced to iodate in the last step.
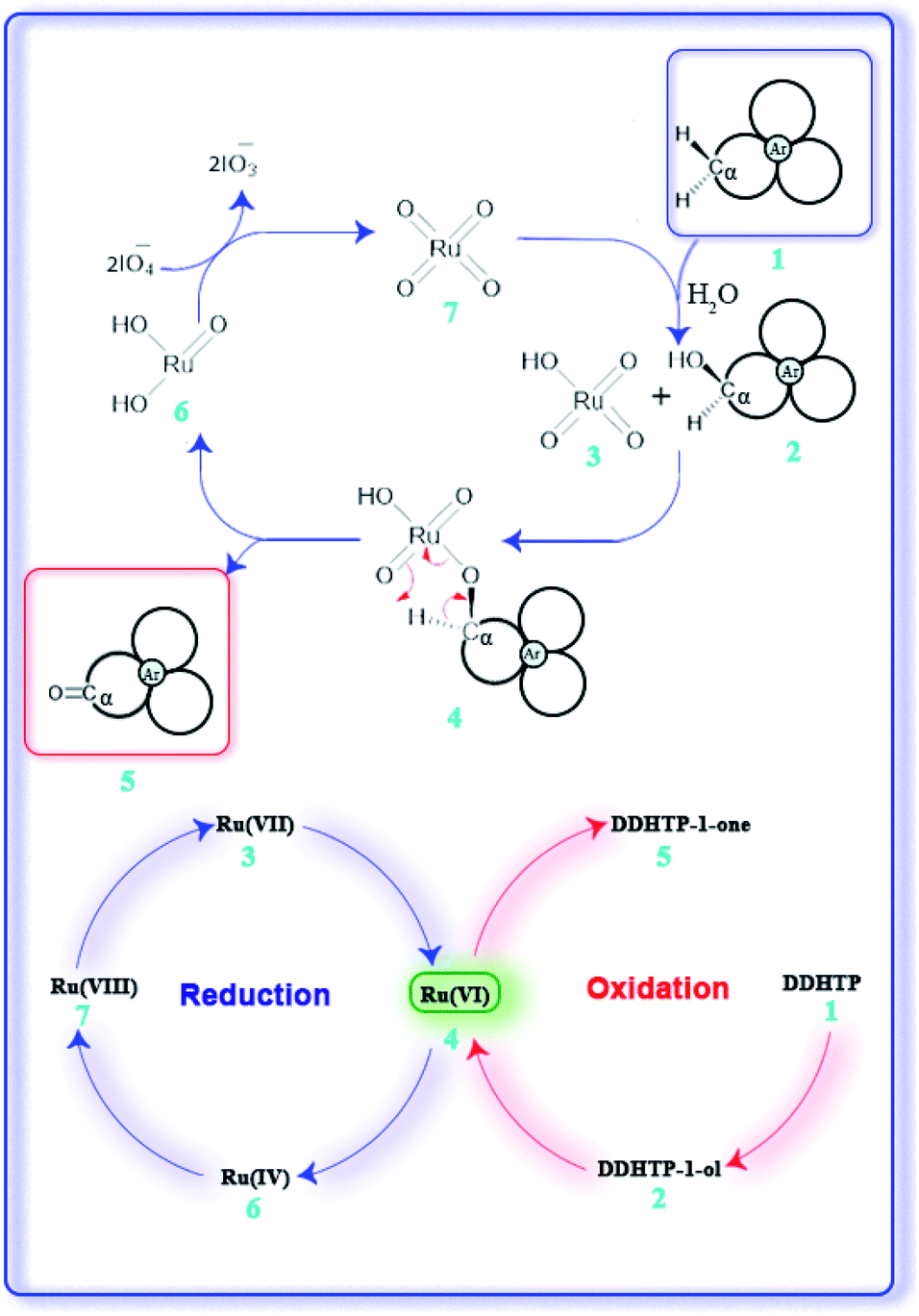 | ||
| Fig. 5 Plausible mechanism of the ruthenium oxidation. | ||
A comparison of various ruthenium assisted oxidations is presented in Scheme 3. α-Diketones are obtained via ruthenium catalyzed sp2 C–H oxidation in which catalyst attacks the activated double bond of alkene.7a To achieve benzylic sp3 C–H oxidation, Ru-complexes have been employed.8a When Ru(VIII) species is used, trindane 1 undergoes complete oxidative cleavage of the central benzene ring to form polycyclic oxygenated product 2.9 Interestingly in spite of structural similarity between 1 and 3, sp3 C–H oxidation is not observed in 1. However we found that the central benzene ring in 3 remains intact and three out of six benzylic positions are chemoselectively oxidized to form a C3v symmetric keto derivative 6 having unidirectional character along with 4 and 5 (Scheme 2).
 | ||
| Scheme 3 An overview of ruthenium mediated oxidations. | ||
DFT calculations
We have performed independent structural optimization of all the possible isomers of diones and triones to determine their minimum energy. Further calculations such as ground-state structural and electronic (HOMO–LUMO) were performed using density functional theory based on first principle calculations using the Gaussian09 suite of program.11The Becke three parameters hybrid functional with Lee–Yang–Perdew correlation functionals (B3LYP)12 are utilized with LanL2DZ basis-set for the Ru-based systems and 6-311G basis-set for rest the systems to accurately predict the minimum energy states. The hybrid functionals are the mixture of Hartree–Fock (HF) exchange along with density functional theory (DFT) exchange–correlation functionals. B3LYP functional uses LYP expression for non-local correlation and VWN function III for local correlation. The optimized structures were visualized using GaussView (version 5).13 The HOMO–LUMO gap is also evaluated as it gives the important parameters such as electronegativity (χ), chemical potential (μ), global hardness (η), global electrophilicity index (ω) and global softness (S) etc.
In order to validate the experimental findings and to understand the atomic level properties we have individually configured and optimized the structural geometries by utilizing the parameters described above. The optimized geometries along with the energies are presented in the Fig. 6. It can be observed that the minimum energies are obtained for the compounds 4, 5 and 6 that can also be observed in experimental results.
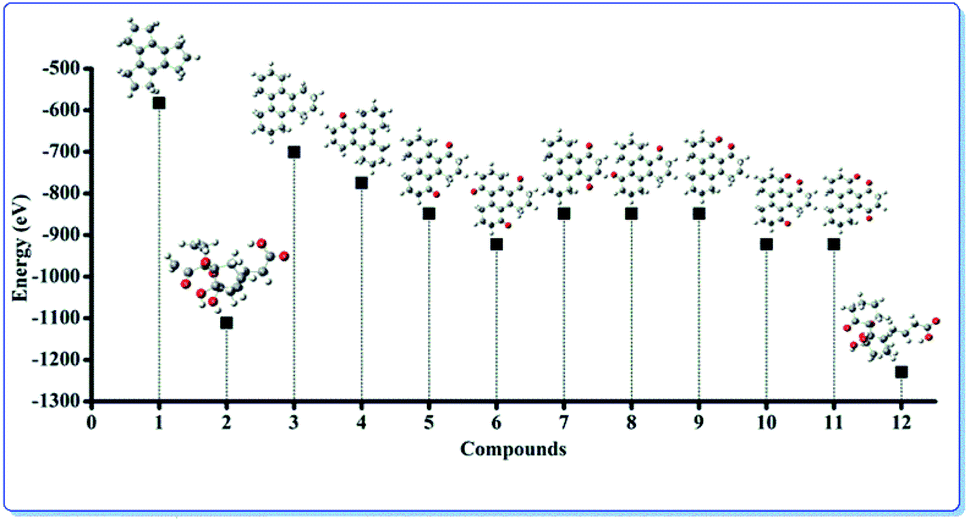 | ||
| Fig. 6 Optimized geometries of the molecules corresponding to their energy state. | ||
Fig. 6 shows the optimized geometry calculated using the DFT method that corresponds to the energy of the molecules. The calculated potential energy of compounds 4, 5 and 6 are −774.392 eV, −848.401 eV and −922.410 eV respectively. Compounds 5 and 6 have lower minimized energy than their corresponding possible diones [7 (−848.398 eV), 8 (−848.398 eV) and 9 (−848.398 eV)] and triones [10 (−922.401 eV), 11 (−922.401 eV)] respectively.
Ru(VIII) is the most stable state among its other prevalent oxidation states. The continuous reduction from Ru(VIII) via ruthenate(VI) as an intermediate state leads to the formation of most common Ru(IV) oxidation state. We have calculated relative energy pathway and energy barrier for each step and plotted the energy diagram (Fig. 7). Initially RuO4 abstracts a hydrogen from 3, leading to its reduction into Ru(VII) with concomitant formation of the hydroxy derivative (IM-2). The calculated energy barrier for this step is −0.036 keV per atom. This is followed by the combination of IM-2 and Ru(VII) species giving rise to the alkoxyruthenium(VI) (IM-4) having −0.144 keV per atom energy barrier for this step. In the key step, ruthenium–oxygen bond cleavage leads the formation of the keto-products along with the generation of Ru(IV). The energy barrier for this last step is 0.12 keV per atom. The calculated HOMO–LUMO energy gap of 4, 5 and 6 are shown in the Fig. 8.
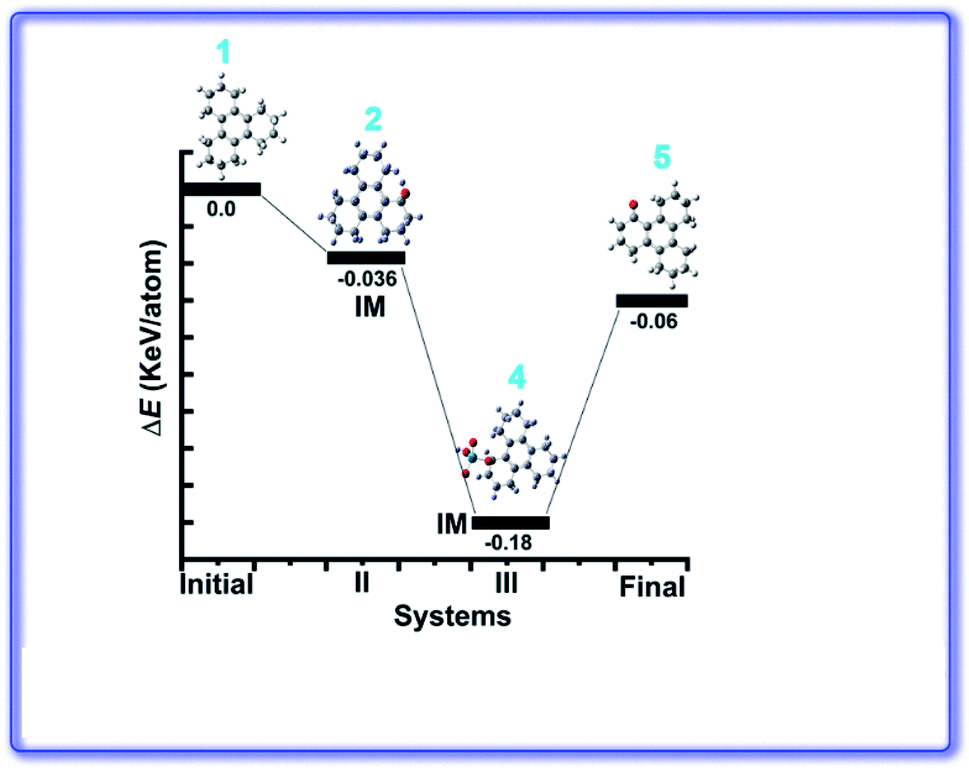 | ||
| Fig. 7 Calculated relative energy pathway of the intermediates (IM) during oxidation reaction. | ||
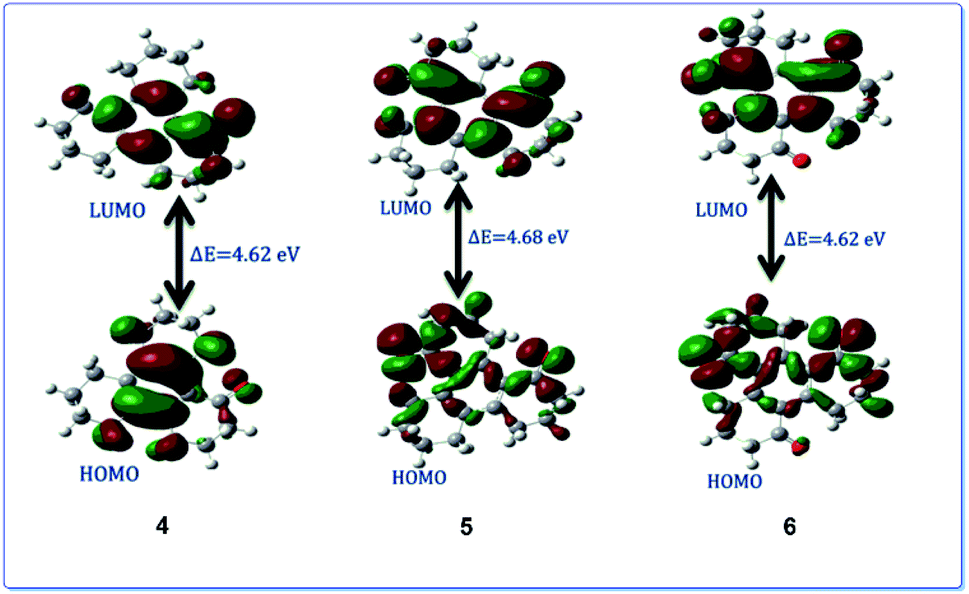 | ||
| Fig. 8 The HOMO–LUMO gap of compounds 4, 5 and 6. | ||
Conclusions
In summary, the central benzene ring of compound 3 is found to remain unaffected by ruthenium and subsequently sp3 C–H benzylic oxidation leading to formation of benzylic ketones 4–6 is observed. The DFT calculations reflect strong agreement with our experimental data. Further investigations on scope of these ketones are currently under process in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GB acknowledges award of a research fellowship under DST-PURSE program. Authors thank M/s Hindustan Platinum Pvt. Ltd, Navi Mumbai & Ravindra Heraeus Pvt Ltd, Jaipur for providing gift samples of RuCl3.Notes and references
-
(a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed
; (b) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed
-
(a) Y. Bonvin, E. Callens, I. Larrosa, D. A. Henderson, J. Oldham, A. J. Burton and A. G. M. Barrett, Org. Lett., 2005, 7, 4549–4552 CrossRef CAS PubMed
-
(a) S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734–5740 CrossRef CAS PubMed
-
(a) C.-M. Ho, W.-Y. Yu and C.-M. Che, Angew. Chem., Int. Ed., 2004, 43, 3303–3307 CrossRef CAS PubMed
-
(a) P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS
-
(a) S. Chen, Z. Liu, E. Shi, L. Chen, W. Wei, H. Li, Y. Cheng and X. Wan, Org. Lett., 2011, 13, 2274–2277 CrossRef CAS PubMed
-
(a) S. K. Gupta and J. Choudhury, ChemCatChem, 2017, 9, 1979–1984 CrossRef CAS
- S. Ranganathan, K. M. Muraleedharan, P. Bharadwaj and K. P. Madhusudanan, Chem. Commun., 1998, 2239–2240 RSC
- I. W. C. E. Arends, T. Kodama and R. A. Sheldon, in Ruthenium Catalysts and Fine Chemistry, Berlin, Heidelberg, 2004, pp. 277–320 Search PubMed
- M. Caricato, Æ. Frisch, J. Hiscocks and M. J. Frisch, Gaussian 09: IOps Reference, Gaussian, Inc, Wallingford, CT 06492, U.S.A., 2nd edn, 2009 Search PubMed
- A. D. Becke, Phys. Rev. A, 1986, 33, 2786–2788 CrossRef CAS PubMed
- A. Frisch, A. B. Nelson and A. J. Holder, Gaussview, Pittsburgh, Pa, USA, 2005 Search PubMed
- M. Farina and G. Audisio, Tetrahedron, 1970, 26, 1827–1837 CrossRef
Footnotes |
| † The authors have dedicated this paper to Prof. Vishwakarma Singh on his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06897k |
| This journal is © The Royal Society of Chemistry 2021 |