
Autocatalytic deoximation reactions driven by visible light†
Hongjia
Li
,
Xiaobi
Jing
*,
Yaocheng
Shi
and
Lei
Yu
 *
*
School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225002, P. R. China. E-mail: xbjing@yzu.edu.cn; yulei@yzu.edu.cn
First published on 8th October 2020
Abstract
It was found that the photocatalytic oxidative deoximation reaction is actually an autocatalytic process catalyzed by the generated ketone products. In contrast to reported deoximation methods, this reaction is metal-free and waste-free. The deoximation reaction is a significant transformation process in the industrial production of a variety of fine chemicals. The use of visible light as the driving force makes this method even more practical for the utilization of sustainable energy from an industrial viewpoint. Moreover, understanding the mechanism of the autocatalytic oxidative deoximation reaction may help chemical engineers to develop related techniques to avoid the decomposition of oximes, which are significant starting materials and intermediates during the production of fine chemicals and medicines.
Introduction
The energy sources in our world include fossil energy, hydropower, wind energy, bioenergy, and even nuclear energy, and most of them come from a single star: the sun. Among all the types of energy, photons are undoubtedly the most direct and mild energy source coming from the sun.1 In line with calls for environmental protection and sustainable development, the use of light, especially visible light, as the energy to drive chemical reactions is regarded as an advanced protocol in modern industrial production. In order to achieve this objective, a variety of photocatalysts and protocols have been developed,2 but these suffer from many issues, such as the use of expensive metal3 or non-metal4 catalysts, which leads to high production costs, high loading of photochemical sensitizers/additives in the reactions, which generates waste,5 and low atom utilization ratios in the reaction processes, which result in the generation of large amounts of by-products,6 among others. Therefore, the development of efficient and low-cost photocatalytic reactions is still a highly demanded research objective from a practical application viewpoint.The deoximation reaction is an important transformation in organic synthesis with profound industrial application potential.7 Because most oximes are stable and have unchanging melting points, the oximation–deoximation protocol can be used to protect carbonyls in total synthesis, as well as in the characterization of carbonyl compounds. For example, this strategy was successfully applied by Corey et al. in the total synthesis of the natural product erythronolide A in 1970s.8 Moreover, since a variety of oximes can be synthesized from non-carbonyl starting materials, deoximation reactions are the key step in many fine chemical production processes to produce carbonyls. For example, the deoximation of carvone oxime is the key reaction to synthesize the high-value-added spice carvone from easily accessible limonene, and this technique has been successfully industrialized (Scheme 1).9
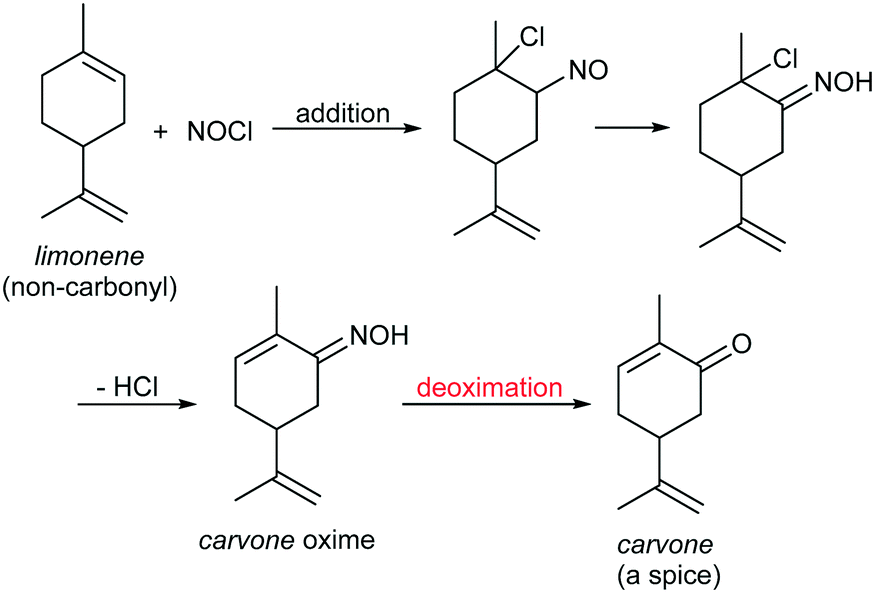 | ||
| Scheme 1 Diagram of the synthetic route to carvone from non-carbonyl limonene in industrial production. | ||
We have continuously focused on green synthetic technologies with industrial application potential.10 Recently, deoximation reactions have attracted our attention because of their great significance in fine chemical production. The present techniques suffer from a series of issues that need to be resolved, such as the use of potentially hazardous cyanide-containing catalysts or chemical oxidants,11 the need for high loadings of irritants such as hexachlorodisilane, SnCl2/TiCl3 or p-toluenesulfonic acid as reagents,12 the employment of halogenated solvents,13 the use of excess hydrochloric acid with iron powder,14 or the employment of nitrite/nitrate-containing catalysts or additives.15 It was found that chalcogen elements such as Se and Te were good oxygen-carrier catalysts for oxidative deoximation reactions under relatively green and mild conditions.16 However, these methods required the explosive oxidant H2O2,16a employed an iron salt as a co-catalyst, which might lead to metal residues in the product,16b or used Te catalysts, which are uncommon and expensive reagents.16c Recently, we unexpectedly found that, under visible light irradiation, the oxidative deoximation reaction could occur in the presence of a few carbonyls (i.e. the products). This process is actually an autocatalytic reaction that occurs without additional metal- or non-metal catalysts and can utilize molecular oxygen as a green, cheap and safe oxidant. Understanding the mechanism of the process may also facilitate the development of related techniques to avoid the decomposition of oximes and reduce the resulting losses in industry. Herein, we wish to report our findings.
Results and discussion
The investigation was started by irradiating an MeCN solution of benzophenone oxime (1a) with blue light LEDs at room temperature in open air. Interestingly, the reaction of 1a that had been stored for a long period produced the deoximation product 2a in good yield, while no reaction occurred in the experiment using freshly prepared 1a (Fig. 1, image (a)). This abnormal phenomenon drew our attention and led us to perform further investigation. Thin-layer chromatography (TLC) analysis was conducted to examine the purity of the samples, and it was found that the stored 1a contained impurities, which were reflected by the additional dot above 1a (ca. 0.19% weight content of impurity materials was obtained after separation) on the TLC plate (Fig. 1, image (b)). The Fourier transform infrared (IR) spectrum of the impurities isolated from stored 1a indicated the existence of a carbonyl group, as reflected by the strong absorption peak at ca. 1700 cm−1 (Fig. 1, image (c), red line). Further analysis of the peaks in the fingerprint area demonstrated that the impurities contained benzophenone (Fig. 1, image (c), red line vs. blue line). The isolated impurity material was confirmed to be pure benzophenone via1H NMR analysis. The spectrum was in accordance with the literature data,17 and no other impurity peaks were observed in the spectrum (Fig. 1, image (d)).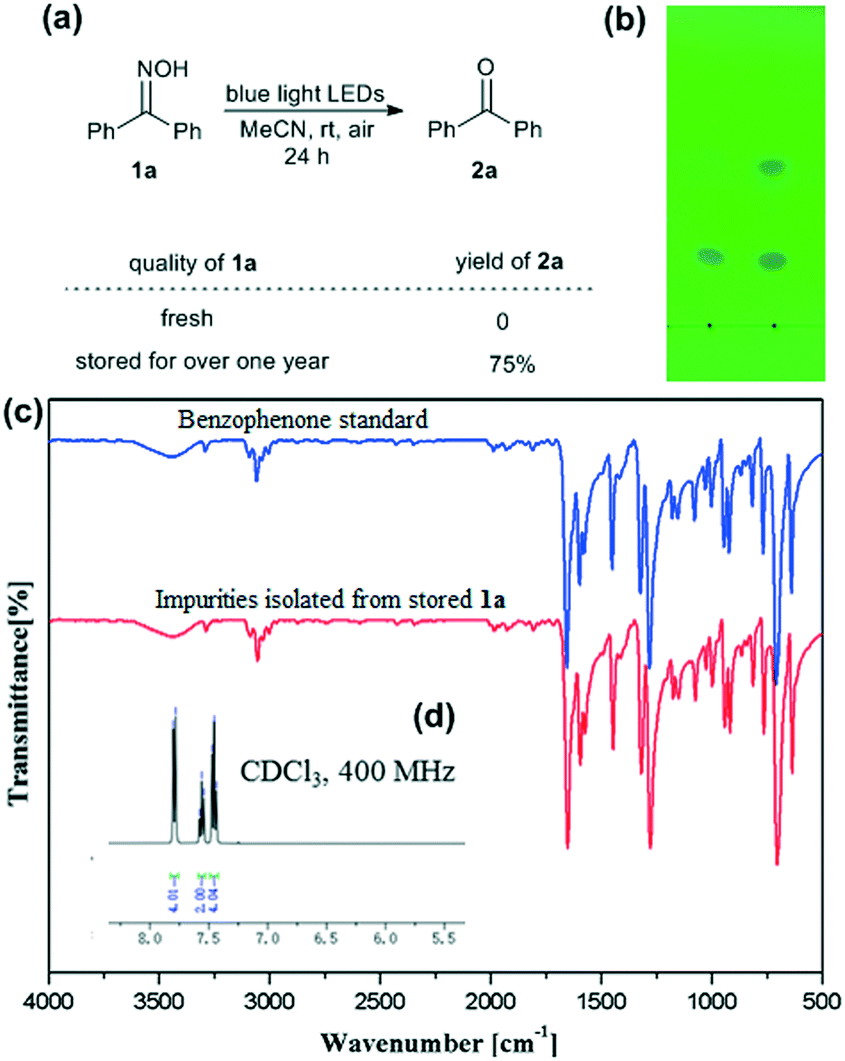 | ||
| Fig. 1 Parallel experiments involving the irradiation of fresh 1a and 1a after long-term storage with blue light LEDs: (a) reaction equation and product yields; (b) photograph of the TLC analysis (eluent: petroleum ether/EtOAc = 10/1) of fresh (left) and stored (right) 1a; (c) IR spectra of the impurities isolated from stored 1a (red) and a benzophenone standard (blue); and (d) the 1H NMR spectrum of the impurities isolated from stored 1a (details given in the ESI†). | ||
The above results demonstrated that the photocatalytic deoximation of 1a was an autocatalytic reaction. Parallel experiments showed that using 0.5 mol% of 2a as the catalyst was the optimum condition for this reaction, affording the product in a good yield of 86% (Table 1, entries 4 vs. 1–3). Increasing the amount of 2a used did not enhance the product yield further (Table 1, entries 5–7 vs. 4). Reactions in the green solvents EtOAc, EtOH, dimethyl carbonate (DMC) and water were tested, but did not generate the desired product (Table 1, entries 8–11). The non-polar solvent p-xylene was also unfavourable for the reaction (Table 1, entry 12). The reaction did not occur under solvent-free conditions (Table 1, entry 13).
|
|
|||
|---|---|---|---|
| Entry | Amount of catalytic 2ab | Solvent | Yieldc |
| a 0.2 mmol of fresh 1a and 1 mL of solvent were employed. b Molar ratio of 2a based on 1a. c Isolated yield based on 1a. | |||
| 1 | 0 | MeCN | 0 |
| 2 | 0.1% | MeCN | 82% |
| 3 | 0.2% | MeCN | 83% |
| 4 | 0.5% | MeCN | 86% |
| 5 | 1.0% | MeCN | 85% |
| 6 | 2.0% | MeCN | 84% |
| 7 | 5.0% | MeCN | 81% |
| 8 | 0.5% | EtOAc | 0 |
| 9 | 0.5% | EtOH | 0 |
| 10 | 0.5% | DMC | 0 |
| 11 | 0.5% | H2O | 0 |
| 12 | 0.5% | p-Xylene | 0 |
| 13 | 0.5% | — | 0 |

A series of oximes (2) were then treated under the optimized conditions to examine the substrate scope of the reaction. Catalyzed by its deoximation product 2b (0.5 mol%), the reaction of phenyl(p-tolyl)methanone oxime (1b) provided 2b in decreased yield (Table 2, entries 2 vs. 1). Using 2a as the catalyst slightly enhanced the product yield of the reaction (Table 2, entry 3). The substrate di-p-tolylmethanone oxime (1c) bearing two methyl groups on each aryl ring gave 2c in only 53% yield in the reaction catalyzed by 2c (Table 2, entry 4). The reaction was improved by using 2a as the catalyst and O2 as oxidant, which afforded an increased product yield of 55–70% (Table 2, entries 5, 6 vs. 4). The substrate 1d bearing a strongly electron-donating group gave the related carbonyl 2d in 42% yield in EtOAc (Table 2, entry 7). The electron-deficient substrate bis(4-chlorophenyl)methanone oxime (1e) was unfavourable for the reaction, no matter what catalyst or oxidant was used (Table 2, entries 8–10). 1-Phenylpropan-1-one oxime (1f) was tested as an example of a substrate bearing an alkyl substituent, and gave the product 2f in 49% yield (Table 2, entry 11). The reaction was significantly enhanced by using 2a as the catalyst (Table 2, entries 12 vs. 11). The use of 1-phenylpentan-1-one oxime (1g), a substrate with a longer alkyl chain, led to decreased product yield (Table 2, entries 13, 14). The reactions using electron-enriched 1-(m-tolyl)ethan-1-one oxime (1h) produced 2h in good yields (Table 2, entries 15, 16), but that of 1-(3-chlorophenyl)ethan-1-one oxime (1i) required 2a as the catalyst and O2 as the oxidant (Table 2, entries 19 vs. 17, 18). Interestingly, the reaction of the similar substrate 1j with a chloro group at the ortho position occurred smoothly with 2j or 2a as the catalyst and using air as the oxidant (Table 2, entries 20, 21). In the deoximation reaction of cyclohexanone oxime 1k, ca. 11% of the starting material was converted to produce cyclohexanone 2k with 92% selectivity (Table 2, entry 22). (E)-3,4-Dihydronaphthalen-1(2H)-one oxime (1l), a substrate bearing a fused phenyl ring, led to the deoximation product 2l in 51% yield (Table 2, entry 23). The deoximation reaction of 9H-fluoren-9-one oxime (1m) did not occur, no matter what catalyst or oxidant was used (Table 2, entries 24–26).
|
|
||||
|---|---|---|---|---|
| Entry | R1, R2 or chemical structure (1)b | Catalytic 2, oxidant | 2, yieldb | Recovery rate of 1 |
| a 0.2 mmol of 1 and 1 mL of MeCN were employed unless otherwise noted. b Isolated yield of 2 based on 1. c Reaction performed in EtOAc. d Selectivity of the product detected via GC-MS. | ||||
| 1 | Ph, Ph (1a) | 2a, air | 2a, 86% | 0 |
| 2 | Ph, 4-MeC6H4 (1b) | 2b, air | 2b, 78% | Trace |
| 3 | 2a, air | 2b, 81% | Trace | |
| 4 | 4-MeC6H4, 4-MeC6H4 (1c) | 2c, air | 2c, 53% | 31% |
| 5 | 2a, air | 2c, 55% | 25% | |
| 6 | 2a, O2 | 2c, 70% | 8% | |
| 7 | 4-MeOC6H4, 4-MeOC6H4 (1d) | 2a, airc | 2d, 42% | 37% |
| 8 | Ph, 4-ClC6H4 (1e) | 2e, air | 2e, 20% | 60% |
| 9 | 2a, air | 2e, 21% | 55% | |
| 10 | 2a, O2 | 2e, 36% | 42% | |
| 11 | Ph, Et (1f) | 2f, air | 2f, 49% | 28% |
| 12 | 2a, air | 2f, 83% | 0 | |
| 13 | Ph, n-Bu (1g) | 2g, air | 2g, 56% | 18% |
| 14 | 2a, air | 2g, 77% | Trace | |
| 15 | 3-MeC6H4, Me (1h) | 2h, air | 2h, 70% | Trace |
| 16 | 2a, air | 2h, 87% | 0 | |
| 17 | 3-ClC6H4, Me (1i) | 2i, air | No reaction | 94% |
| 18 | 2a, air | No reaction | 92% | |
| 19 | 2a, O2 | 2i, 93% | 0 | |
| 20 | 2-ClC6H4, Me (1j) | 2j, air | 2j, 73% | Trace |
| 21 | 2a, air | 2j, 85% | 0 | |
| 22 | –(CH2)5– (1k) | 2a, air | 2k, 10 (92%d) | 89% |
| 23 |
 (1l) (1l) |
2a, O2c | 2l, 51% | 37% |
| 24 |
 (1m) (1m) |
2m, air | No reaction | 95% |
| 25 | 2a, air | No reaction | 92% | |
| 26 | 2a, O2 | No reaction | 92% | |
Control experiments were performed to study the mechanism of the reaction. First, the deoximation reaction of 1a (with 0.5 mol% of 2a) did not occur without light irradiation (in the dark),18 except when 100 mol% azodiisobutyronitrile (AIBN) was added as a free radical initiator; under these conditions, heating 1a at 80 °C in the dark gave 2a in 55% yield.19 Without AIBN, the thermal reaction afforded 2a in only 13% yield. The visible-light-promoted deoximation reaction was completely inhibited by the free radical scavenger 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO),16c and it did not occur under a N2 atmosphere. Detailed procedures of the control experiments are given in the experimental section (vide infra). Moreover, the reaction of 1a in anhydrous MeCN (treated with P2O5) could produce 2a in an elevated yield (91%). The above results demonstrated that the deoximation reaction occurred via a free radical mechanism and that oxygen was the crucial oxidant for the transformation.
Interestingly, ketones 2a–j showed different catalytic performances in the photocatalytic deoximation reactions (Table 2). UV-vis analyses of the ketones were performed to probe the possible reason for this phenomenon, and the results are summarized in Table 3. In MeCN solution, the maximum absorption peak of 2a was located at 350 nm (Table 3, entry 1), while for 2b–c and 2e, the absorption peaks ranged from 350–360 nm, and were thus very similar to that of 2a (Table 3, entries 2–4). Indeed, the catalytic activities of 2b–c and 2e were similar (Table 2, entries 2 vs. 3, 4 vs. 5, 8 vs. 9). A significant blue shift of the maximum absorption peak relative to that of 2a occurred in the spectrum of 2f (Table 3, entries 5 vs. 1), and the catalytic activity of 2f was obviously lower (Table 2, entries 11 vs. 12). Similar trends were also observed between the UV-vis adsorption and the catalytic activities of 2g–j (Table 3, entries 6–9). It was supposed that the light adsorption of 2b–c and 2e near the visible light region facilitated more efficient utilization of light energy than that of the other ketone catalysts, affording higher photocatalytic activity in the deoximation reactions as a result.
| Entry | Ketone | Maximum UV-vis absorption peak |
|---|---|---|
| a 0.5 mmol of 2 was dissolved in 10 mL of MeCN for UV-vis testing (spectra given in the ESI†). | ||
| 1 | 2a | 350 nm |
| 2 | 2b | 350 nm |
| 3 | 2c | 360 nm |
| 4 | 2e | 351 nm |
| 5 | 2f | 298 nm |
| 6 | 2g | 316 nm |
| 7 | 2h | 311 nm |
| 8 | 2i | 313 nm |
| 9 | 2j | 319 nm |
A plausible mechanism was proposed based on the above experiments and reports in the literature (Scheme 2).16c,20–22 First, when irradiated by light, a ketone catalyst such as 2a was activated to its excited state 2a*,20 which could react with oxime 1 to generate the intermediate bi-radical 3.16c,21 Oxidation of the radical moiety in 3 by O2 (or air) furnished 4,22 which soon led to the cyclic intermediate 5. Decomposition of 5 afforded the deoximation product 2, HNO and the catalyst 2a. The by-product HNO could be oxidized to nitrate,16c while the regenerated catalyst 2a initiated the next catalytic cycle. Because the attack of the free radical at the sp2-C site of the oxime was crucial, the reaction was unfavourable for substrates with large steric hindrance (Table 2, entries 24–26). The reduced yield of the reaction of 1gversus that of 1f could also be attributed to retardation of the free radical attack due to the enhanced steric hindrance of the substrate (Table 2, entries 14 vs. 12). The generation of the ketone accelerated the reaction process. The reaction of 1a afforded 2a in only 29% yield after 12 h, which was far less than half of the product yield after 24 h of reaction, showing that the generation of the ketone accelerated the reaction.
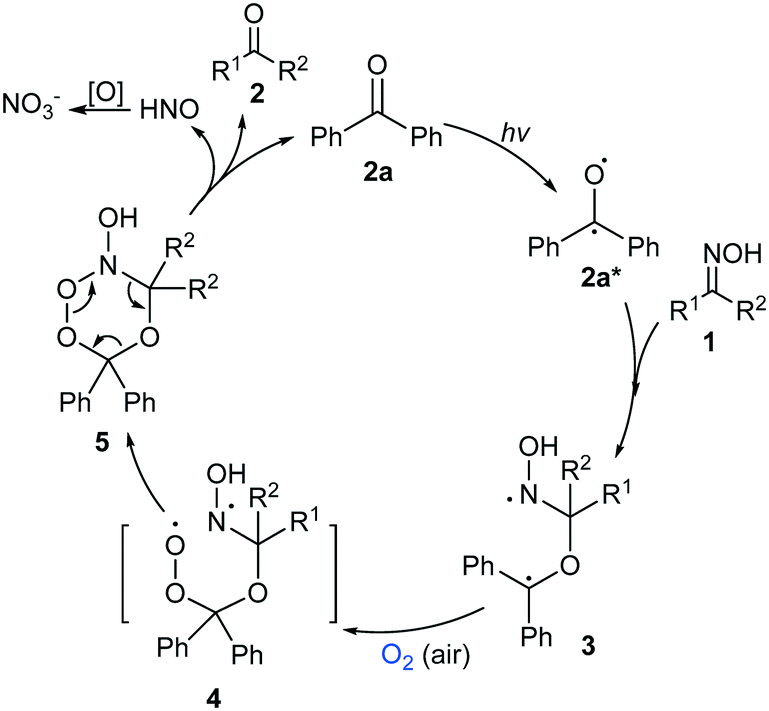 | ||
| Scheme 2 Possible mechanism of the reaction. | ||
Conclusions
In conclusion, the photocatalytic deoximation reaction driven by visible light is actually an autocatalytic process. In contrast with reported deoximation methods, this reaction is metal-free and waste-free. The deoximation reaction is a significant transformation process to produce a variety of fine chemicals. The utilization of visible light as the driving force makes this method even more practical, with the use of sustainable energy. This method is also in accordance with the developing trend of metal-free catalysis from an industrial viewpoint.23 The protocol may be effective for certain examples in industrial production, while others will require a photocatalyst to promote the process. In addition, disclosure of the mechanism of the visible-light-promoted autocatalytic oxidative deoximation reaction may aid chemical engineers in developing related techniques to avoid the decomposition of important oxime starting materials and intermediates and to reduce the resulting losses in industry. Continuing investigations into deoximation reactions, including both catalyst development and mechanistic studies, are ongoing in our laboratory.Experimental
General methods
Chemicals and solvents were purchased from a reagent merchant and were directly used without special treatment. Thin layer chromatogram (TLC) experiments were employed to monitor the reactions, while preparative TLC separation was used to isolate the products. Infrared (IR) spectra were measured using a Bruker Tensor 27 Infrared spectrometer. Proton nuclear magnetic resonance (1H NMR) spectra were recorded using a Bruker Avance instrument (400 MHz). Chemical shifts for 1H NMR are relative to internal Me4Si (0 ppm) and J-values are shown in Hz. The UV-vis spectra were measured using Hanon i5 UV-vis absorption spectrometer. Melting points (m.p.) were measured with a WRS-2A digital melting point measuring instrument.General procedure for the photocatalytic deoximation reaction
0.2 mmol of oxime 1 was added to a reaction tube. A solution of 0.18 mg of 2a as a catalyst (0.5 mol%) in 1 mL of MeCN was then injected by syringe. The mixture was irradiated by blue light LEDs (8 W) in open air for 24 h. The solvent was removed by distillation under reduced pressure with a rotary evaporator, and the residue was isolated using preparative TLC (silica gel plate, eluent: petroleum/EtOAc = 10/1) to afford the related product 2.Control experiments
(1) 0.2 mmol of oxime 1a was added to a reaction tube. A solution of 0.18 mg of 2a as a catalyst (0.5 mol%) in 1 mL of MeCN was then injected by syringe. The above mixture was kept in the dark for 24 h, and no reaction occurred.(2) 0.2 mmol of oxime 1a, 0.2 mmol of AIBN, a magnetic bar and 1 mL of MeCN were added to a reaction tube. The mixture was heated in the dark at 80 °C in open air for 24 h. The solvent was removed via distillation under reduced pressure with a rotary evaporator, and the residue was isolated by preparative TLC (silica gel plate, eluent: petroleum/EtOAc = 10/1), affording 2a in 55% yield.
(3) 0.2 mmol of oxime 1 and 0.2 mmol of TEMPO were added to a reaction tube. A solution of 0.18 mg of 2a as a catalyst (0.5 mol%) in 1 mL of MeCN was then injected by syringe. The mixture was irradiated by blue light LEDs (8 W) in open air for 24 h. No reaction occurred.
(4) 0.2 mmol of oxime 1 was added to a reaction tube. A solution of 0.18 mg of 2a as a catalyst (0.5 mol%) in 1 mL of MeCN was then injected by syringe. The mixture was irradiated by blue light LEDs (8 W) under a protective N2atmosphere for 24 h. No reaction occurred.
Characterization data from the products
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Jiangsu Province (No. BK20181449), Jiangsu Provincial Six Talent Peaks Project (No. XCL-090), and Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- C. Chen, Y. Cao, X. Wu, Y. Cai, J. Liu, L. Xu, K. Ding and L. Yu, Chin. Chem. Lett., 2020, 31, 1078 CrossRef CAS.
- (a) L.-Y. Xie, T.-G. Fang, J.-X. Tan, B. Zhang, Z. Cao, L.-H. Yang and W.-M. He, Green Chem., 2019, 21, 3858 RSC; (b) L.-Y. Xie, J.-L. Hu, Y.-X. Song, G.-K. Jia, Y.-W. Lin, J.-Y. He, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 19993 CrossRef CAS; (c) Q. Guo, Z. Ma, C. Zhou, Z. Ren and X. Yang, Chem. Rev., 2019, 119, 11020 CrossRef CAS; (d) C. Xu, P. Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868 RSC; (e) C. Dong, J. Ji, Z. Yang, Y. Xiao, M. Xing and J. Zhang, Chin. Chem. Lett., 2019, 30, 853 CrossRef CAS; (f) T. Shang, L. Lu, Z. Cao, Y. Liu, W. He and B. Yu, Chem. Commun., 2019, 55, 5408 RSC.
- (a) Y. Wu, M. Yan, Z. Gao, J. Hou, H. Wang, D. Zhang, J. Zhang and Z. Li, Chin. Chem. Lett., 2019, 30, 1383 CrossRef CAS; (b) W. Deng, W. Feng, Y. Li and H. Bao, Org. Lett., 2018, 20, 4245 CrossRef CAS; (c) Y. Huang, Z. Liu, G. Gao, Q. Xiao, W. Martens, A. Du, S. Sarina, C. Guo and H. Zhu, Catal. Sci. Technol., 2018, 8, 726 RSC.
- (a) W. Ou, R. Zou, M. Han, L. Yu and C. Su, Chin. Chem. Lett., 2020, 31, 1899 CrossRef CAS; (b) K. Luo, Y. Chen, W. Yang, J. Zhu and L. Wu, Org. Lett., 2016, 18, 452 CrossRef CAS.
- (a) J. Zelenka, E. Svobodová, J. Tarábek, I. Hoskovcová, V. Boguschová, S. Bailly, M. Sikorski, J. Roithová and R. Cibulka, Org. Lett., 2019, 21, 114 CrossRef CAS; (b) X. Huang, H. Chen, Z. Huang, Y. Xu, F. Li, X. Ma and Y. Chen, J. Org. Chem., 2019, 84, 15283 CrossRef CAS; (c) Y. Ge, P. Diao, C. Xu, N. Zhang and C. Guo, Chin. Chem. Lett., 2018, 29, 903 CrossRef CAS.
- W. Guo, M. Zhao, C. Du, L. Zheng, L. Li, L. Chen, K. Tao, W. Tan, Z. Xie, L. Cai, X. Fan and K. Zhang, J. Org. Chem., 2019, 84, 15508 CrossRef CAS.
- (a) Y. Zheng, A. Wu, Y. Ke, H. Cao and L. Yu, Chin. Chem. Lett., 2019, 30, 937 CrossRef CAS; (b) G. Zhang, X. Wen, Y. Wang, W. Mo and C. Ding, Prog. Chem., 2012, 24, 361 CAS.
- E. J. Corey, P. B. Hopkins, S. Kim, S. E. Yoo, K. P. Nambiar and J. R. Falck, J. Am. Chem. Soc., 1979, 101, 7131 CrossRef CAS.
- (a) C. Isart, D. Bastida, J. Burés and J. Vilarrasa, Angew. Chem., Int. Ed., 2011, 50, 3275 CrossRef CAS; (b) A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 268, 350 CrossRef CAS; (c) R. Reitsema, J. Org. Chem., 1958, 23, 2038 CrossRef CAS; (d) E. E. Royals and S. E. Horne Jr., J. Am. Chem. Soc., 1951, 73, 5856 CrossRef CAS.
- (a) L. Yu, H. Cao, X. Zhang, C. Yang and L. Yu, Sustainable Energy Fuels, 2020, 4, 730 RSC; (b) J. Liu, Y. Cai, C. Xiao, H. Zhang, F. Lv, C. Luo, Z. Hu, Y. Cao, B. Cao and L. Yu, Ind. Eng. Chem. Res., 2019, 58, 20491 CrossRef CAS; (c) S. Zhao, B. Xu, L. Yu and Y. Fan, Chin. Chem. Lett., 2018, 29, 884 CrossRef CAS; (d) H. Cao, B. Zhu, Y. Yang, L. Xu, L. Yu and Q. Xu, Chin. J. Catal., 2018, 39, 899 CrossRef CAS; (e) S. Zhao, B. Xu, L. Yu and Y. Fan, Chin. Chem. Lett., 2018, 29, 475 CrossRef CAS; (f) D. Zhang, Z. Wei and L. Yu, Sci. Bull., 2017, 62, 1325 CrossRef CAS.
- (a) M. Waheed, N. Ahmed, M. A. Alsharif, M. I. Alahmdi and S. Mukhtar, ChemistrySelect, 2019, 4, 7572 CrossRef CAS; (b) A. A. Manesh and B. S. Shaghasemi, J. Chem. Sci., 2015, 127, 493 CrossRef CAS; (c) A. García-Ortiz, A. Grirrane, E. Reguera and H. García, J. Catal., 2014, 311, 386 CrossRef.
- (a) L. Du, J. Gao, S. Yang, D. Wang, X. Han, Y. Xu and Y. Ding, Russ. J. Gen. Chem., 2014, 84, 2200 CrossRef CAS; (b) M.-H. Lin, H.-J. Liu, C.-Y. Chang, W.-C. Lin and T.-H. Chuang, Molecules, 2012, 17, 2464 CrossRef CAS; (c) Y. Liu, N. Yang, C. Chu and R. Liu, Chin. J. Chem., 2015, 33, 1011 CrossRef CAS.
- G. Zhang, X. Wen, Y. Wang, W. Mo and C. Ding, J. Org. Chem., 2011, 76, 4665 CrossRef CAS.
- P. K. Pradhan, S. Dey, P. Jaisankar and V. S. Giri, Synth. Commun., 2005, 35, 913 CrossRef CAS.
- (a) Y. Li, N. Xu, G. Mei, Y. Zhao, Y. Zhao, J. Lyu, G. Zhang and C. Ding, Can. J. Chem., 2018, 96, 810 CrossRef CAS; (b) H. Firouzabadi, N. Iranpoor and K. Amani, Synth. Commun., 2004, 34, 3587 CrossRef CAS; (c) G.-C. Arash, S. Lotfi and Z. Javad, Bull. Korean Chem. Soc., 2008, 29, 2496 CrossRef.
- (a) X. Jing, D. Yuan and L. Yu, Adv. Synth. Catal., 2017, 359, 1194 CrossRef CAS; (b) C. Chen, X. Zhang, H. Cao, F. Wang, L. Yu and Q. Xu, Adv. Synth. Catal., 2019, 361, 603 CrossRef CAS; (c) X. Deng, H. Cao, C. Chen, H. Zhou and L. Yu, Sci. Bull., 2019, 64, 1280 CrossRef CAS.
- Spectral Database for Organic Compounds, SDBS: http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi.
- (a) M. Liu, Y. Li, L. Yu, Q. Xu and X. Jiang, Sci. China: Chem., 2018, 61, 294 CrossRef CAS; (b) L. Yu and X. Huang, Synlett, 2006, 2136 CAS.
- (a) F. Wang, J. Huang, Y. Yang, L. Xu and L. Yu, Ind. Eng. Chem. Res., 2020, 59, 1025 CrossRef CAS; (b) X. Huang, N. Rong, P. Li, G. Shen, Q. Li, N. Xin, C. Cui, J. Cui, B. Yang, D. Li, C. Zhao, J. Dou and B. Wang, Org. Lett., 2018, 20, 3332 CrossRef CAS.
- J. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS.
- Y. Ban, J. Dai, X. Jin, Q. Zhang and Q. Liu, Chem. Commun., 2019, 55, 9701 RSC.
- (a) L. Yu, Y. Huang, Z. Bai, B. Zhu, K. Ding, T. Chen, Y. Ding and Y. Wang, J. Chin. Chem. Soc., 2015, 62, 479 CrossRef CAS; (b) L. Yu, L. Ren, R. Yi and R. Guo, Synth. Commun., 2011, 41, 2530 CrossRef CAS.
- (a) W. Bao, M. He, J. Wang, X. Peng, M. Sung, Z. Tang, S. Jiang, Z. Cao and W. He, J. Org. Chem., 2019, 84, 6065 CrossRef CAS; (b) H. Zhang, M. Han, C. Yang, L. Yu and Q. Xu, Chin. Chem. Lett., 2019, 30, 263 CrossRef CAS; (c) L. Yu, R. Qian, X. Deng, F. Wang and Q. Xu, Sci. Bull., 2018, 63, 1010 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and UV-vis spectra of the products. See DOI: 10.1039/d0re00333f |
| This journal is © The Royal Society of Chemistry 2021 |