
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical control of peptide material phase transitions†
Junjun
Tan‡
 a,
Li
Zhang‡
a,
Ming-Chien
Hsieh
ab,
Jay T.
Goodwin
a,
Martha A.
Grover
b and
David G.
Lynn
*a
a,
Li
Zhang‡
a,
Ming-Chien
Hsieh
ab,
Jay T.
Goodwin
a,
Martha A.
Grover
b and
David G.
Lynn
*a
aDepartment of Chemistry and Biology, Emory University, Atlanta, Georgia 30322, USA. E-mail: dlynn2@emory.edu
bSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
First published on 7th January 2021
Abstract
Progressive solute-rich polymer phase transitions provide pathways for achieving ordered supramolecular assemblies. Intrinsically disordered protein domains specifically regulate information in biological networks via conformational ordering. Here we consider a molecular tagging strategy to control ordering transitions in polymeric materials and provide a proof-of-principle minimal peptide phase network captured with a dynamic chemical network.
Introduction
The properties of polymeric materials are often defined by molecular disorder. Dense liquid states of silicates form glasses that acquire the mechanical properties of a solid while maintaining the disorder of a liquid. Naturally occurring silks contain ordered β-sheet domains for strength dispersed within a flexible disordered protein matrix.1 Such disorder in polymeric materials may arise from subtle quantum defects and/or large scale conformational and covalent heterogeneities,2,3 all contributing to the material properties. Living systems have evolved special strategies to exploit this property. Intrinsically Disordered Protein (IDP) domains, which occur in 30–40% of eukaryotic proteins,4 exist in a condensed liquid state that orders in response to distinct binding partners.5 This signaling function has evolved to amplify and extend the information encoded in genetic networks.6 Such signaling information is also implicated in protein misfolding diseases7 through the oligomer cascade hypothesis where disordered proteins mediate cellular transfection.8 We imagined transforming conformational disorder by chemically triggering ordering of polymeric materials.9We began with our initial kinetic model for peptide phase transitions incorporating the Flory–Huggins parameter (χ), oligomer growth rate constants (kg), and peptide stacking energy (ΔG),10 when validated with the amyloid peptide Ac-KLVFFAE-NH2, identified solubility of the initial dense liquid state as a critical point of control. As with general glass transitions, significant hysteresis can accompany phase changes of peptide assemblies,11 consistent with the recent demonstration of dynamic nuclei pre-existing within the solute-rich phases.12 The ability to nucleate the growth of distinct structures from these phases with other polymers13 highlights the potential to externally regulate the growth of specific structural order in polymeric materials14 and thereby capture the encoded conformational preferences.
Given the sophisticated process by which eukaryotic cells enzymatically tag disordered proteins for clearance,15 we sought an analogous process, a chemical tagging strategy to capture the ordering potential of disordered polymer phases. Here we report a proof-of-principle approach where oligomer peptide phase transitions16,17 are initiated by direct chemical tagging.
Results and discussion
a. Phase network design
The low complexity domain (LCD) proteins regulating RNA processing18–21 contain repetitive amino acid dyads facilitating ready access to both the solute-rich liquid phases and the cross-β assemblies of RNP granules.18–21 The amino acid threonine (T) is a polar amino acid whose solubility should limit initial condensation, but maintain a propensity22,23 for stabilizing β-sheets. To evaluate concatemer assembly, a series of TT dyads (H–TTF)n–NH2 (n = 2, 3) which included phenylalanine (F) to stabilize stacking in cross-β assemblies, were constructed. When these peptides (2 mM) are incubated in 40% acetonitrile (ACN) with an apparent pH of 8 at 24 °C, the nonapeptide H–TTFTTFTTF–NH2, (TTF)3, undergoes an initial condensation to form particles that subsequently transition to nanotubes with diameters of 38.4 ± 7.4 nm (Fig. 1a–c and Fig. S1†). FT-IR analyses of these (TTF)3 assemblies (Fig. S2a†) confirmed amide I stretches at 1621 cm−1, characteristic of extended β-sheets24,25 with a weaker transition at 1693 cm−1 supporting anti-parallel strand arrangements.26 The cross-β architecture was confirmed by powder X-ray diffraction (XRD) d-spacings at 4.7 Å and 10.1 Å (Fig. S2b†).27 | ||
| Fig. 1 Transmission electron microscopy analyses of (TTF)3 and (TTF)2 peptides. The peptides were incubated at 2 mM in 40% ACN/water, 24 °C, and pHapp 8. TEM micrographs of (a) (TTF)3 particles at 1 h, (b) growing ribbons at 24 h that transform into (c) mature nanotubes after 2 weeks compared with (d) (TTF)2 solution at 1 week. | ||
In contrast, the hexapeptide H–TTFTTF–NH2, (TTF)2, did not transition to particles or transition to higher-order assemblies under these conditions (Fig. 1d). These two concatemers, (TTF)3 and (TTF)2, then provide phase networks positioned on either side of the particle solubility threshold, and the (TTF)2 peptide concatemer provides a scaffold to explore N-terminal tagging strategies that direct liquid–liquid phase transitions.
b. Chemical network design
Building on successful imine polymerization chemistry,28–32 we sought a monomer capable of condensation but not polymerization. Based on the LCD designs above, the di- and tri-peptide aldehydes TF–CHO and TTF–CHO were envisioned as potential bifunctional tagging reagents for either C-terminal aldehydes or N-terminal Ser, Thr, Asn, or Cys residues via the cyclic N, X-backbone acetal linkage shown for Thr in Fig. 2a. We anticipated that the 5-membered N,O-acetal of Thr would be compatible with β-sheet assembly but with greater solubility, making it less available for initial condensation and subsequent template-directed polymerization.17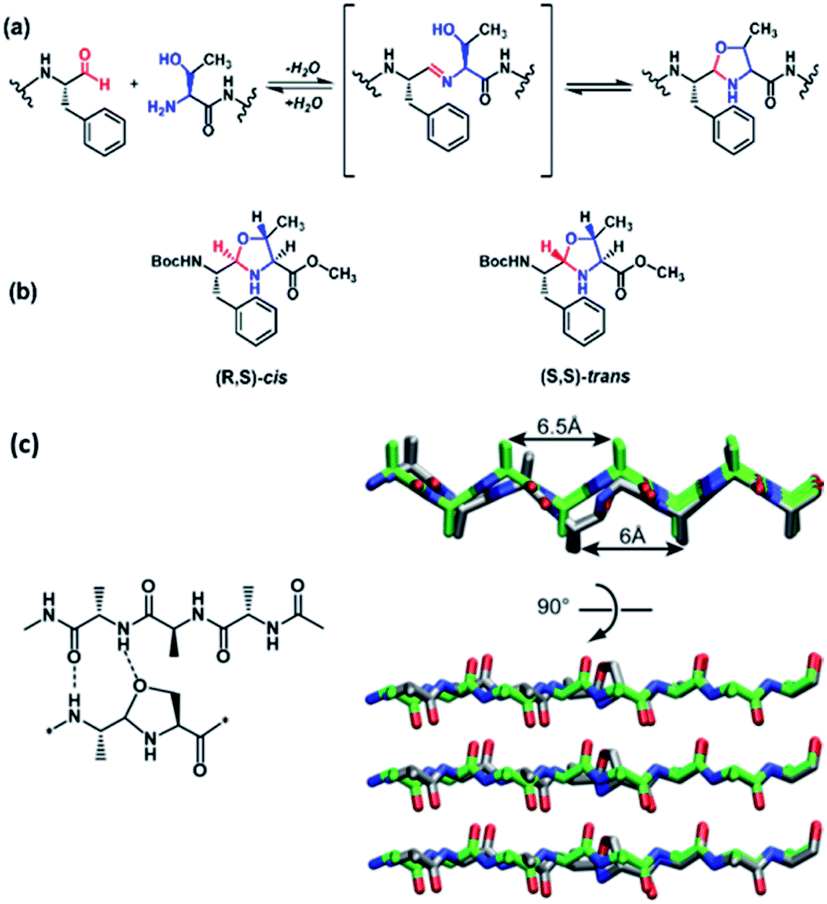 | ||
| Fig. 2 N,O-acetal linkage on peptide backbone. (a) N,O-acetal linkage generated by condensation of the phenylalanine aldehyde and with the threonine N-terminus amine and sidechain alcohol. (b) Of the two possible cyclic N,O-acetals, the (R,S)-cis configuration places the ring substituents at C-2 and C-4 in quasi-equatorial orientations. (c) Modelling N,O-acetal associations on the minimal scaffold of AAAAoxAAAA and H–AAAAAAAA–H showing H-bonding (left) and parallel in-register strands (right). | ||
As shown in Scheme S1† for the TTF–CHO monomer, preparation occurs via the N-Boc phenylalanine Weinreb amide33 to allow for reduction to the aldehyde34 and protection as the dimethyl acetal. Peptide coupling follows standard solution-phase activation protocols with Fmoc-protected amino acids35 allowing the dynamic chemical network (DCN) to be generated with HCl deprotection of the acetal. This reaction mixture can be dried in vacuo, and the TTF–CHO monomer re-suspended in 40% ACN/water at an apparent pH of 8 for dynamic assembly (Fig. 2a). While the TF–CHO monomer reversibly cyclizes to a product that is susceptible to irreversible oxidation to the pyrazine (Scheme S2†), the TTF–CHO monomer does not suffer this side reaction. Rather, TTF–CHO generates a dynamic chemical network (DCN) dominated by the cyclic dimer with minor amounts of cyclic trimer and cyclic tetramer (Fig. S3 and Table S1†), but no higher-order oligomeric assemblies are detected. Consistent with the design, this system does not access liquid–liquid phases or template paracrystalline assembly under these conditions.
c. Structural models for peptide backbone N,O-acetals
There are two possible cyclic N,O-acetals as shown in Fig. 2b. The (R,S)-cis configuration places the ring substituents at C-2 and C-4 in quasi-equatorial orientations, and the acetal stereochemical preference for the N,O-derivatives were analyzed via condensation of N-Boc–L-Phe–CHO with L-Ser and L-Thr methyl ester in benzene to approximate the hydrophobic particle environment. A single N,O-acetal hydrogen resonance is observed by NMR, and irradiation of this acetal H-2 resonance in the L-Thr product gives significant Overhauser enhancement of H-4 intensity relative to H-5 (Fig. S4a†), consistent with the acetal hydrogen and H-4 located on the same face of the acetal ring. The L-Ser product also gives a single acetal proton, and again irradiation of the acetal proton significantly enhances the H-4 resonance (Fig. S4b†). These results are consistent with a single dominant conformation adopting a ring pucker positioning the backbone substituents in pseudo-equatorial positions in the (R,S)-cis stereoisomer (Fig. 2b). Initial model building with this structure in a peptide backbone suggests that an H-bond acceptor and amine donor are both suitably positioned for stabilizing β-sheet H-bonding (Fig. 2c), showing no electrostatic or steric constraints that would violate cross-β associations even in parallel strand arrangements.d. DCN peptide tagging
Based on our current understanding, the energetic specificity of the N,O-acetal may contribute positively or negatively to intercepting the chemical network of H–TTFTTF–NH2. Providing access of this TTF–CHO DCN to the peptide-rich liquid phase (Fig. 3a) with average particle widths of 29.0 ± 3.0 nm after 24 h (Fig. S5†) allows particles to form and grow in size, nucleate, and grow fibers (Fig. 3b–d). HPLC and mass spectrometry analyses confirm the appearance of a new product, TTFoxTTFTTF (Fig. S6†), where H–TTFTTF–NH2 has been covalently tagged N-terminally with a single aldehyde monomer to give the 5-membered N,O-acetal linkage (ox = oxazolidine). Unlike the final assembly of (TTF)3, which assembles as nanotubes, this network generates fibrils which bundle into larger fibers over time (Fig. 3b–d).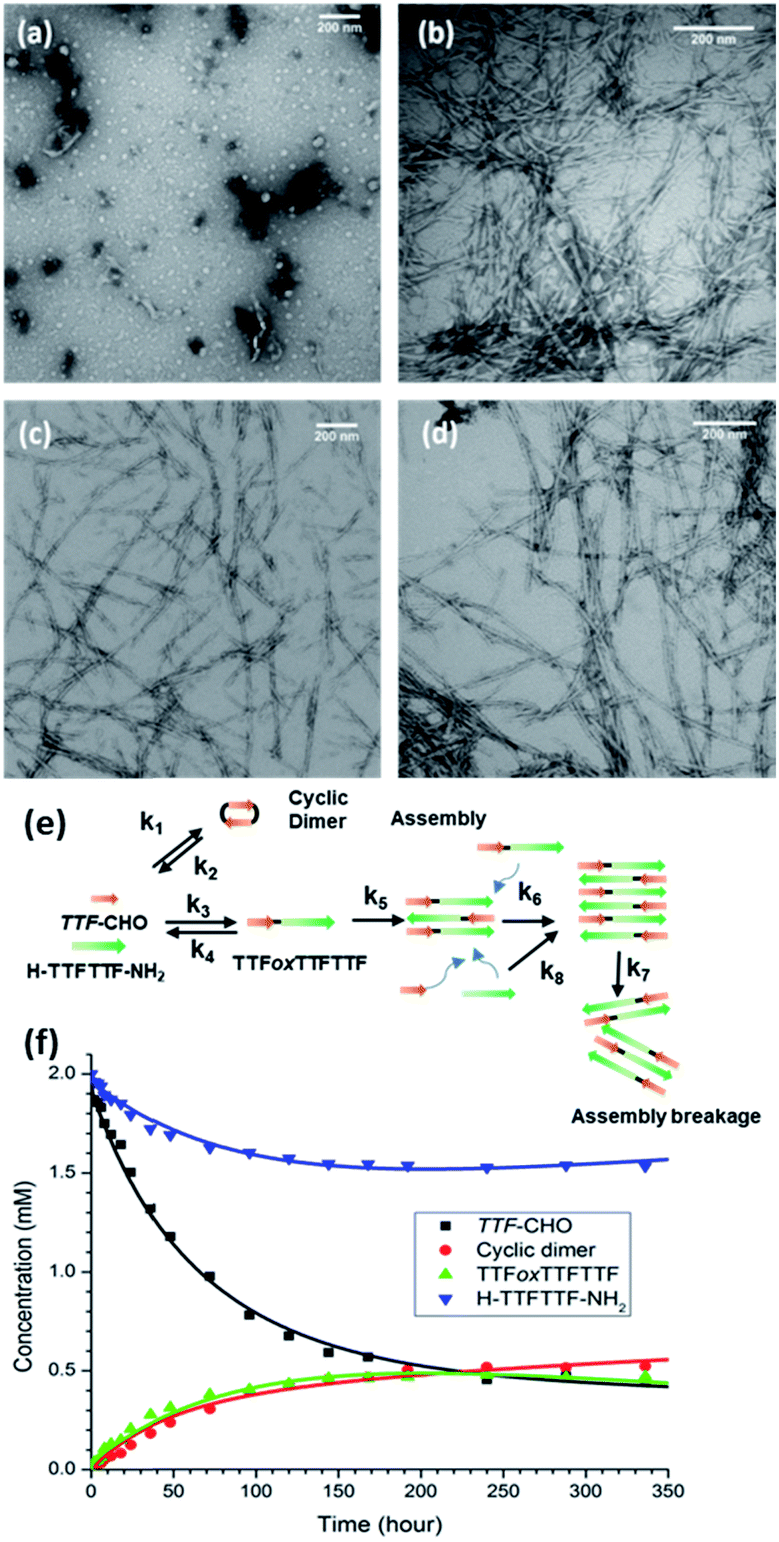 | ||
| Fig. 3 TEM analyses and kinetic modelling of the TTF–CHO network containing H–TTFTTF–NH2. Electron micrographs of (a) particles at 24 h, (b) fibers after 3 days, (c), mature fibers after 1 week, and (d) higher order assemblies after 10 days. Mechanistic model (e) for the DCN and (f) mathematic fits for kinetic data described in Experimental section. | ||
e. Network distribution and assembly
The fibrillar assembly can be pelleted at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 30 min, but quantitative enrichment proved to be limited by the gel-like character of the network. Reversed-phase HPLC analyses (Fig. S6†) do show Gaussian peak shapes, consistent with aggregation and hydrolysis during elution not limiting the use of the chromatograms to estimate network composition (Table 1). Accordingly, the supernatant is dominated by the peptide H–TTFTTF–NH2 and the cyclic TTF–CHO dimer, while the TTFoxTTFTTF condensation product is enriched in the pellet (Table 1). The positively charged surfaces of these assemblies restricted thioflavin staining of the pelleted fiber, and the presence of cross-β assembly36–38 was confirmed with powder diffraction d-spacings of 4.7 Å and 10 Å (Figure S7b†).39,40 The amide I IR band was blue shifted to 1645 cm−1, relative to the 1621 cm−1 observed for the model (TTF)3 peptide nanotubes (Fig. S7a†), consistent with the expected reduction in normal mode delocalization for β-sheets containing backbone amide replacements.41
000 × g for 30 min, but quantitative enrichment proved to be limited by the gel-like character of the network. Reversed-phase HPLC analyses (Fig. S6†) do show Gaussian peak shapes, consistent with aggregation and hydrolysis during elution not limiting the use of the chromatograms to estimate network composition (Table 1). Accordingly, the supernatant is dominated by the peptide H–TTFTTF–NH2 and the cyclic TTF–CHO dimer, while the TTFoxTTFTTF condensation product is enriched in the pellet (Table 1). The positively charged surfaces of these assemblies restricted thioflavin staining of the pelleted fiber, and the presence of cross-β assembly36–38 was confirmed with powder diffraction d-spacings of 4.7 Å and 10 Å (Figure S7b†).39,40 The amide I IR band was blue shifted to 1645 cm−1, relative to the 1621 cm−1 observed for the model (TTF)3 peptide nanotubes (Fig. S7a†), consistent with the expected reduction in normal mode delocalization for β-sheets containing backbone amide replacements.41
| DCN | TTF–CHO/H–TTFTTF–NH2, unseeded | TTF–CHO/H–TTFTTF–NH2, (TTF)3 nanotubes seeded | ||
|---|---|---|---|---|
| Species | Molar% | Molar% | ||
| Supernatant | Pellet | Supernatant | Pellet | |
| Monomer | 4.6 | 2.9 | 7.0 | 1.7 |
| H–TTFTTF–NH2 | 65.8 | 18.9 | 64.5 | 17.1 |
| TTFoxTTFTTF | 9.1 | 59.6 | 8.1 | 66.3 |
| Cyclic dimer | 20.5 | 18.6 | 20.4 | 14.9 |
f. Kinetic analysis of the TTF–CHO/H–TTFTTF–NH2 network
The introduction of H–TTFTTF–NH2 into the TTF–CHO network expands the previously developed kinetic models for these networks17 to include the intermolecular N,O-acetal linkage between TTF–CHO and H–TTFTTF–NH2 (k3 & k4), the cyclic dimerization of TTF–CHO (k1 & k2), the phase transitions with TTFoxTTFTTF (k5), and the templated assembly of TTFoxTTFTTF (k6, k7, k8; Fig. 3e, S8, Table S2, and Scheme S3†). As illustrated in Fig. 3f, the equilibrated network (at 250 hours) includes unreacted TTF–CHO monomer, cyclic dimer, H–TTFTTF–NH2, and TTFoxTTFTTF. As shown in Scheme S3,† where Akaike Information Criterion (AIC) is used to select the model most consistent with the experimental data,42 the growth of TTFoxTTFTTF appears as a simple second-order reaction with the assemblies reaching equilibrium without consuming all the TTF–CHO and H–TTFTTF–NH2 substrates. These analyses suggest that a simpler model without the assembly reactions may be sufficient. However, removal of the assembly events from the model results in poor data fitting and Akaike weights (Scheme S3 and Table S4†), suggesting that templated-assembly has an important impact on the kinetics and ultimate distribution in the DCN. Given the slow kinetics along with growth of metastable particles, the network dynamics are most consistent with two-step nucleation and template-directed assembly, with the caveat that generalization of the kinetic constants at this point may risk overinterpretation and will require further substantiation.g. Control by alternative cross-β templates
Neither H–TTFTTF–NH2 nor the TTF–CHO chemical network undergoes condensation until they are combined. After mixing, particles form early and grow slowly with no apparent limit on assembly as the growing particles enable the nucleation and propagation of fibrils that mature into bundled fibers. This additional peptide condensation, with some analogy to nucleic acid recombination events,43 here changes the solubility of the particles. The creation of this new environment in the particles opens the nucleation and propagation of fibers just as seen commonly with other peptides.44–46 To evaluate the susceptibility of the particles to external templates, nanotube seeds prepared by mild sonication of mature (TTF)3 peptide assemblies were added to the TTF–CHO/H–TTFTTF–NH2 network (Fig. 4). These (TTF)3 nanotube seeds rapidly induce nanotube growth to give diameters indistinguishable from the parent (TTF)3 nanotube with no observable fiber product (Fig. 4). Enriching the assemblies at 16000 × g (Fig. S9, Table S5†) confirm the presence of TTFoxTTFTTF products in the seeded network. XRD of these enriched assemblies show the typical cross-β d-spacings of 4.7 Å and 10.1 Å (Fig. S10b†) with FT-IR amide I stretches at 1622 cm−1 and 1645 cm−1, supporting the presence of both (TTF)3 seeds and TTFoxTTFTTF in the observed nanotubes (Fig. S10a†). The weak but diagnostic 1693 cm−1 transition for anti-parallel β-sheet structures is also apparent (Fig. S10†), consistent with antiparallel strand orientations being retained. While the N,O-acetal is compatible with both nanotube and fiber morphologies, the extent of reaction is the same with both templates (Table 1). Moreover, no evidence for TTFoxTTFoxTTF products is obtained with either template, suggesting that N,O-acetal stacking energy of this isostere within the cross-β configuration limits multiple tagging events.
 | ||
| Fig. 4 Adding (TTF)3 nanotube seeds to the TTF–CHO/H–TTFTTF–NH2 network. TEM images of (a) the mature (TTF)3 nanotubes fragmented by sonication for 2 h prior to being added at 0.5 mM to the fresh TTF–CHO/H–TTFTTF–NH2 (2 mM/2 mM) network; (b) nanotubes induce the growth of the network within 10 min that (c) grow into robust nanotubes within 24 h and (d) continue to mature into homogeneous nanotubes. Scale bars = 200 nm. | ||
Conclusions
Initial synthetic dynamic polymerization networks have been extended from reductive amination ligation to acetal formation for amine nucleoside polymers.28 The template-directed approach achieves sequence specific translation via base complementarity,29,30 and expands nature's chain-growth processes to sequence selective step-growth polymerization.31,32 Distinctly different ligation chemistries continue to be developed,47–50 opening the possibility for other reaction networks21 to be used for regulating phase behaviour51,52 in polymer assembly pathways.27,44 Our kinetic modelling of peptide assembly predicts several limiting states of assembly,10 and H–TTFTTF–NH2, which does not transition to higher order assembly, and H–TTFTTFTTF–NH2, which transitions through particle phases to achieve higher order nanotube assembly, provided a framework for exploring the critical initial phase transitions in multi-nucleation processes.17,47,48,53 Distinct from oxidative disulfide cross-linking in protein folding54 and native chemical ligation of peptide fragments,55 our reversible tagging strategy targets N-terminal Asn, Thr, and likely Ser and Cys residues, to regulate biopolymer assembly10,28–32,48–50 by creating a peptide isostere compatible with cross-β peptide supramolecular assembly.19,45,56Congeners of LCD proteins are used to populate the limiting extremes of our model, and dynamic coupling of the TTF–CHO tag with H–TTFTTF–NH2 generates TTFoxTTFTTF, which induces the liquid transition to the solute-rich particle phase that makes possible nucleation and propagation of a new cross-β supramolecular assembly. The resulting network is an equilibrium mixture of soluble peptides and fibers (Table 1). The ordered composition of this mixed state is predicted by our model to be limited by peptide stacking energies.10 Replacement of the amide with a cyclic N,O-acetal isostere then is able to access the liquid–solid transition as it is accommodated within cross-β architectures to produce heterologous nanotubes following nanotube seeding. However, no oligomers containing two backbone N,O-acetals, TTFoxTTFoxTTF, are detected, even in the presence of template with excess TTF–CHO. This finding suggests that the N,O-acetal is less well accommodated in cross-β assemblies than 6-membered cyclic N,N-acetals,17,31 thus enabling selective mono-tagging. Control of the molecular disorder in diverse materials36,57 biased for aggregation58,59 may find its greatest potential in ribonucleoprotein granules and intrinsically disordered protein domains regulating biological information flow through conformational ordering.60,61 This basic tagging strategy is nicely demonstrated in phosphorylation mediated liquid–liquid phase transitions of reflectin proteins,62 however this chemical tagging strategy can now be more broadly explored across polymeric materials, triggering phase changes and nucleating crystallization for molecular computation, catalysis, and information processing.
Experimental
General material and methods
All chemical reagents and solvents are purchased from Sigma-Aldrich, Fisher, AnaSpec, Alfa-Aesar, and Cambridge Isotope Laboratories without further purification. All chemical reactions are performed under inert N2 atmospheres unless otherwise noted. All NMR experiments are performed on INOVA 400 MHz or INOVA 600 MHz NMR spectrometers.H–TTFTTF–NH2 (TTF)2 and H–TTFTTFTTF–NH2 (TTF)3 peptides are synthesized using standard Fmoc solid-phase synthesis protocols on CEM Liberty Microwave Automated Peptide Synthesizer on rink amide resin. The peptides are cleaved from dried resin (resulting in C-terminal amine capped peptide) by addition of cleavage cocktail (90 vol% TFA, 5 vol% thioanisole, 3 vol% ethanedithiol and 2 vol% anisole) for 4 h at room temperature, precipitated and washed in cold diethyl ether then purified by reverse-phase HPLC (Waters Delta 600) using a Waters Atlantis C-18 preparative column (19 × 250 mm) with acetonitrile–water gradient and lyophilized. The molecular identities of purified peptides are confirmed by MALDI-TOF mass spectrometry. Structural models were constructed by energy minimizing (MacroModel v9.9. Schrodinger, LLC63) H-bonded peptides (OPLS 2005 force field64) arranged as β-strands.
Transmission electron microscopy (TEM)
All TEM analyses are performed with a Hitachi H-7500 transmission electron microscope at 75 kV. A TEM copper grid with a 200-mesh carbon support (Electron Microscopy Sciences) is covered with 10 μL of DCN or peptide solution for 1 min before wicking the excess solution with filter paper. 10 μL of 2% uranyl acetate is added and incubated for 2 min, excess solution wicked away, and the grids are placed in desiccators to dry under vacuum overnight. Widths measured from TEM are converted into diameters by setting 2× width = the tube circumference and solving for the diameter.Attenuated total reflectance fourier transform infrared (FT-IR)
FT-IR spectra are acquired using a Jasco FT-IR 4100 at room temperature and averaging 500 to 1000 scans with 2 cm−1 resolution, using an MCT detector with a 5 mm aperture and a scanning speed of 4 mm s−1. Aliquots (10 μL) of DCN or peptide solution are dried as thin films on a Pike Galdi ATR diamond crystal.X-ray powder diffraction (XRD)
The samples are lyophilized to yield dry powder and the spectra are obtained by loading the powder into a 0.2 mm mylar capillary. The diffraction patterns are acquired on a Bruker APEX-II diffractometer with graphite monochromated Cu radiation K-alpha radiation, λ = 1.54184 Å, 40 kV and 35 mA, with a 0.5 pinhole collimator. Exposure times are typically 300 s per frame, and the data integration software XRD2DSCAN65 is used to convert the two-dimensional data into a theta-2theta scan.Network generation
TTF–CHO monomer is generated by deprotecting TTF–dimethyl acetal with 4% HCl in H2O. The solvent is removed in vacuo at 20 °C, with the TTF–CHO residue re-dissolved with H–TTFTTF–NH2 peptide to give an equimolar (2 mM) solution in 40% ACN/H2O. The solution pH is adjusted by titrating with aliquots of 5% Et3N in H2O using a Fisher Scientific Accumet Basic AB15 pH meter, and is then incubated at 4 °C. For the template seeding experiments, (TTF)3 nanotube seeds are generated by sonicating (TTF)3 nanotubes for 1 h in a bath sonicator (Branson ultrasonic Model 3510R-MT, 42 kHz) at room temperature and the resulting seeds are added to TTF–CHO + H–TTFTTF–NH2 network after pH adjustment. Solution pH is re-measured and readjusted if necessary. For HPLC analysis of assembly, the network is pelleted at 16000 × g for 30 min at 4 °C, and the pellet is resuspended in 40% acetonitrile/water for analysis.
Kinetic analyses
The DCN is analyzed by sampling the DCN solution by HPLC over 2–4 weeks. All HPLC analyses use a Waters Delta 600 HPLC equipped with a photodiode array UV/Vis detector and a reverse-phase HPLC column (Kromasil 100-5C18, 4.6 × 250 mm). HPLC solvent gradient: acetonitrile: 10 mM TEAA in H2O: 0–90 min: 10% to 100%, 90–100 min: 100%, 100–101 min: 100% to 10%, 101–110 min: 10%. UV absorbance at 258 nm is recorded and integrated for kinetics analysis, using the absorption coefficient obtained with phenylalanine standards. Species identification is assigned by ESI mass spectrometry at the Emory University Mass Spectrometry Center.To analyse the chemical reaction and physical assembly of the network, all reactions for the TTF–CHO network as arranged in Table S2,† including the reversible dimer and trimer formation, primary and secondary nucleation of assembly, assembly growth, and templating of trimer formation at the end of assembly ends, are included in kinetic modelling. The rate equations for the individual species are then arranged in Scheme S3.† The kinetic equations were fit to the species concentrations, and the optimal parameter set is gained by minimizing the sum of square error (SSE) between the calculated and experimental values10 with the fmincon function in MATLAB 2012a (The Mathworks, Inc). The optimized parameters (Table S3†) and the further model discrimination are included in the ESI† (SI).
Relative quantification of network components
Based on previous network analyses, the absorbance (A) for each peptide is determined by integration of the peak area at 258 nm, where the aromatic phenyl ring is responsible for the absorption. The absorbance increase is linear with the number of Phe (F) residues and provides the relative extinction coefficient, based on the Lambert–Beer Law A = εcl, for the network members as the number of monomer units increase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Hong Yi and Jeanette Taylor from the Robert P. Apkarian Electron Microscopy Core of Emory University for Electron Microscopy assistance, Dr Bing Wang and Dr Shaoxiong Wu from the Emory NMR Center for NMR assistance, and Dr Fred Strobel for mass spectrometry analysis. We acknowledge Pieter Burger for access to MacroModel and the processing power to run minimizations. The research was supported by grants from NSF CHE-1507932 and NSF/DMR-BSF 1610377, and NIH Alzheimer's Disease Research Center: P50AG025688.References
- N. J.-A. Chan, D. Gu, S. Tan, Q. Fu, T. G. Pattison, A. J. O'Connor and G. G. Qiao, Nat. Commun., 2020, 11, 1630 CrossRef.
- C. Scalliet, L. Berthier and F. Zamponi, Nat. Commun., 2019, 10, 5102 CrossRef.
- D. Khomenko, C. Scalliet, L. Berthier, D. R. Reichman and F. Zamponi, Phys. Rev. Lett., 2020, 124, 225901 CrossRef CAS.
- K. J. Niklas, A. K. Dunker and I. Yruela, J. Exp. Bot., 2018, 69, 1437–1446 CrossRef CAS.
- A. Toto, C. Camilloni, R. Giri, M. Brunori, M. Vendruscolo and S. Gianni, Sci. Rep., 2016, 6, 21994 CrossRef CAS.
- I. Lindström and J. Dogan, ACS Chem. Biol., 2018, 13, 1218–1227 CrossRef.
- S. Auer, P. Ricchiuto and D. Kashchiev, J. Mol. Biol., 2012, 422, 723–730 CrossRef CAS.
- L. C. Walker, D. G. Lynn and Y. O. Chernoff, Prion, 2018, 12, 261–265 CrossRef.
- A. Majumdar, P. Dogra, S. Maity and S. Mukhopadhyay, J. Phys. Chem. Lett., 2019, 10, 3929–3936 CrossRef CAS.
- M.-C. Hsieh, D. G. Lynn and M. A. Grover, J. Phys. Chem. B, 2017, 121, 7401–7411 CrossRef CAS.
- F. Garcia Quiroz, N. K. Li, S. Roberts, P. Weber, M. Dzuricky, I. Weitzhandler, Y. G. Yingling and A. Chilkoti, Sci. Adv., 2019, 5, eaax5177 CrossRef.
- R. F. Rengifo, A. Sementilli, Y. Kim, C. Liang, N. X. A. Li, A. K. Mehta and D. G. Lynn, ChemSystemsChem, 2020, 2, e2000007 Search PubMed.
- A. K. Rha, D. Das, O. Taran, Y. Ke, A. K. Mehta and D. G. Lynn, Angew. Chem., Int. Ed., 2020, 59, 358–363 CrossRef CAS.
- L.-W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry, Nat. Commun., 2017, 8, 1273 CrossRef.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS.
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC.
- C. Chen, J. Tan, M.-C. Hsieh, T. Pan, J. T. Goodwin, A. K. Mehta, M. A. Grover and D. G. Lynn, Nat. Chem., 2017, 9, 799–804 CrossRef CAS.
- W. M. Aumiller and C. D. Keating, Nat. Chem., 2016, 8, 129–137 CrossRef CAS.
- S. Calabretta and S. Richard, Trends Biochem. Sci., 2015, 40, 662–672 CrossRef CAS.
- M. Feric, N. Vaidya, T. S. Harmon, D. M. Mitrea, L. Zhu, T. M. Richardson, R. W. Kriwacki, R. V. Pappu and C. P. Brangwynne, Cell, 2016, 165, 1686–1697 CrossRef CAS.
- Y. Lin, D. S. W. Protter, M. K. Rosen and R. Parker, Mol. Cell, 2015, 60, 208–219 CrossRef CAS.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95–120 CrossRef CAS.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS.
- T. Miyazawa, J. Chem. Phys., 1960, 32, 1647–1652 CrossRef CAS.
- A. K. Mehta, K. Lu, W. S. Childers, Y. Liang, S. N. Dublin, J. Dong, J. P. Snyder, S. V. Pingali, P. Thiyagarajan and D. G. Lynn, J. Am. Chem. Soc., 2008, 130, 9829–9835 CrossRef CAS.
- J. T. Goodwin and D. G. Lynn, J. Am. Chem. Soc., 1992, 114, 9197–9198 CrossRef CAS.
- Z.-Y. J. Zhan and D. G. Lynn, J. Am. Chem. Soc., 1997, 119, 12420–12421 CrossRef CAS.
- Z.-y. J. Zhan, J. Ye, X. Li and D. G. Lynn, Curr. Org. Chem., 2001, 5, 885–902 CrossRef CAS.
- X. Li, A. F. Hernandez, M. A. Grover, N. V. Hud and D. G. Lynn, Heterocycles, 2010, 82, 1477–1488 CrossRef.
- X. Li, Z.-Y. J. Zhan, R. Knipe and D. G. Lynn, J. Am. Chem. Soc., 2002, 124, 746–747 CrossRef CAS.
- M. Mentzel and H. M. R. Hoffmann, Chem.-Ztg., 1997, 339, 517–524 CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- W. S. Childers, N. R. Anthony, A. K. Mehta, K. M. Berland and D. G. Lynn, Langmuir, 2012, 28, 6386–6395 CrossRef CAS.
- D. Lin, Y. Luo, S. Wu, Q. Ma, G. Wei and X. Yang, Langmuir, 2014, 30, 3170–3175 CrossRef CAS.
- J. Wang, K. Tao, P. Zhou, E. Pambou, Z. Li, H. Xu, S. Rogers, S. King and J. R. Lu, Colloids Surf., B, 2016, 147, 116–123 CrossRef CAS.
- E. D. Eanes and G. G. Glenner, J. Histochem. Cytochem., 1968, 16, 673–677 CrossRef CAS.
- A. J. Geddes, K. D. Parker, E. D. T. Atkins and E. Beighton, J. Mol. Biol., 1968, 32, 343–358 CrossRef CAS.
- R. C. Elgersma, T. Meijneke, R. de Jong, A. J. Brouwer, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, Org. Biomol. Chem., 2006, 4, 3587–3597 RSC.
- W.-F. Xue, S. W. Homans and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8926–8931 CrossRef CAS.
- S. L. Andersen and J. Sekelsky, BioEssays, 2010, 32, 1058–1066 CrossRef CAS.
- M.-C. Hsieh, C. Liang, A. K. Mehta, D. G. Lynn and M. A. Grover, J. Am. Chem. Soc., 2017, 139, 17007–17010 CrossRef CAS.
- C. Liang, R. Ni, J. E. Smith, W. S. Childers, A. K. Mehta and D. G. Lynn, J. Am. Chem. Soc., 2014, 136, 15146–15149 CrossRef CAS.
- J. E. Smith, C. Liang, M. Tseng, N. Li, S. Li, A. K. Mowles, A. K. Mehta and D. G. Lynn, Isr. J. Chem., 2015, 55, 763–769 CrossRef CAS.
- Y. Altay, M. Tezcan and S. Otto, J. Am. Chem. Soc., 2017, 139, 13612–13615 CrossRef CAS.
- Y. Bai, A. Chotera, O. Taran, C. Liang, G. Ashkenasy and D. G. Lynn, Chem. Soc. Rev., 2018, 47, 5444–5456 RSC.
- W. Hordijk, S. Shichor and G. Ashkenasy, ChemPhysChem, 2018, 19, 2437–2444 CrossRef CAS.
- J. H. van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC.
- Y. Peng, F. Wang, Z. Wang, A. M. Alsayed, Z. Zhang, A. G. Yodh and Y. Han, Nat. Mater., 2015, 14, 101–108 CrossRef CAS.
- M. A. Vorontsova, D. Maes and P. G. Vekilov, Faraday Discuss., 2015, 179, 27–40 RSC.
- A. Chotera, H. Sadihov, R. Cohen-Luria, P.-A. Monnard and G. Ashkenasy, Chem.–Eur. J., 2018, 24, 10128–10135 CrossRef CAS.
- D. Fass and C. Thorpe, Chem. Rev., 2018, 118, 1169–1198 CrossRef CAS.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem., 2018, 2, 0122 CrossRef CAS.
- S. Li, A. K. Mehta, A. N. Sidorov, T. M. Orlando, Z. Jiang, N. R. Anthony and D. G. Lynn, J. Am. Chem. Soc., 2016, 138, 3579–3586 CrossRef CAS.
- Y. Liang, D. G. Lynn and K. M. Berland, J. Am. Chem. Soc., 2010, 132, 6306–6308 CrossRef CAS.
- A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson, T. P. J. Knowles and E. Gazit, Nat. Commun., 2014, 5, 5219 CrossRef CAS.
- J. Nývlt, Cryst. Res. Technol., 1995, 30, 443–449 CrossRef.
- P. E. Wright and H. J. Dyson, Curr. Opin. Struct. Biol., 2009, 19, 31–38 CrossRef CAS.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- R. Levenson, C. Bracken, C. Sharma, J. Santos, C. Arata, B. Malady and D. E. Morse, J. Biol. Chem., 2019, 16804–16815 CrossRef CAS.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- A. Rodriguez-Navarro, J. Appl. Crystallogr., 2006, 39, 905–909 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03666h |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2021 |