
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenated salt assisted Cu-catalyzed asymmetric 1,4-borylstannation of 1,3-enynes: enantioselective synthesis of allenylstannes†
Jialin
Ye
a,
Yang
Liao
bc,
Hao
Huang
bc,
Yang
Liu
bc,
Dongmei
Fang
bc,
Min
Wang
*bc,
Lianrui
Hu
*d and
Jian
Liao
 *abc
*abc
aCollege of Chemical Engineering, Sichuan University, Chengdu 610065, China
bNatural Products Research Center, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China. E-mail: jliao@cib.ac.cn; wangmin@cib.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 10049, China
dSchool of Science and Research Center for Advanced Computation, Xihua University, Chengdu 610039, China. E-mail: hulianrui@iccas.ac.cn
First published on 22nd December 2020
Abstract
An enantioselective 1,4-borylstannation of 1,3-enynes employed a chiral sulfoxide phosphine (SOP)/Cu complex as a catalyst, and the desired products, chiral allenylstannes, were first synthesized by asymmetric catalysis with satisfactory yields and enantioselectivies. In this protocol, a catalytic amount of additive, a halogenated salt, plays a crucial role in the success. Control experiments and theoretical studies disclosed that the four-membered ring transmetallation transition states which were stabilized by a halide anion are the key to yields and stereochemical outcomes.
Introduction
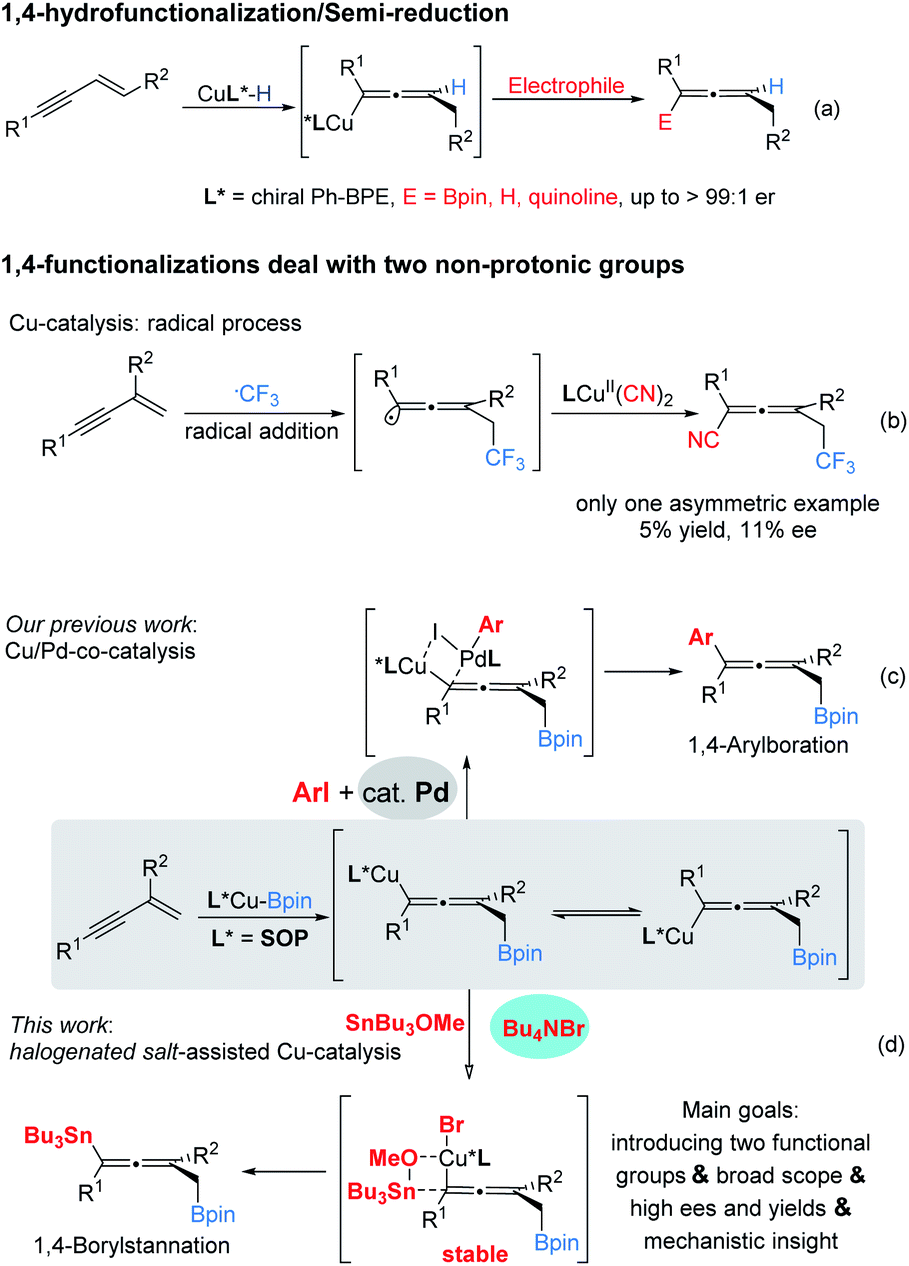
Developing novel and efficient methods to prepare axially chiral allenes, an important class of skeletons broadly present in natural products, pharmaceuticals, and other materials and which serve as synthetic intermediates, is of great interest.1 Compared to classically predominant strategies, e.g. resolution2 and stereospecific transformation of chiral substrates,3 accessing chiral axial allenes via efficient asymmetric catalysis (transition metal catalysis and organocatalysis) of prochiral substrates is a long-standing challenge and highly desirable.Transition metal-catalyzed 1,4-functionalizations of unactivated 1,3-enynes is becoming a valuable and straightforward option to prepare allenes,4 mainly because of the ready availability of the starting materials, and two (mostly, different) functional groups can be installed for the construction of more complex multisubstituted allenes in a step-economic way. However, to date, the majority of reported methods have focused on racemic versions and there are only sporadically successful examples of asymmetric catalysis.5,6 Until more recently, Cu-catalyzed 1,4-functionalization of unactivated 1,3-enynes has attracted significant attention with respect to the synthesis of axially chiral allenes. Representative examples by Hoveyda, Ge, Engle, Buchwald and their co-workers showed that, via the addition of chiral LCu–H species to 1,3-enynes, the in situ generated enantioenriched Cu-allenyl intermediates were immediately trapped by appropriate electrophiles (Bpin, proton and quinolone) to afford the corresponding chiral allenes efficiently (Scheme 1a).6 These protocols could be termed enantioselective 1,4-hydrofunctionalization/semi-reduction;6e that is, one proton and another functional group were introduced into the allenyl frameworks. Despite these eminent advances, practical enantioselective 1,4-difunctionalization of 1,3-enynes dealing with two non-protonic functional groups has rarely been achieved. In 2018, Liu and co-workers disclosed an efficient radical 1,4-addition to access CF3-containing allenyl nitriles. However, in their enantioselective version, an unsatisfactory stereochemical outcome, which could be caused by poor remote stereocontrol, was delivered (Scheme 1b).7 We considered that a protocol involving trapping enantioenriched copper allenyl species generated by the addition of chiral Cu–Bpin species to the carbon–carbon double bond of 1,3-enynes, once appropriate electrophiles had been engaged, would afford chiral allenes.8 In general, pursuing suitable electrophiles, particularly non-protonic electrophiles, is a challenging task.9,10
 | ||
| Scheme 1 Cu-catalyzed asymmetric 1,4-functionalization of 1,3-enynes. | ||
Recently, our lab reported a cooperative Cu/Pd-catalyzed enantioselective 1,4-borylarylation to synthesise chiral allenes. In that catalysis, apart from the exclusive 1,4-selectivities, it is notable that the stereochemical outcome benefited from the excessive usage of palladium catalyst (15 mol% Pd vs. 5 mol% Cu), due to ready racemization of enantioenriched copper–allenyl species (Scheme 1c).8 This racemization process represents a serious challenge to access polyfunctional chiral allenes via this strategy. Instead of bimetal catalysis, we envisaged that the enantioenriched copper–allenyl species could be stabilized and, as a consequence, chiral allenes would be afforded with highly stereochemical outcomes. We imagined that this goal could be achieved through the identification of an appropriate additive, which is frequently presented in various asymmetric catalysis11 and metal-catalyzed cross-couplings12 and plays critical roles in stereocontrol, transformation efficiency and other outcomes. Meanwhile, a tin reagent was demonstrated to be a suitable electrophile in Cu-catalyed borylstannylation of alkynes (by Takaki et al.) and alkenes (by our group).13
Herein we report the first example of salt additive assisted copper catalyzed highly enantioselective 1,4-borylstannation of 1,3-enynes which was promoted by a chiral sulfoxide phosphine (SOP)14 ligand. According to the present protocol, chiral allenylstannes,15 which are very important synthetic intermediates due to their versatile transformation in many sophisticated reactions, were first synthesized via asymmetric catalysis; further, an added catalytic amount of additive plays a critical role in the stereochemical outcome; in addition, mechanistic investigations, including experimental and theoretical studies, which distinctly elucidate the role of additive salt (to date, an investigation of the mechanistic roles of additive remains a challenge)11 are also presented in this article (Scheme 1d).
Results and discussion
1,3-Enyne 1a, Bu3SnOMe and bis(pinacolato)diboron (B2pin2) were chosen as reaction partners to commence our investigation on the enantioselective 1,4-borylstannation (Table 1; for detailed information about condition screening, please see ESI†). By employing an in situ prepared chiral P-di-iPr-sulfoxide phosphine (SOP-1)/CuCl complex (10 mol%) as a pre-catalyst, the reaction was carried in THF and at 0 °C for 12 hours. The desired product 2a′ was afforded in a moderate NMR yield and stereochemical outcome (73% NMR yield, 76% ee; entry 1; an unidentified side product was observed). Comparing this to our previous study,8 we reasoned that the moderate result here was not a result of poor stereocontrol over the chiral ligand but was caused by racemization of the enantioenriched copper–allenyl intermediate. To find a solution to this issue, salt additives, which have been proven to play an important role in some metal-catalyzed crossing-couplings,12 were employed in this reaction. To our delight, in the presence of LiCl, both yield and enantioselectivity were significantly enhanced to an excellent level (97% yield, 94% ee, entry 2), and it is notable that a catalytical amount of LiCl enables this efficiency. Encouraged by this result, we then turned our attention to ligand evaluation. All reactions with P-di-iPr-sulfoxide phosphine ligands (SOP-1–3) bearing a benzodioxole skeleton produced 2a′ in excellent yields and the good to excellent stereocontrol outcomes were determined from the ring size (94–97% yield, 88–94% ee, entries 2–4). Similarly, P-di-Ph-(SOP-4) and P-di-cyclopentyl-sulfoxide phosphine (SOP-5) ligands also work well but slightly lower yields and enantioselectivities were afforded (75–80% yield, 82–88% ee, entries 5–6). A commercially available ligand (R,R)-Ph-BPE, which has been successfully employed in the asymmetric 1,4-hydrofunctionalization of 1,3-enynes, failed to generate the desired product (entry 7).16 We next examined the impact of salt additives on the reaction. LiBr furnished a similar result to LiCl. Ammonium salts (Bu4NX, X = F, Cl, Br and I) were obviously beneficial both for yields and ees (entries 8–12). These results highlight the importance of chiral sulfoxide phosphine ligands (SOPs) and halogenated salts for significant outcomes (yield and ee). Other copper salts like CuBr and CuI had a slight influence on ee value (entries 13–14). Thus, SOP-1, CuBr and Bu4NBr were selected as preferential ligand, copper source and additive, respectively. And this catalytic system worked well when the catalyst loading was reduced to 1 mol%: 2a′ was obtained with 99% NMR yield and 97% ee (entry 15). The absolute configuration of the product was assigned to be an (S)-configuration by comparing its electronic circular dichroism (CD) spectrum with a theoretical simulated CD spectrum by the time-dependent DFT method (for details, see ESI†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | CuX | Additive | Yieldb (%) | eec (%) |
| a Unless otherwise noted, reactions were carried out with 1a (0.2 mmol), Bu3SnOMe (0.3 mmol), (Bpin)2 (0.3 mmol), L (12 mol%), Cu salt (10 mol%), additive (10 mol%) in THF (1.0 mL) at 0 °C for 12 h. b Determined by crude 1H NMR analysis with dimethyl terephthalate as internal standard, n.r. = no reaction, n.d. = not determined. c The ee was determined by chiral HPLC analysis of compound 2a, which was prepared via oxidation of the C–Bpin group of 2a′ to the corresponding primary alcohol (C–OH). d (CuBr1 mol%), SOP-1 (1.2 mol%), Bu4NBr (1 mol%). e In parentheses, isolated yield of 2a. | |||||
| 1 | SOP-1 | CuCl | None | 73 | 76 |
| 2 | SOP-1 | CuCl | LiCl | 97 | 94 |
| 3 | SOP-2 | CuCl | LiCl | 95 | 91 |
| 4 | SOP-3 | CuCl | LiCl | 94 | 88 |
| 5 | SOP-4 | CuCl | LiCl | 75 | 82 |
| 6 | SOP-5 | CuCl | LiCl | 80 | 88 |
| 7 | (R,R)-Ph-BPE | CuCl | LiCl | n.r. | n.d. |
| 8 | SOP-1 | CuCl | LiBr | 95 | 92 |
| 9 | SOP-1 | CuCl | Bu4NF·3H2O | 74 | 94 |
| 10 | SOP-1 | CuCl | Bu4NCl·H2O | 95 | 96 |
| 11 | SOP-1 | CuCl | Bu4NBr | 99 | 96 |
| 12 | SOP-1 | CuCl | Bu4NI | 99 | 95 |
| 13 | SOP-1 | CuBr | Bu4NBr | 99 | 97 |
| 14 | SOP-1 | CuI | Bu4NBr | 99 | 96 |
| 15d | SOP-1 | CuBr | Bu4NBr | 99 (76)e | 97 |

With the optimized conditions in hand, we then explored the scope of this enantioselective 1,4-borylstannation (Table 2). Firstly, we examined the aromatic 1,3-enynes. A wide range of enynes with different functional groups like alkyl (Me, tBu, CF3), halogens (F, Cl, Br), ether (OMe), aryl (Ph), ester (COOEt) and cyanide, at para-, meta-, and ortho-positions, could be readily converted into the corresponding products with generally good to excellent enantioselectivities (68–97% ee) (Table 2, A). Among these, a sensitive functional group (COOEt) was tolerated in this borylstannation process, with modest results (2i, 78% yield, 68% ee). Enynes with electron-withdrawing groups (4-CF3, 2-Cl, 2-Br) showed inferior reactivities, giving the corresponding products with slightly lower ee values (2j, 2l, 2m). In addition, 2-naphthyl (2r) and heteroaromatic 1,3-enynes (2s, 2t) were accommodated with excellent results (85–94% yields, 90–94% ee).
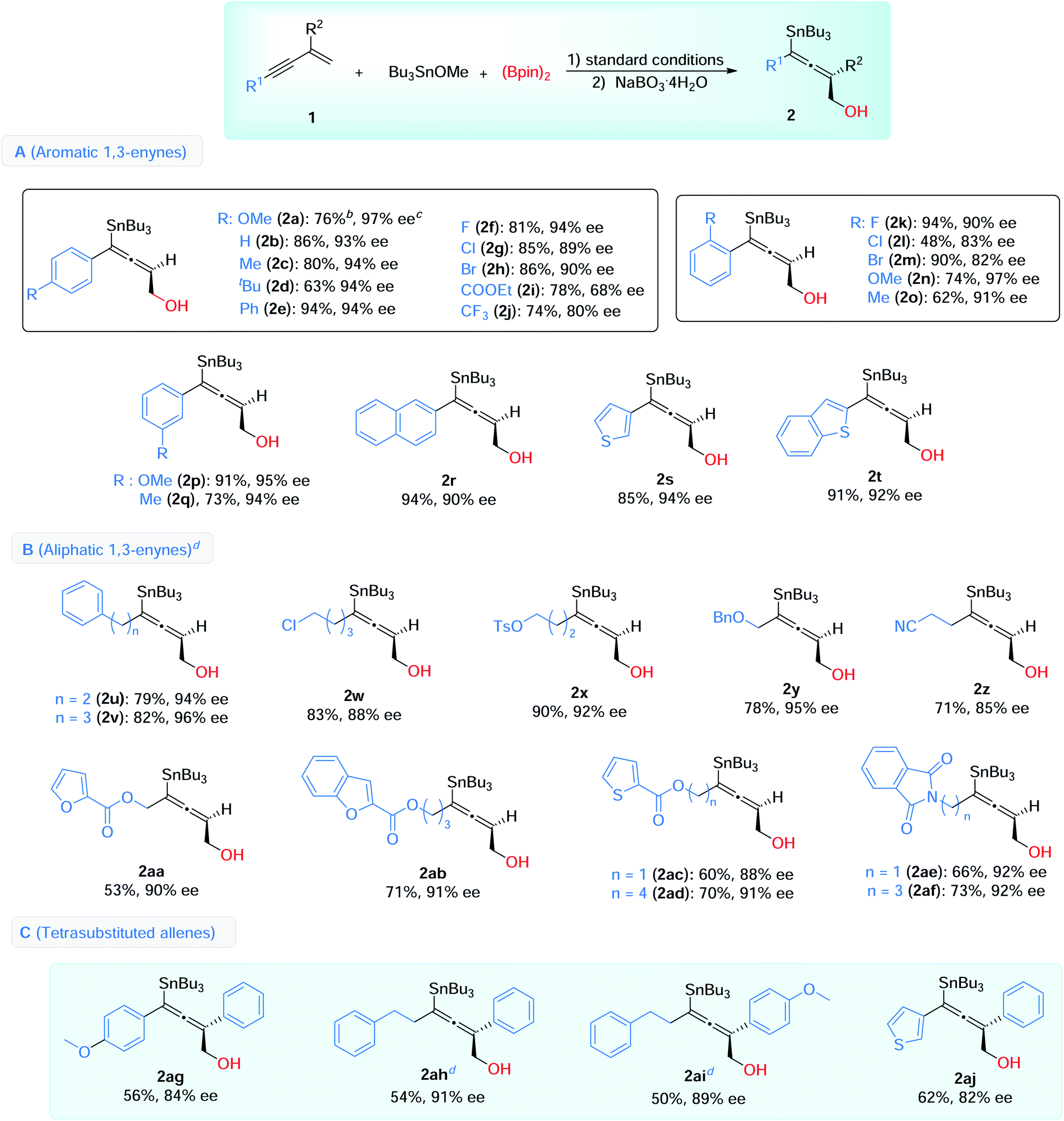
For the aliphatic scope of 1,3-enyne, different chain lengths and functional groups were investigated, and good enantioselectivities were observed in the process (85–96% ee) (Table 2, B). The chain lengths have little effect on the results (2uvs.2v, 2acvs.2ad, 2aevs.2af). Various functional groups, including halogen (2w), ether (2y), esters (2x, with aromatic heterocycles, 2aa–2ad), nitrile (2z) and imides (2ae and 2af), were compatible under these conditions, and the desired products were afforded with high enantioselectivities.
We further showed that this reaction could be used to access chiral tetrasubstituted allenes (Table 2, C). 2-Arylsubstituted 1,3-enynes were employed as substrates, under the standard conditions, and the desired tetrasubstituted chiral allenes were successfully obtained with satisfactory yields and enantioselectivities (2ag–2aj, 54–62% yields, 82–91% ee).
Finally, a gram-scale experiment and transformation of chiral allenylstanne product was carried out (Scheme 2). This protocol can be readily scaled up: with 8 mmol of 1a, in the presence of 0.25 mol% of catalyst (S/C = 400) and a longer reaction time (24 h), the desired product 2a (3.45 g) was obtained in 92% isolated yield and 96% ee. In the presence of SnCl4 as Lewis acid,17 the chiral allenylstanne 2a′ was transformed into allenylcarbinol, which was oxidized by NaBO3·4H2O to give the corresponding chiral allenyldiol 3a with high enantioselectivity but moderate diastereoselectivity. Interestingly, treating the chiral allenylstanne 2a with Ac2O, the acetyl protected allene was obtained in quantitative yield, which could be stereospecifically transformed into allenylcarbinol 4a with excellent diastereoselectivity (100% ee and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) under the same conditions. The Still-coupling of the Sn–C bond also worked with a moderate stereospecific transformation (see ESI† for details).
:1 dr) under the same conditions. The Still-coupling of the Sn–C bond also worked with a moderate stereospecific transformation (see ESI† for details).
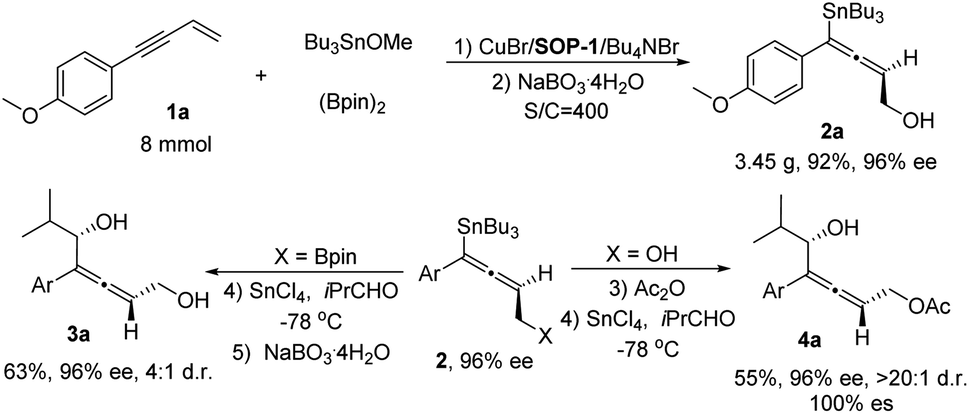 | ||
| Scheme 2 Gram-scale experiment and transformation of allenylstanne. Conditions: (1) 1a (8 mmol), Bu3SnOMe (12 mmol), (Bpin)2 (12 mmol), SOP-1 (0.024 mmol, 0.3 mmol%), CuBr (0.02 mmol, 0.25 mol%), Bu4NBr (0.02 mmol, 0.25 mol%) in THF (4.0 mL) at 0 °C for 24 h. (2) Na BO3·4H2O, THF-H2O, rt, 2 h. (3) Ac2O, TEA, DMAP, Et2O, rt, 20 min. (4) −78 °C, SnCl4, 30 min, then, iPrCHO, 30 min. (5) NaBO3·4H2O, THF-H2O, rt, 2 h. Ar = 4-Me-Ph. | ||
What concerned us next was the mechanism, particularly the mechanistic role of an additive in this metal-catalyzed asymmetric protocol. It is worth noting again that the additive (halogenated salts) was added in a catalytic amount in this protocol, which indicates that the role of the additive should be different from that in the previous transition metal-catalyzed cross-couplings.12 An important task is to determine whether it is the cation or anion of Bu4NBr, or the synergy between cation and anion that promotes the excellent stereochemical outcome in this protocol. We set up two control experiments (Scheme 3). Experiment a was conducted by adding KBr (10 mol%, equal to the Cu-catalyst) into this catalytic system and product 2a was obtained with 48% NMR yield and 78% ee (similar to the results obtained by the reaction without an additive, see Table 1, entry 1); experiment b was conducted by adding KBr (10 mol%) and the equivalent crown ether (10 mol% 18-C-6), it is interesting that in the presence of 18-C-6, the yield and ee of product 2a were greatly enhanced (87%, 96% ee). The difference between these two experiments (a and b) could be reasoned to be the solubility of the additive (KBr is poorly soluble in THF). In experiment b, the entrapment of potassium cation by 18-C-6 enables KBr to be soluble in THF. According to these data (experiments a, b, c and d), we could reason that, in this protocol, in the presence of a soluble halide anion, the racemization process could be avoided or slowed down dramatically via the coordination of a halide anion to the copper(I) intermediate, generating the desired chiral allene with high yields and excellent stereochemical outcomes.
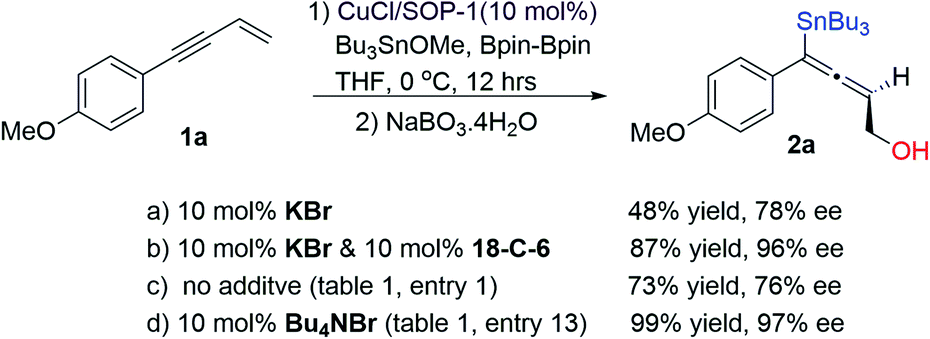 | ||
| Scheme 3 Control experiments. | ||
Next, we conducted a series of experiments under synthetically relevant conditions. The progress of the reaction was monitored by in situ1H NMR spectroscopy (Fig. 1, left plot) (see ESI† for details). We first conducted a standard experiment, and we plotted the formation of product 2a′ as a function of time (blue line). We then performed the same experiment with changed amounts of 1a, (Bpin)2, and Bu3SnOMe, respectively. The rate of product formation in this experiment was represented. When the amount of 1a and (Bpin)2, was changed, the rate of product formation (green line and red line) was almost identical to that in the standard reaction (blue line), while the rate of product formation was sensitive to the concentration of Bu3SnOMe (purple line), suggesting that this reaction has a zeroth order of dependence on 1a and (Bpin)2, and has a first order of dependence on Bu3SnOMe. We next carried out initial rate experiments to probe kinetic information about the catalyst for this reaction, and the data demonstrated that the reaction can be promoted faster when the catalyst loading is increased, and the reaction rate showed a negative order on the catalyst (Fig. 2, right plot, and see ESI† for details). We then investigated the effect of salt (Bu4NBr) on the reaction (Fig. 2, left plot) and an inhibiting effect on the reaction was observed when Bu4NBr was overloaded (compared to CuBr). The nonlinear effect experiment indicated that this reaction follows a single-metal catalyzed model (Fig. 2, right plot). According to these results, we speculated that the inhibiting effect of the salt and the aforementioned negative order is probably caused by partial dimerization of the copper complex.18
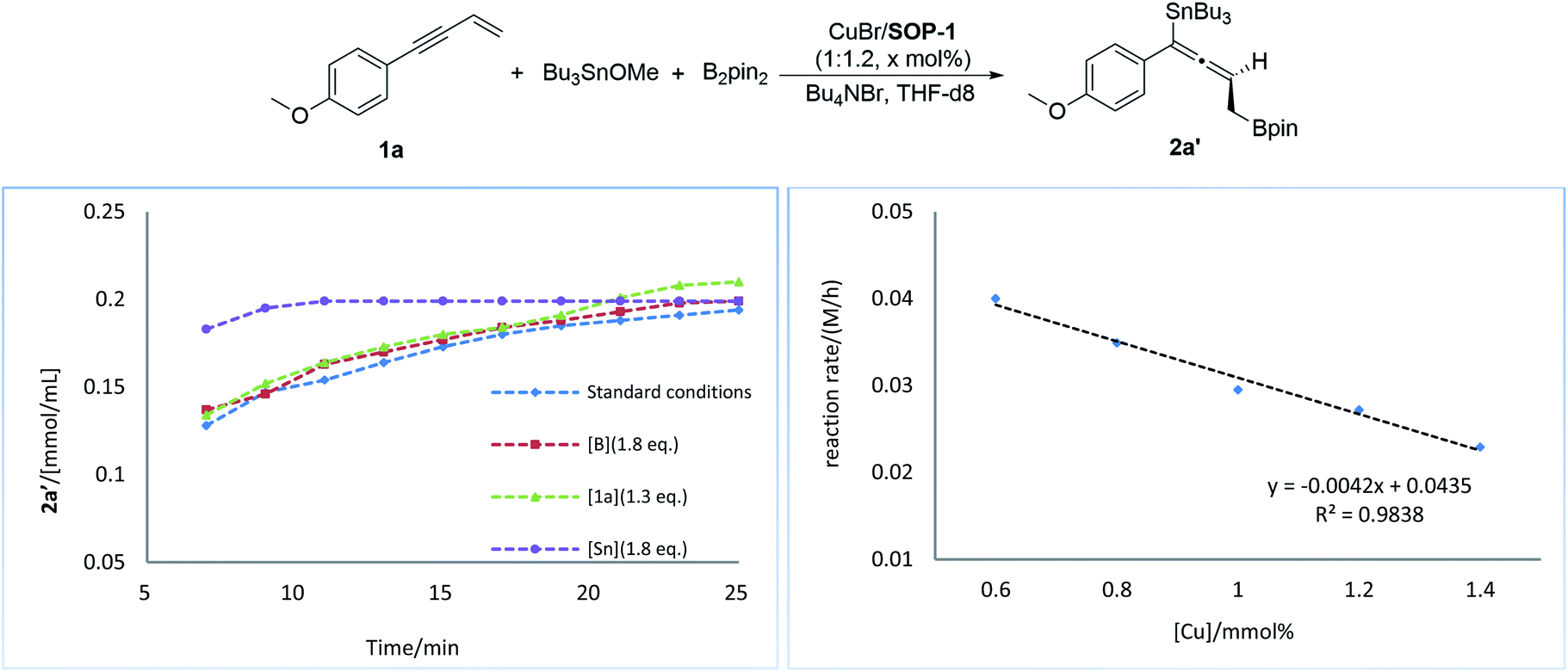 | ||
| Fig. 1 Kinetic profiles of the reaction (left plot). Rate dependence on catalyst loading (right plot). | ||
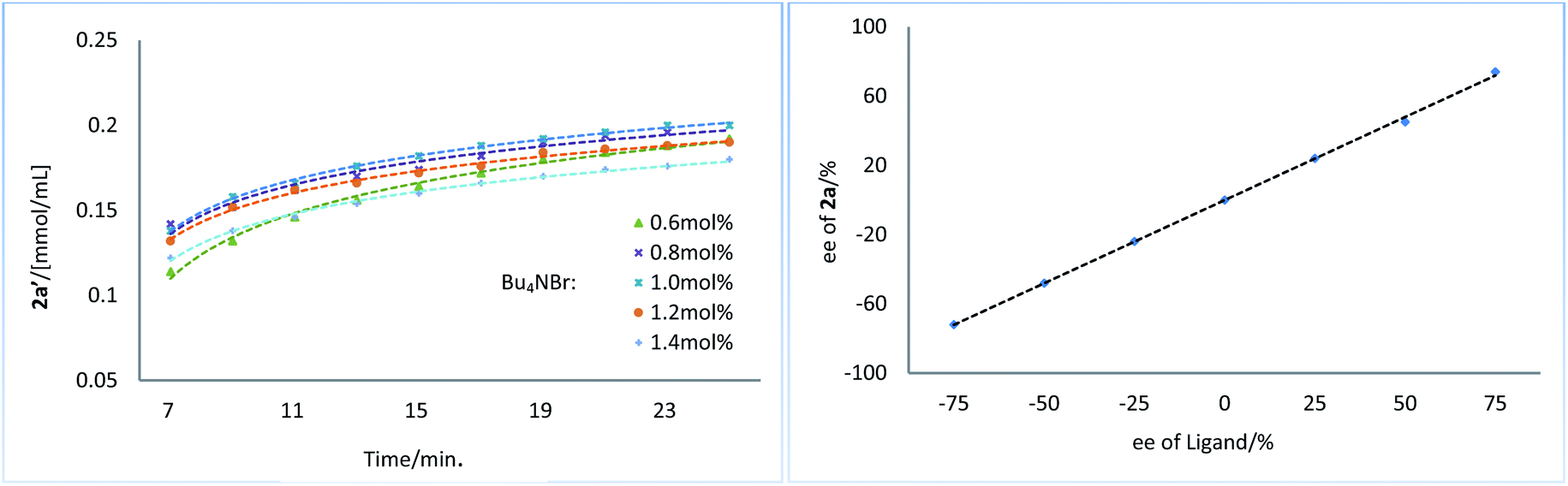 | ||
| Fig. 2 Kinetic profiles of Bu4NBr on reaction (left plot). Nonlinear effect experiment (right plot). | ||
Based on experimental and theoretical studies (see ESI† for details), a catalytic cycle of this protocol is proposed in Fig. 3. It is notable that, first, the bromide ion of the copper source (CuBr) was consumed in the process of formation of SOPCu–OMe species with Bu3SnBr as the side product; second, the added bromide ion (from Bu4NBr) quickly coordinates with copper–allenyl species I-4a and Bu3SnOMe to form a stable I-5a′ intermediate via a four-membered ring transition state, before the racemization process (I-4a to I-4b) happened; third, the chirality-preserving transmetallation intermediate I-6a′ released the bromide ion and desired product 2, and subsequently turned over the catalytic cycle.
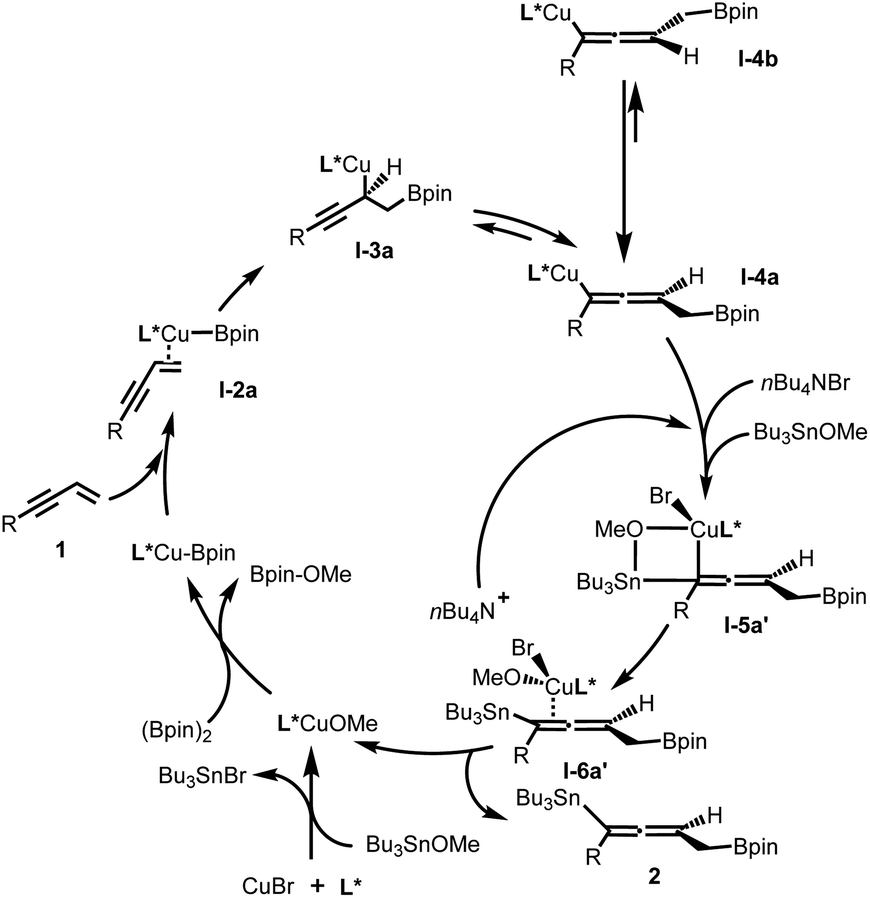 | ||
| Fig. 3 Proposed catalytic cycle. | ||
Conclusions
In summary, we reported an enantioselective Cu-catalyzed 1,4-borylstannation of unactivated 1,3-enynes, as the desired products, chiral allenylstannes, for the first time, were achieved via asymmetric catalysis. The achievements (introducing two functional groups, excellent yields and ees, broad substrate scope and high reactivity) of this protocol could be highlighted by chiral sulfoxide phosphine ligands (SOPs) and salt additives (halogenated salts). Mechanistic investigations, including control experiments and DFT calculations, disclosed that the halide anion played an important role in this protocol for stereochemical outcome by stabilizing the key copper–allenyl intermediate via a four-membered ring transition state to avoid the facile racemization of enantioenriched copper–allenyl species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Nature Science Foundation of China (No. 21871251), the Biological Resources Programme, Chinese Academy of Sciences (KFJ-BRP-008) and Sichuan Sci & Tech Department (2016JZ0022 and 2019YJ0471) and the Chunhui Program of the Ministry of Education of China (No. 191655) for financial support.References
- For selected reviews, see: (a) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 ( Angew. Chem. , 2004 , 116 , 1216 ) CrossRef; (b) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef; (c) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC; (d) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS; (e) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 ( Angew. Chem. , 2012 , 124 , 3128 ) CrossRef CAS; (f) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210 RSC; (g) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS; (h) R. K. Neff and D. E. Frantz, ACS Catal., 2014, 4, 519 CrossRef CAS; (i) W.-D. Chu, Y. Zhang and J. Wang, Catal. Sci. Technol., 2017, 7, 4570 RSC.
- Selected examples via resolution of racemic allenes, see: (a) J. Yu, W. Chen and L. Gong, Org. Lett., 2010, 12, 4050 CrossRef CAS; (b) C. M. Sapu, J.-E. Bäckvall and J. Deska, Angew. Chem., Int. Ed., 2011, 50, 9731 ( Angew. Chem. , 2011 , 123 , 9905 ) CrossRef CAS; (c) T. Inokuma, M. Furukawa, Y. Suzuki, T. Kimachi, Y. Kobayashi and Y. Takemoto, ChemCatChem, 2012, 4, 983 CrossRef CAS; (d) W. F. Zheng, W. Zhang, C. Huang, P. Wu, H. Qian, L. Wang, Y. L. Guo and S. Ma, Nat. Catal., 2019, 2, 997 CrossRef CAS.
- Selected examples via chirality transfer, see: (a) H. Ohmiya, U. Yokobori, Y. Makaida and M. Sawamura, Org. Lett., 2011, 13, 6312 CrossRef CAS; (b) M. R. Uehling, S. T. Marionni and G. Lalic, Org. Lett., 2012, 14, 362 CrossRef CAS; (c) M. Yang, N. Yokokawa, H. Ohmiya and M. Sawamura, Org. Lett., 2012, 14, 816 CrossRef CAS; (d) X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956 RSC; (e) S. Wu, X. Huang, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2015, 6, 7946 CrossRef; (f) J. Ruchti and E.-M. Carreira, Org. Lett., 2016, 18, 2174 CrossRef CAS.
- L. Fu, S. Greßies, P. Chen and G. Liu, Chin. J. Chem., 2020, 38, 91 CrossRef CAS . And references cited therein.
- Pioneering works accomplished by Hayashi, see: (a) Y. Matsumoto, M. Naito, Y. Uozumi and T. Hayashi, J. Chem. Soc., Chem. Commun., 1993, 1468 RSC; (b) J.-M. Han, N. Tokunaga and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 12915 CrossRef CAS.
- Asymmetric Cu-catalyzed 1,4-hydrofunctionalization of 1,3-enynes, see: (a) Y. Huang, J. Pozo, S. Torker and A.-H. Hoveyda, J. Am. Chem. Soc., 2018, 140, 2643 CrossRef CAS; (b) H.-J. Sang, S. Yu and S. Ge, Org. Chem. Front., 2018, 5, 1284 RSC; (c) D.-W. Gao, Y. Xiao, M. Liu, M.-K. Karunananda, J.-S. Chen and K.-M. Engle, ACS Catal., 2018, 8, 3650 CrossRef CAS; (d) S. Yu, H.-L. Sang, S.-Q. Zhang, X. Hong and S. Ge, Commun. Chem., 2018, 1, 64 CrossRef; (e) L.-B. Romero and S.-L. Buchwald, J. Am. Chem. Soc., 2019, 141, 13788 CrossRef.
- (a) F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140 ( Angew. Chem. , 2018 , 130 , 7258 ) CrossRef CAS; When this work was under review, two enantioselective radical 1,4-difunctionalizations of 1,3-enynes were reported: (b) Y. H. Zeng, M. F. Chiou, X. T. Zhu, J. Cao, D. Q. Lv, W. J. Jian, Y. J. Li, X. H. Zhang and H. L. Bao, J. Am. Chem. Soc., 2020, 142, 18014 CrossRef CAS; (c) X. Y. Dong, T. Y. Zhan, S. P. Jiang, X. D. Liu, L. Ye, Z. L. Li, Q. S. Gu and X. Y. Liu, Angew. Chem., Int. Ed. DOI:10.1002/anie.202013022.
- Y. Liao, X. Yin, X. Wang, W. Yu, D. Fang, L. Hu, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2020, 59, 1176 ( Angew. Chem. , 2020 , 132 , 1192 ) CrossRef CAS.
- Examples of aldehydes/ketones as electrophiles and enantioenriched propargylic products were afforded, see: (a) F. Meng, F. Haeffner and A.-H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304 CrossRef CAS; (b) X.-C. Gan and L. Yin, Org. Lett., 2019, 21, 931 CrossRef CAS; (c) X.-C. Gan, Q. Zhang, X.-S. Jia and L. Yin, Org. Lett., 2018, 20, 1070 CrossRef CAS.
- Recent example of enantioselective 1,4-hydroborylation/hydrosilylation, see: C. Yang, Z. Liu, D. Dai, Q. Li, W. Ma, M. Zhao and Y. Xu, Org. Lett., 2020, 22, 1360 CrossRef CAS.
- For reviews, see: (a) E. M. Vogl, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 1570 ( Angew. Chem. , 1999 , 111 , 1672 ) CrossRef CAS; (b) L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006 CrossRef CAS.
- Selected examples for salt additives were employed in metal-catalyzed cross-couplings, see: (a) L. Huang, L. K. G. Ackerman, K. Kang, A. M. Parsons and D. J. Weix, J. Am. Chem. Soc., 2019, 141, 10978 CrossRef CASQ. Cao, J. L. Howard, E. Wheatley and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 11339 ( Angew. Chem. , 2018 , 130 , 11509 ) CrossRef CAS; (b) L. C. McCann and M. G. Organ, Angew. Chem., Int. Ed., 2014, 53, 4386 ( Angew. Chem. , 2014 , 126 , 4475 ) CrossRef CAS; (c) B. J. Li, L. Xu, Z. H. Wu, B. T. Guan, C. L. Sun, B. Q. Wang and Z. J. Shi, J. Am. Chem. Soc., 2009, 131, 14656 CrossRef CAS; (d) Y. Chen, M. Sun and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 2236 ( Angew. Chem. , 2009 , 121 , 2270 ) CrossRef CAS; (e) W. Liu, D. Chen, X.-Z. Zhu, X.-L. Wan and X.-L. Hou, J. Am. Chem. Soc., 2009, 131, 8734 CrossRef CAS; (f) L. E. Overman and D. J. Poon, Angew. Chem., Int. Ed., 1997, 36, 518 ( Angew. Chem. , 1997 , 109 , 536 ) CrossRef CAS.
- (a) H. Yoshida, M. Kimura, I. Osaka and K. Takaki, Organometallics, 2017, 36, 1345 CrossRef CAS; (b) T. Jia, P. Cao, D. Wang, Y. Z. Lou and J. Liao, Chem.–Eur. J., 2015, 21, 4918 CrossRef CAS.
- Selected examples of SOP ligands in asymmetric catalysis, see: (a) F. Lang, G. Chen, L. Li, J. Xing, F. Han, L. Cun and J. Liao, Chem.–Eur. J., 2011, 17, 5242 CrossRef CAS; (b) D. Wang, P. Cao, B. Wang, T. Jia, Y. Lou, M. Wang and J. Liao, Org. Lett., 2015, 17, 2420 CrossRef CAS; (c) T. Jia, P. Cao, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760 CrossRef CAS; (d) Y. Lou, P. Cao, T. Jia, Y. Zhang, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2015, 54, 12134 ( Angew. Chem. , 2015 , 127 , 12302 ) CrossRef CAS; (e) B. Chen, P. Cao, X. Yin, Y. Liao, L. Jiang, J. Ye, M. Wang and J. Liao, ACS Catal., 2017, 7, 2425 CrossRef CAS; (f) X. Wang, B. Wang, X. Yin, W. Yu, Y. Liao, J. Ye, M. Wang, L. Hu and J. Liao, Angew. Chem., Int. Ed., 2019, 58, 12264 ( Angew. Chem. , 2019 , 131 , 12392 ) CrossRef CAS; (g) X. Yin, B. Chen, F. Qiu, X. Wang, Y. Liao, M. Wang, X. Lei and J. Liao, ACS Catal., 2020, 10, 1954 CrossRef CAS.
- (a) J. A. Marshall, Chem. Rev., 1996, 96, 31 CrossRef CAS; (b) S. R. Chemler, W. R. Roush, Recent Applications of the Allylation Reaction to the Synthesis of Natural Products. p403–490, ed. J. Otera, Modern Carbonyl Chemistry, 2000, Selected examples, see Search PubMed; (c) J. A. Marshall and X. Wang, J. Org. Chem., 1992, 57, 1242 CrossRef CAS; (d) P. B. D. Ranslow, L. S. Hegdus and C. Rios, J. Org. Chem., 2004, 69, 105 CrossRef CAS; (e) R. Villa, A. L. Mandel, B. D. Jones, J. Clair and M. D. Burkart, Org. Lett., 2012, 14, 5396 CrossRef CAS; (f) J. K. Belardi and G. C. Micalizio, Angew. Chem., Int. Ed., 2008, 47, 4005 CrossRef CAS.
- (a) C. Y. He, Y. X. Tan, X. Wang, R. Ding, Y. F. Wang, F. Wang, D. D. Gao, P. Tian and G. Q. Lin, Nat. Commu., 2020, 11, 4293 CrossRef CAS; (b) X. W. Chen, L. Zhu, Y. Y. Gui, K. Jing, Y. X. Jiang, Z. Y. Bo, Y. Lan, J. Li and D. G. Yu, J. Am. Chem. Soc., 2019, 141, 18825 CrossRef CAS; (c) C. X. Li, R. Y. Liu, L. T. Jesikiewicz, Y. Yang, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 5062 CrossRef CAS.
- J. A. Marshall and J. Perkins, J. Org. Chem., 1994, 59, 3509 CrossRef CAS.
- For selected examples of dimer or trimer of Rh-complexes see: (a) Q. P. Hu, Z. F. Zhang, Y. G. Liu, T. Imamoto and W. B. Zhang, Angew. Chem., Int. Ed., 2015, 54, 2260 CrossRef CAS; (b) P. Krüger and H. Werner, Eur. J. Inorg. Chem., 2004, 4811 Search PubMed; (c) S. A. Colebrooke, S. B. Duckett, J. A. B. Lohman and R. Eisenberg, Chem.–Eur. J., 2004, 10, 2459 CrossRef CAS . And for a structure of [SOP-CuCl]2, see ref. 14b.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05425a |
| This journal is © The Royal Society of Chemistry 2021 |