
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven palladium-catalyzed Dowd–Beckwith ring expansion/C–C bond formation cascade†
Li
Chen
a,
Li-Na
Guo
 a,
Shuai
Liu
a,
Le
Liu
a and
Xin-Hua
Duan
*ab
a,
Shuai
Liu
a,
Le
Liu
a and
Xin-Hua
Duan
*ab
aSchool of Chemistry, Xi'an Key Laboratory of Sustainable Energy Material Chemistry, MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter Xi'an Jiaotong University, Xi'an, Shaanxi 710049, China
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou730000, P. R. China. E-mail: duanxh@xjtu.edu.cn
First published on 9th December 2020
Abstract
A visible-light-induced palladium-catalyzed Dowd–Beckwith ring expansion/C–C bond formation cascade is described. A range of six to nine-membered β-alkenylated cyclic ketones possessing a quaternary carbon center were accessed under mild conditions. Besides styrenes, the electron-rich alkenes such as silyl enol ethers and enamides were also compatible, providing the desired β-alkylated cyclic ketones in moderate to good yields.
Introduction
The medium-sized carbocycles constitute the core frameworks of many natural products, valuable pharmaceutical compounds and so on.1 Given their widely recognized importance, continuous efforts from chemists have been devoted to construct the related skeletons.1,2 In addition to the cyclization reactions, the ring-expansion reactions represent complementary and useful strategy to access these frameworks.3 Different methods including fragmentation of the zero bridge in fused bicyclic units, inter- and intramolecular carbon-insertion reactions and others have been developed to achieve the ring-expansion.4 In this field, the ring expansion of cycloalkanones provided an efficient strategy for the medium-sized cyclic ketones' construction.3c–e The ring expansion of unstrained cyclic ketones, particularly five- and six-membered rings is more challenging and attractive due to the relatively much weaker ring strain. Recently, Dong's group presented an elegant Rh-catalyzed strategy based on the installation of temporary directing group to activate the C–C bond, followed by carbon insertion to enlarge the cyclic ketones.3b,3f,5 Unfortunately, this strategy is limited to cyclopentanones. The development of ring-expansion reactions to form functionalized medium-sized carbonyl compounds still remains highly demanding.The Dowd–Beckwith reaction and variants represent an important class of radical ring-expansion reaction, which offers useful alternative to obtain the medium-sized cyclic ketones. Since the seminal pioneering works of Dowd and Beckwith,3,6 a variety of radical initiating systems, especially photocatalytic systems have been established for this transformation (Scheme 1, eqn a).7 Although many progress have been made in the reaction system, the ring expansion/H-atom abstraction is still the mainstream transformation, which limited its application in the radical cascade.8 Recently, the light-induced transition-metal catalysis has emerged as a versatile platform to realize transformations that are otherwise difficult to achieve. In this field, the groups of Gevorgyan, Fu, Yu and others recently demonstrated a series of fantastic chemical transformations through visible light-induced Pd-catalyzed alkyl couplings, wherein the hybrid alkyl Pd(I)-radical species enabled the alkyl couplings achievable.9 As a part of our interest in radical chemistry and transition-metal catalysis,10 we hope to explore an intermolecular Dowd–Beckwith ring-expansion/Heck-type coupling cascade (Scheme 1, eqn b). Challenges for developing such cascade include the fast competitive H-atom abstraction, the possible β-H elimination and other side reactions due to several different reactive radical species involved. Herein, we report the first visible light induced, Pd-catalyzed intermolecular Dowd–Beckwith ring-expansion/C–C bond formation cascade. A range of medium-sized β-alkenylated cyclic ketones bearing a quaternary carbon centre were obtained under mild conditions with good yields and excellent stereoselectivity.
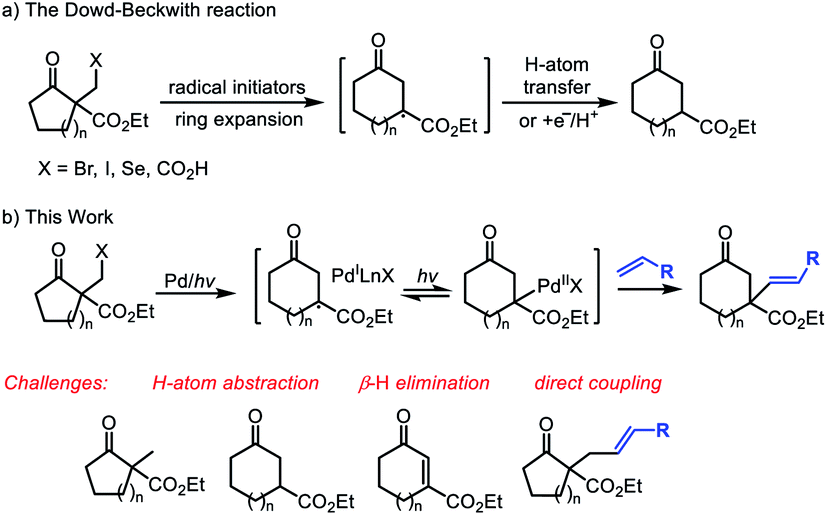 | ||
| Scheme 1 The classical Dowd–Beckwith reactions and our design on the Dowd–Beckwith ring-expansion/Heck-type coupling cascade. | ||
Results and discussion
Inspired by the pioneering works on visible-light induced Pd-catalyzed alkyl-Heck coupling reactions,11 we envisioned that photoirradiation might facilitate the SET event between Pd(0) complex and Dowd–Beckwith halides to initiate the ring expansion process. The interaction of the tertiary radical species and Pd(I) complex generated in situ would provide opportunities for further transformation (Scheme 1, eqn b). To check this hypothesis, we set to examine the reaction of α-bromomethyl β-keto ester 1a and styrene 2a in the presence of Pd(0) complex under visible light irradiation. To our delight, treatment of 1a and 2a with 10 mol% of Pd(OAc)2, 20 mol% of 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) and K2CO3 (2.0 equiv.) in toluene under blue LEDs irradiation afforded the desired ring-expansion/alkenylation product 3a in 66% yield (Table 1, entry 1). Other palladium catalysts such as Pd(TFA)2, PdCl2 and Pd(PPh3)4 also displayed some catalytic activity, but gave lower yields than Pd(OAc)2 (entries 2 and 3). When Ni(OAc)2 was used instead of Pd(OAc)2 as the catalyst, no reaction occurred (entry 4). The ligand proved to be crucial for this reaction. Except for Xantphos, other phosphine ligands neither bidentate (DPEPhos, BINAP) nor monodentate (P(o-MeOC6H4)3) were effective (entries 5 and 6). Both inorganic and organic bases were examined, and K2CO3 turned out to be the optimal one (entries 7 and 8). Solvent screening reveals that PhCF3, xylene and DCE were inferior to toluene, while polar solvents such as CH3CN and DMF were completely unsuitable (entries 9 and 10). Lowering the loading of Pd/Xantphos to 5/10 mol% resulted in a diminished yield of 3a (entry 1). Control experiments indicated that catalyst, ligand and base were all essential for this reaction (entry 11). Furthermore, visible light irradiation was also required (entry 12). Without irradiation, the reaction did not work even heating up to 80 °C (entry 13). It is noteworthy that some amount of the direct alkyl-Heck coupling product 3a′ (without ring-expansion) and ring-expansion/β-H elimination product 1a′ were also observed during the reaction. Under the optimal conditions, the byproduct 3a′ was isolated in 10% yield and the 1a′ was obtained in 5% yield. Finally, to demonstrate the utility of this procedure, the reaction of 1a and 2a was conducted on a 1.0 mmol scale, and 62% yield of 3a was still obtained.|
|
||
|---|---|---|
| Entry | Variants from standard conditions | Yield (%) |
| a Reaction conditions A: 1a (0.2 mmol, 1.0 equiv.), 2a (0.4 mmol, 2.0 equiv.), Pd(OAc)2 (10 mol%), Xantphos (20 mol%) and K2CO3 (0.4 mmol, 2.0 equiv.) in toluene (2.0 mL) were irradiated with 30 W blue LEDs at room temperature for 24 h under N2. Yields of isolated product were given. b 5 mol% of Pd(OAc)2 and 10 mol% of Xantphos were used. c n.r. = no reaction. | ||
| 1 | None | 66 (55)b |
| 2 | Pd(TFA)2, or PdCl2 as the catalyst | 14, 16 |
| 3 | Pd(PPh3)4 as the catalyst | 33 |
| 4 | Ni(OAc)2 as the catalyst | n.r.c |
| 5 | DPEPhos or BINAP as the ligand | Trace, n.r.c |
| 6 | P(o-MeOC6H4)3 | n.r.c |
| 7 | Cs2CO3, Li2CO3 or KOAc as the base | 50, 16, 48 |
| 8 | Et3N or DIPEA as the base | 12, 28 |
| 9 | PhCF3, xylene or DCE as the solvent | 39, 43, 20 |
| 10 | CH3CN or DMF as the solvent | n.r.c |
| 11 | Without catalyst or ligand or base | n.r.c or trace |
| 12 | In the dark | n.r.c |
| 13 | Without irradiation, heating to 80 °C | n.r.c |
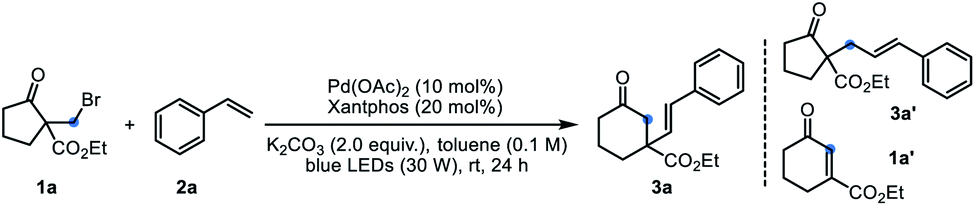
With the optimized reaction conditions in hand, the generality of this reaction with respect to alkenes was evaluated (Table 2). A variety of para-, meta-, and ortho-substituted styrenes reacted with 1a smoothly to afford the desired six-membered products 3b–3p in 15–76% yields. In general, styrenes with electron-donating groups gave better yields than those bearing strong electron-withdrawing groups (3b–3gvs.3k and 3l), probably due to the polarity-matching of radicals. In these cases, most of the substrate 1a was recovered. Remarkably, excellent stereoselectivity was observed for all these reactions, and only E-isomers were obtained as sole products. 2-Vinylpyridine was also efficient for this reaction, providing the desired product 3r in 27% yield. 1,1-Disubstituted alkenes participated in this reaction to deliver the corresponding products 3s–3z in moderate yields. Satisfactorily, the exocyclic alkenes derived from 1-tetralone and 1-indanones furnished the products 3x–3z in 60–80% yields. It is worth noting that the bromo groups in styrenes were retained in this palladium catalytic system (3j, 3o, 3z), thereby providing opportunity for further derivatization. Besides terminal alkenes, the 1H-indene containing an internal alkene also gave the product 4a, albeit with somewhat low yield due to poor conversion. Notably, the estrone-derived olefins underwent this reaction efficiently to give the products 4b and 4c in satisfied yields, which highlighted the potential of this method in late-stage functionalization of complex molecules. Unfortunately, aliphatic alkenes, allylbenzene and acrylate esters were inert under the standard conditions.
| a Reaction conditions A: 1a (0.2 mmol, 1.0 equiv.), 2 (0.4 mmol, 2.0 equiv.), Pd(OAc)2 (10 mol%), Xantphos (20 mol%) and K2CO3 (0.4 mmol, 2.0 equiv.) in toluene (2.0 mL) were irradiated with 30 W blue LEDs at room temperature for 24 h under N2. Yields of isolated product were given. E/Z and t/l ratios of products were determined by 1H NMR analysis: t = terminal and l = linear. |
|---|

|
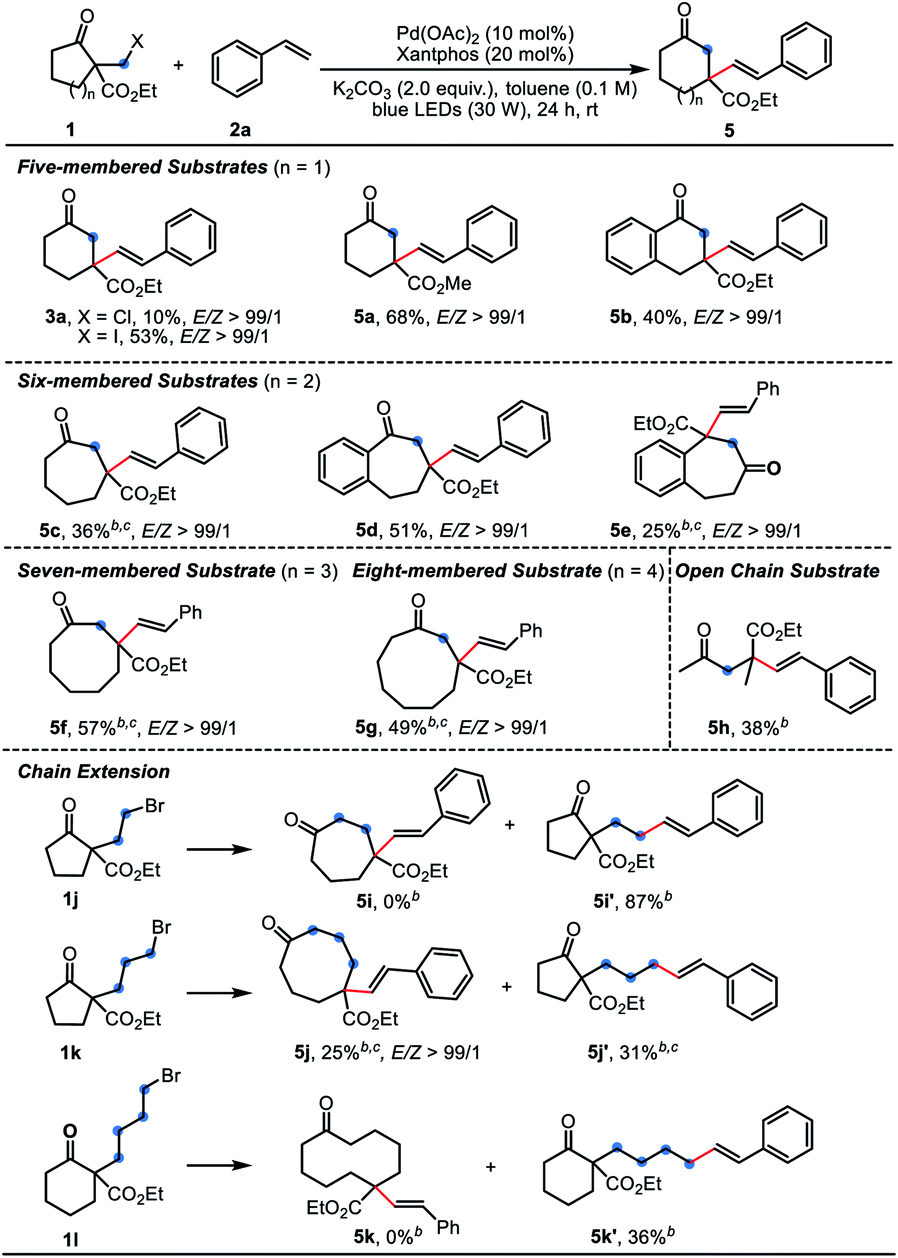
Subsequently, the scope and limitations of β-keto esters 1 were examined using 2a as the coupling partner (Table 3). Besides the α-bromomethyl substrate 1a, the homologous chloride and iodide gave the product 3a in 10% and 53% yield, respectively. A range of α-bromomethyl substrates containing five-to eight-membered ring could react smoothly to afford the one-carbon extended products 5a–5g. For six-, seven-, eight-membered or other substrates, PdCl2(PPh3)2 was found as optimal catalyst (conditions B). The relatively strained five-membered substrates displayed much better reaction efficiency, giving the six-membered products 5a and 5b in good yields. It is a pity that some of the yields were unsatisfied due to the low conversion even after prolonged reaction time. Therein, the Dowd–Beckwith bromides could be recovered. When the substrate 1k bearing a three-carbon side chain was subjected to the reaction, the anticipated product 5j was formed in a low yield, along with the by-product 5j’ in 31% isolated yield. Substrates with two- or four-carbon side chain (1j and 1l) failed to give any expected products, delivering 87% and 36% of the direct coupling by-products, respectively.3a The 2-phenyl cycloheptanone only afforded trace amount of the desired product (not shown).
| a Reaction conditions A: 1 (0.2 mmol, 1.0 equiv.), 2a (0.4 mmol, 2.0 equiv.), Pd(OAc)2 (10 mol%), Xantphos (20 mol%) and K2CO3 (0.4 mmol, 2.0 equiv.) in toluene (2.0 mL) were irradiated with 30 W blue LEDs at room temperature for 24 h under N2. Yields of isolated product were given. b Reaction conditions B: 1 (0.2 mmol, 1.0 equiv.), 2a (0.4 mmol, 2.0 equiv.), PdCl2(PPh3)2 (5 mol%), PPh3 (12 mol%), Xantphos (6 mol%) and KOAc (0.3 mmol, 1.5 equiv.) in 1,4-dioxane (2.0 mL) were irradiated with 30 W blue LEDs at room temperature for 24 h under N2. Yields of isolated product were given. c The reactions were irradiated for 48 h. |
|---|
|
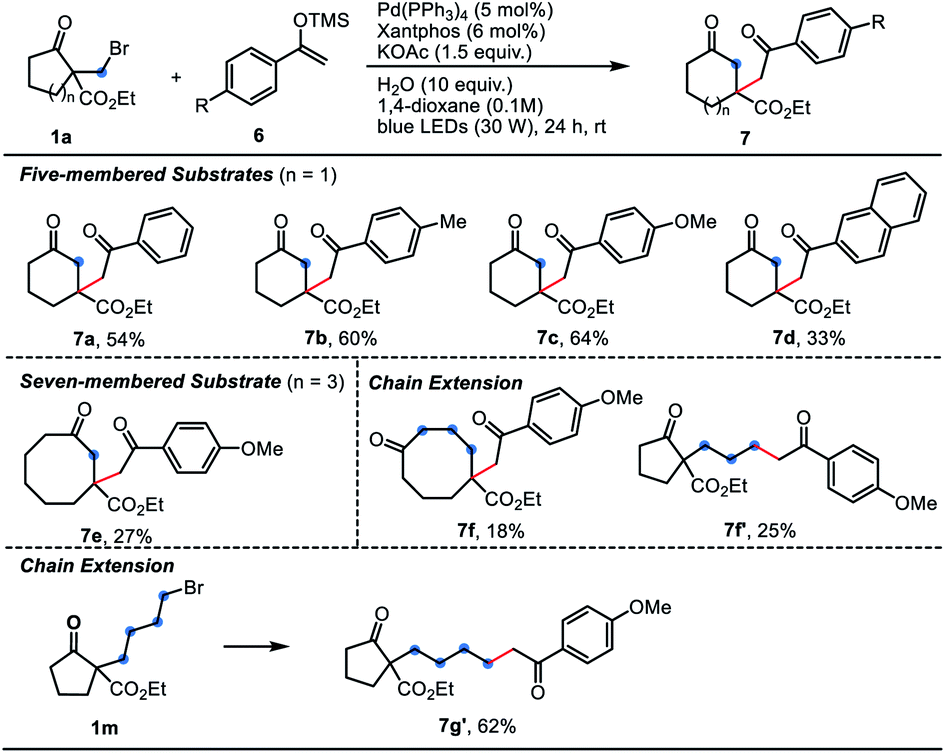
As we know, the Dowd–Beckwith ring-expansion led to a tertiary alkyl radical species bearing an ester group, which is quite electrophilic in nature. Thus, we thought the electron-rich alkenes would be compatible for this tandem transformation. Satisfactorily, the Dowd–Beckwith bromide 1a reacted smoothly with a series of silyl enol ethers to provide the expected β-alkylated cyclic ketones 7a–7d in moderate to good yields. Substrate with a three-carbon side chain afforded the desired product 7f in 18% yield, along with by-product 7f′ in 25% yield. Substrate 1m with four-carbon side chain didn't undergo the ring-expansion process, only delivering the direct alkylated product 7g′ in 62% yield (Table 4). In addition, the enamides also worked with 1a, yielding the β-alkylated cyclic ketones 9 in moderate yields (Table 5).
| a Reaction conditions C: 1 (0.2 mmol, 1.0 equiv.), 6 (0.4 mmol, 2.0 equiv.), Pd(PPh3)4 (5 mol%), Xantphos (6 mol%) and KOAc (0.3 mmol, 1.5 equiv.) in 1,4-dioxane (2.0 mL) were irradiated with 30 W blue LEDs at room temperature for 24 h under N2. Yields of isolated product were given. |
|---|
|
| a Reaction conditions B: 1a (0.2 mmol, 1.0 equiv.), 8 (0.4 mmol, 2.0 equiv.), PdCl2(PPh3)2 (5 mol%), PPh3 (12 mol%), Xantphos (6 mol%) and KOAc (0.3 mmol, 1.5 equiv.) in 1,4-dioxane (2.0 mL) were irradiated with 30 W blue LEDs at room temperature for 24 h under N2. Yields of isolated product were given. |
|---|

|
Notably, the byproduct 1a′ formed through ring-expansion/β-H elimination was always observed in above reactions, which is a valuable synthetic intermediate but is hard to obtain in organic synthesis.12 Thus we hope to amplify this useful transformation (Fig. 1). After several trails, we were delighted to find that the substrate 1a delivered the desired product 1a′ in 80% yield by using TMEDA as the base and PhH as the solvent. While, the analogue 1d only afforded the product 1d′ in 26% isolated yield due to incomplete conversion.
 | ||
| Fig. 1 Ring expansion/β-H elimination reaction. | ||
To shed light on the mechanism of this reaction, a series of control experiments were performed (Fig. 2). The addition of TEMPO and BTH, well-known radical scavengers, both inhibited the model reaction significantly (eqn (1) and (2)). When 1.0 equiv. of TEMPO was added, the yield of 3a was reduced to 20%. Meanwhile, the TEMPO-adduct 10 was isolated in 13% yield. Further increase of the amount of TEMPO led to a higher yield of 10. These results implied that a radical intermediate might be involved in this reaction. In addition, the reaction of 1a with 1-(1-cyclopropylvinyl)benzene 2d′, a radical clock substrate afforded 3x as the sole product in 55% yield (eqn (3)), which provided clear evidence for radical pathway. Moreover, light on-off studies revealed that constant photoirradiation is essential for this transformation (for details, see the ESI†). The UV-vis analysis indicated that the Pd0 complex formed in the catalytic system is the photoabsorbing species (for details, see the ESI†).
 | ||
| Fig. 2 Control experiments. | ||
Based on the above results and literature,11 a plausible mechanism for this ring-expansion/alkenylation reaction is proposed (Scheme 2). Firstly, the LnPd0 complex is photoexcited to form an excited state LnPd0* species upon irradiation, which promotes a single-electron transfer event with 1a to generate the putative PdI species and a primary alkyl radical I. Subsequently, intermediate I undergoes intramolecular cyclization of the carbon radical on the carbonyl group, followed by alkoxyl radical triggered C–C bond cleavage to yield the ring extended radical intermediate III. Finally, radical addition of III to styrene 2a provides the benzyl radical IV, which produces the target product 3a and regenerates the Pd0 catalyst through β-H elimination. Due to the equilibrium between hybrid alkyl Pd(I)-radical species and alkyl Pd(II) species,13 the intermediate III could also deliver the product 1a′ and regenerate the Pd0 catalyst through β-H elimination. In addition, the direct coupling product 3a′ could also be formed during the reaction.
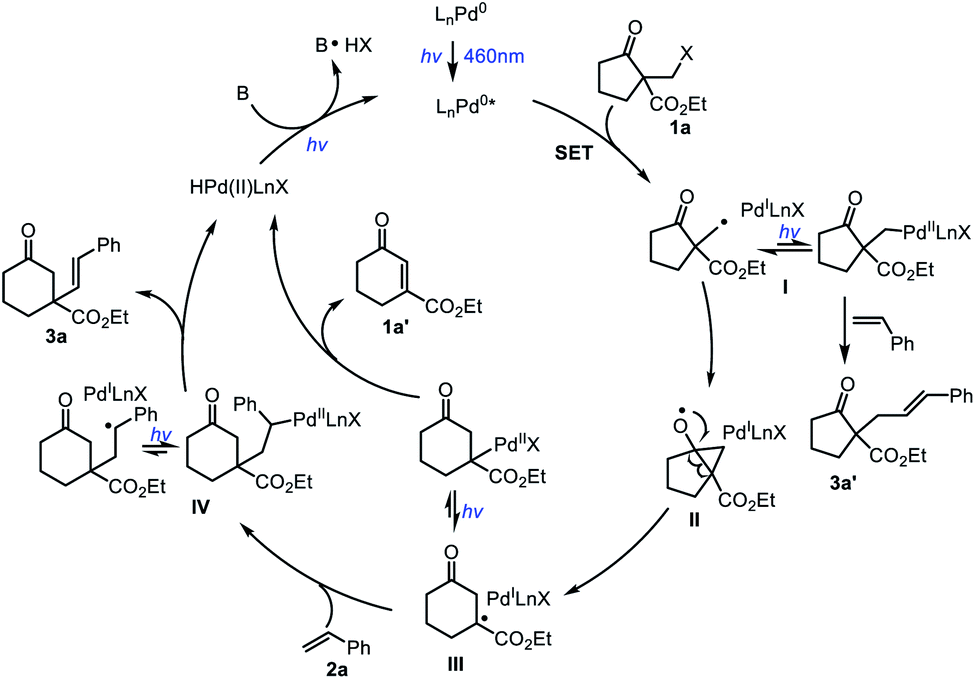 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a Dowd–Beckwith ring expansion/C–C bond formation cascade by light-induced palladium catalysis. A range of five to eight-membered Dowd–Beckwith halides reacted smoothly with styrenes to afford the enlarged β-alkenylated cyclic ketones in moderate to good yields with excellent stereoselectivity. Besides styrenes, the electron-rich alkenes such as silyl enol ethers and enamides were also compatible, providing the desired β-alkylated cyclic ketones. This research would arouse the application of the Dowd–Beckwith reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21971201), Natural Science Basic Research Plan in Shaanxi Province of China (No. 2019JM-299) and Key Laboratory Construction Program of Xi'an Municipal Bureau of Science and Technology (No. 201805056ZD7CG40). We also thank Mr Chang, Miss Bai and Miss Zhang at the Instrument Analysis Center of Xi'an Jiaotong University for their assistance with NMR and HRMS analysis.Notes and references
- For selected examples, see: (a) D. G. I. Kingston, J. Org. Chem., 2008, 73, 3975–3984 CrossRef CAS PubMed; (b) K. Takao, H. Kai, A. Yamada, Y. Fukushima, D. Komatsu, A. Ogura and K. Yoshida, Angew. Chem., Int. Ed., 2019, 58, 9851–9855 CrossRef CAS PubMed.
- For selected reviews, see: (a) L. Yet, Chem. Rev., 2000, 100, 2963–3007 CrossRef CAS PubMed; (b) A. Hussain, S. K. Yousuf and D. Mukherjee, RSC Adv., 2014, 4, 43241–43257 RSC; (c) Y. Wang and Z.-X. Yu, Acc. Chem. Res., 2015, 48, 2288–2296 CrossRef CAS PubMed; (d) M. Yu, S. Lou and F. Gonzalez-Bobes, Org. Process Res. Dev., 2018, 22, 918–946 CrossRef CAS; (e) Y.-J. Hu, L.-X. Li, J.-C. Han, L. Min and C.-C. Li, Chem. Rev., 2020, 120, 5910–5953 CrossRef CAS PubMed.
- For representative reviews, see: (a) P. Dowd and W. Zhang, Chem. Rev., 1993, 93, 2091–2115 CrossRef CAS; (b) P. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed; (c) J. R. Donald and W. P. Unsworth, Chem.–Eur. J., 2017, 23, 8780–8799 CrossRef CAS PubMed; (d) T. Huber, R. E. Wildermuth and T. Magauer, Chem.–Eur. J., 2018, 24, 12107–12120 CrossRef CAS PubMed; (e) A. K. Clarke and W. P. Unsworth, Chem. Sci., 2020, 11, 2876–2881 RSC; (f) L. Deng and G. Dong, Trends in Chemistry, 2020, 2, 183–198 CrossRef CAS.
- For some examples, see: (a) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490–3493 CrossRef CAS PubMed; (b) R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692–12696 CrossRef CAS PubMed; (c) R. Samineni, P. Srihari and G. Mehta, Org. Lett., 2016, 18, 2832–2835 CrossRef CAS PubMed; (d) J. Strehl, C. Kahrs, T. Müller, G. Hilt and J. Christoffers, Chem.–Eur. J., 2020, 26, 3222–3225 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Xia, S. Ochi and G. Dong, J. Am. Chem. Soc., 2019, 141, 13038–13042 CrossRef CAS PubMed; (b) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260–16265 CrossRef CAS PubMed.
- (a) P. Dowd and S.-C. Choi, J. Am. Chem. Soc., 1987, 109, 3493–3494 CrossRef CAS; (b) P. Dowd and S.-C. Choi, J. Am. Chem. Soc., 1987, 109, 6548–6549 CrossRef CAS; (c) A. L. J. Beckwith, D. M. O'Shea, S. Gerba and S. W. Westwood, J. Chem. Soc., Chem. Commun., 1987, 666–667 RSC; (d) A. L. J. Beckwith, D. M. O'Shea and S. W. Westwood, J. Am. Chem. Soc., 1988, 110, 2565–2575 CrossRef CAS.
- (a) E. Hasegawa, Y. Tamura and E. Tosaka, Chem. Commun., 1997, 1895–1896 RSC; (b) K. Nishikawa, T. Ando, K. Maeda, T. Morita and Y. Yoshimi, Org. Lett., 2013, 15, 636–638 CrossRef CAS PubMed; (c) E. Hasegawa, M. Tateyama, T. Hoshi, T. Ohta, E. Tayama, H. Iwamoto, S. Takizawa and S. Murata, Tetrahedron, 2014, 70, 2776–2783 CrossRef CAS; (d) E. Hasegawa and S. Takizawa, Aust. J. Chem., 2015, 68, 1640–1647 CrossRef CAS; (e) M. Deguchi, A. Fujiya, E. Yamaguchi, N. Tada, B. Uno and A. Itoh, RSC Adv., 2018, 8, 15825–15830 RSC.
- (a) D. P. Curran and B. Yoo, Tetrahedron Lett., 1992, 33, 6931–6934 CrossRef CAS; (b) J. Hierold and D. W. Lupton, Org. Lett., 2012, 14, 3412–3415 CrossRef CAS PubMed.
- For some selected reviews, see: (a) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC; (b) W.-J. Zhou, Y. Zhang, G. Cao, H. Liu and D.-G. Yu, Chin. J. Org. Chem., 2017, 37, 1322–1337 CrossRef CAS; (c) W.-J. Zhou, G.-M. Cao, Z.-P. Zhang and D.-G. Yu, Chem. Lett., 2019, 48, 181–191 CrossRef CAS. For representative examples, see: (d) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS PubMed; (e) D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212–14216 CrossRef CAS PubMed; (f) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307–18312 CrossRef CAS PubMed; (g) W.-J. Zhou, G.-M. Cao, G. Shen, X.-Y. Zhu, Y.-Y. Gui, J.-H. Ye, L. Sun, L.-L. Liao, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed; (h) P. Chuentragool, D. Yadagiri, T. Morita, S. Sarkar, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 1794–1798 CrossRef CAS PubMed.
- (a) H. Wang, L.-N. Guo and X.-H. Duan, J. Org. Chem., 2016, 81, 860–867 CrossRef CAS PubMed; (b) J.-J. Zhang, J.-C. Yang, L.-N. Guo and X.-H. Duan, Chem.–Eur. J., 2017, 23, 10259–10263 CrossRef CAS PubMed; (c) J.-F. Zhao, P. Gao, X.-H. Duan and L.-N. Guo, Adv. Synth. Catal., 2018, 360, 1775–1779 CrossRef CAS; (d) J.-F. Zhao, X.-H. Duan, Y.-R. Gu, P. Gao and L.-N. Guo, Org. Lett., 2018, 20, 4614–4617 CrossRef CAS PubMed; (e) J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161–166 RSC; (f) L. Chen, L.-N. Guo, Z.-Y. Ma, Y.-R. Gu, J. Zhang and X.-H. Duan, J. Org. Chem., 2019, 84, 6475–6482 CrossRef CAS PubMed; (g) P. Gao, H. Wu, J.-C. Yang and L.-N. Guo, Org. Lett., 2019, 21, 7104–7108 CrossRef CAS PubMed.
- For recent reports on catalysis with palladium complexes photoexcited by visible light, see: (a) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed and references cited therein; (b) M. Koy, P. Bellotti, F. Katzenburg, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 2375–2379 CrossRef CAS PubMed; (c) B. Zhao, R. Shang, G.-Z. Wang, S. Wang, H. Chen and Y. Fu, ACS Catal., 2020, 10, 1334–1343 CrossRef CAS; (d) M. Li, Y.-F. Qiu, C.-T. Wang, X.-S. Li, W.-X. Wei, Y.-Z. Wang, Q.-F. Bao, Y.-N. Ding, W.-Y. Shi and Y.-M. Liang, Org. Lett., 2020, 22, 6288–6293 CrossRef CAS PubMed.
- (a) W. C. Agosta and W. W. Lowrance Jr, J. Org. Chem., 1970, 35, 3851–3856 CrossRef CAS; (b) F. X. Webster and R. M. Silverstein, Synthesis, 1987, 922–924 CrossRef CAS; (c) N. G. Turrini, R. C. Cioc, D. J. H. Niet, E. Ruijter, R. V. A. Orru, M. Halla and K. Faber, Green Chem., 2017, 19, 511–518 RSC.
- (a) C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed; (b) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111–6137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of new compounds are provided. See DOI: 10.1039/d0sc04399k |
| This journal is © The Royal Society of Chemistry 2021 |