
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterfacial growth of free-standing PANI films: toward high-performance all-polymer supercapacitors†
Fuyao
Zhong
ab,
Mingyu
Ma
b,
Zhuoran
Zhong
b,
Xinrong
Lin
 *a and
Mao
Chen
*b
*a and
Mao
Chen
*b
aKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan Provincial Center for Research & Development of Natural Products, School of Chemical Science and Technology, Yunnan University, Kunming, 650091, P. R. China. E-mail: xrlin@ynu.edu.cn
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai, 200433, P. R. China. E-mail: chenmao@fudan.edu.cn
First published on 8th December 2020
Abstract
Along with high power capability and energy density, long cycle life is regarded an essential performance requirement for energy storage devices. The rapid capacitance decline of conducting polymer-based electrodes remains a major technical challenge and precludes their practical applications in supercapacitors. In this work, a polyaniline (PANI) network is synthesized via interfacial Buchwald–Hartwig polymerization for the first time, facilitating the construction of covalently connected PANI networks by ligand-promoted C–N bond formation. Particularly, the interfacial synthesis and subsequent gas release from pre-anchored protecting groups allow bottom-up and efficient access to porous cross-linked PANI (PCL-PANI) films that are free-standing and solvent-resistant. Upon assembling into supercapacitors, the PCL-PANI material enables an unprecedent long-term charge–discharge cycling performance (>18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times) without clear capacitance loss for an additive-free pseudocapacitive system. In addition, this synthesis affords electrodes entirely consisting of conducting polymers, yielding highly reversible gravimetric capacitance at 435 F gelectrode−1 in a two-electrode system, and a high gravimetric energy of 12.5 W h kgelectrode−1 while delivering an outstanding power density of 16000 W kgelectrode−1, which is 10-fold higher than those of conventional linear PANI composite supercapacitors. This synthetic approach represents a novel and versatile strategy to generate additive/binder-free and high-performance conducting thin-films for energy storage.
000 times) without clear capacitance loss for an additive-free pseudocapacitive system. In addition, this synthesis affords electrodes entirely consisting of conducting polymers, yielding highly reversible gravimetric capacitance at 435 F gelectrode−1 in a two-electrode system, and a high gravimetric energy of 12.5 W h kgelectrode−1 while delivering an outstanding power density of 16000 W kgelectrode−1, which is 10-fold higher than those of conventional linear PANI composite supercapacitors. This synthetic approach represents a novel and versatile strategy to generate additive/binder-free and high-performance conducting thin-films for energy storage.
Introduction
Intrinsically conducting polymers which exhibit electrical, optical and magnetic properties of metals while maintaining good processability and diversified structural functionality of polymers are known as “synthetic metals” and have been appealing materials applied in electronics, sensing and energy storage.1–3 To this end, massive efforts have been dedicated in search of novel synthetic methods and processing techniques to obtain high-performance conducting polymers,4–10 along with various strategies for better incorporating performances of such materials into electrochemical energy storage (EES) devices,11,12 such as supercapacitors13–20 and batteries21–24 in recent years. However, it remains a long-standing challenge for conducting polymers, such as PANI-based electrodes, in providing long life-span and high power/energy density, simultaneously.25 Typically, the doping and de-doping transformation during the charge–discharge cycling could result in large volume changes and strain variation, causing the conducting polymers to suffer from structural collapse and capacitance degradation during repeated cycles, precluding them from many practical applications.26,27Synthetic routes fundamentally determine chemical structures of conducting polymers, and further influence the physicochemical and morphological characteristics of polymer electrodes.28,29 To synthesize PANI, chemical and electrochemical oxidations have been most frequently employed to construct C–N bonds via polymerization of anilines.30,31 These approaches involve the generation of radicals and radical cations during the initiation and subsequent chain-growth processes, which could lead to undesired side reactions (i.e., C–N bond formation on the ortho-position of aniline, and C–C and N–N bond formation, Scheme S1A†), thus causing defects or diverged chemical structures from targeted PANI.32 The transition-metal-catalyzed Buchwald–Hartwig reaction has been heavily used to construct C–N bonds in pharmaceuticals, agrochemicals, etc.33 In comparison with (electro)chemical oxidation, such catalysis enables regiospecific C–N bond formation via the cleavage of the carbon–halide bond (Scheme S1†), and avoids the generation of the C–C or N–N bond. Well-defined PANI with linear, hyperbranched structures, and monodisperse oligoanilines have been synthesized via Pd-catalyzed processes.34–36 While considerable efforts have focused on the synthesis of non-cross-linked PANI backbones, an interfacial catalysis toward a covalently connected network, which could provide resulting materials with improved mechanical stability, has been rarely achieved. In addition, although researchers have shown the importance of ligand selection in Pd-catalyzed C–N bond formation with primary and secondary amines,37 the one-step construction of a PANI network using both types of N-nucleophiles (i.e., terminal aryl-NH2 and diphenyl amine in PANI) remains unexploited.
On the other hand, to fulfill application demands in different devices, it's highly valuable to fabricate materials with desirable shapes/dimensions.38 For example, free-standing, conducting polymeric films have been investigated as promising electrodes for energy storage.39–44 However, conventional processes to obtain such films require steps including synthesis and isolation of polymers, mixing with additives and binders, compressing or coating into films, etc. These multi-step operations could lead to problems such as structural collapse of porous films and detachment of additives from electrodes during long-term usage, jeopardizing energy delivery and life-span.25,27 The growth of polymeric films via in situ polymerization presents an attractive pathway to engineering materials at the molecular level, which has advanced gas separation, water purification, energy storage and so on.45–48 We envision that the in situ generation of cross-linked thin-films would integrate the synthesis of conducting polymers and formation of mechanically stable films into one single step, overcoming the limitations in obtaining electrodes of conducting polymers.
Herein, we report a bottom-up synthetic approach to prepare additive-free, highly porous conducting thin-films that consist of covalently cross-linked PANI backbones via a two-step sequence of interfacial Buchwald–Hartwig polymerization/deprotection. The obtained free-standing PANI films could be readily used as high-performance electrodes, which exhibit superior cycling stability and enhanced gravimetric energy/power in all-polymer supercapacitors.49 Specifically, we report: (1) the development of a novel interfacial transition-metal-catalyzed polymerization reaction based on ligand-determined chemical selectivity, enabling the generation of well-defined PANI networks; (2) the design of a BOC-containing (BOC = tert-butyloxycarbonyl protecting group) PANI backbone that allows a convenient formation of highly porous cross-linked PANI (PCL-PANI) films via gas release; (3) that the PCL-PANI films display outstanding long-term stability versus linear counterparts, circumventing structural collapse and enhancing the cycle life to >18000 charge–discharge cycles with high coulombic efficiency; (4) that the free-standing conducting films avoid the use of extra additives/binders and yield highly reversible specific capacitance at 435 F gelectrode−1 when normalized to the entire electrode weight, and deliver a power density of about 10 times higher (16000 W kgelectrode−1) relative to linear PANI-based electrodes. Overall, this work combines the construction of a polymeric backbone and fabrication of PCL-PANI films, and offers a novel approach to prepare conducting polymer-based electrodes, creating opportunities to improve energy materials by merging polymer synthesis and material engineering synergistically.
Results and discussion
During our investigation, BOC-protected N-(4-bromophenyl)-1,4-benzenediamine was employed as a monomer in the Pd-catalyzed step-growth polymerization (Fig. S1–S8†). To realize interfacial polymerization via C–N bond formation, toluene and water were used to provide a heterogeneous system using sodium hydroxide as a base (Fig. 1). At the beginning of reaction, the monomer, palladium catalyst and ligand were dissolved in the upper organic layer, and the base was dissolved in the lower water phase. To provide a stable interface between two layers, the polymerization was conducted under thermal conditions without stirring (Fig. S9†). To optimize reaction conditions, bidentate and monodentate dialkylbiaryl phosphines,33 which have been demonstrated as highly efficient ligands in the Pd-catalyzed C–N bond formation, were employed in the interfacial polymerization. As shown in Fig. 1A (entries 1 to 4), when the bidentate ligand of BINAP and monodentate phosphines were examined, although excellent monomer conversions (92–100%) were obtained at 110 °C, most generated oligomers and polymers were dissolved in organic layers (Table S1†). These reactions afforded cross-linked PANI materials of only 3–16% isolated yields at the interface, suggesting that most monomers were converted into non-cross-linked linear PANI dissolving in toluene.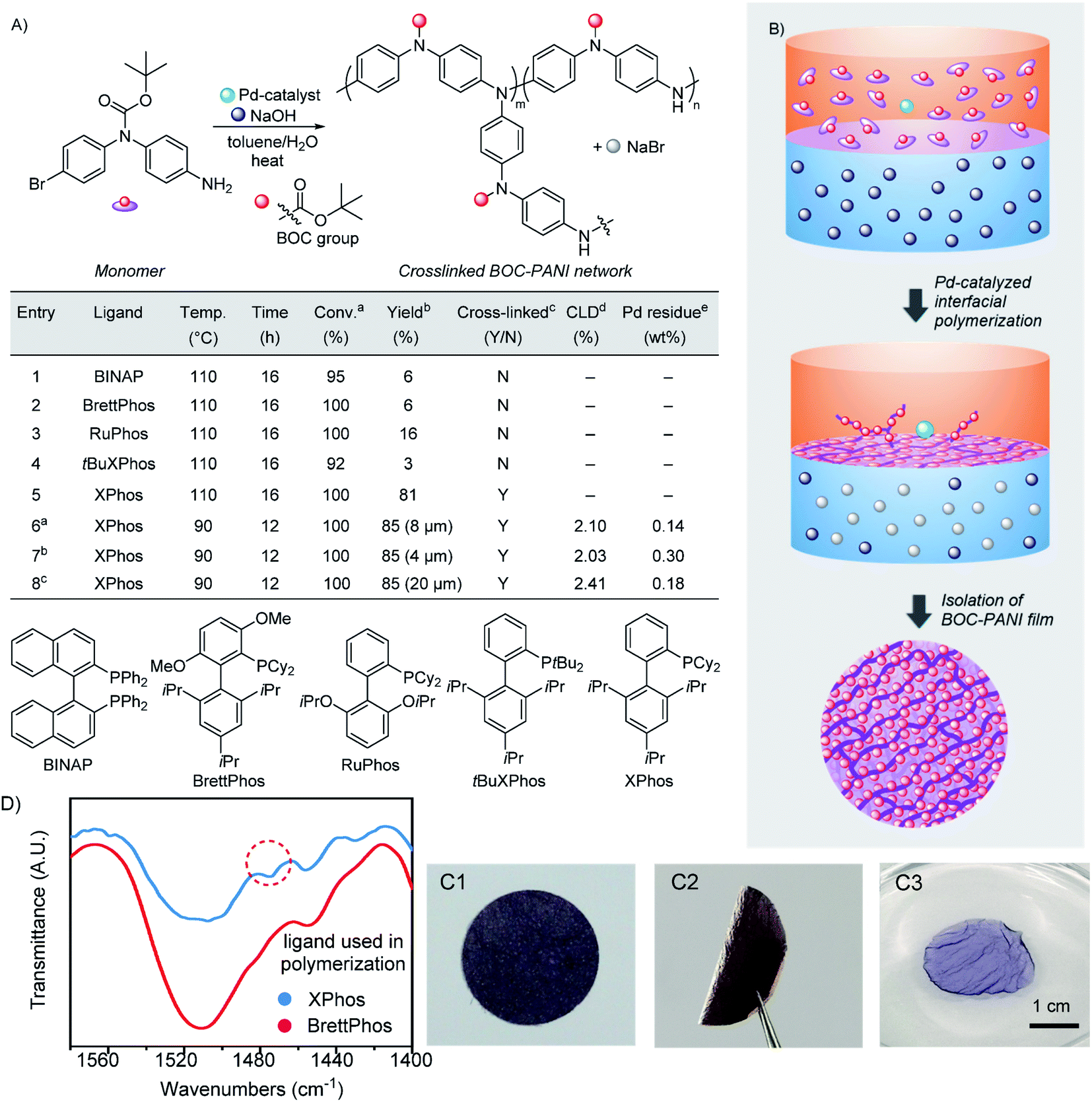 | ||
| Fig. 1 Synthesis of BOC-PANI films. (A) Conditions and results of interfacial polymerization. See Table S1† for details. aConversion determined by 1H NMR. bIsolated yield of cross-linked PANI formed at the interface. cY (yes) and N (no) represent whether the reaction is suitable to generate cross-linked PANI films. dFor the calculation of cross-linking density (CLD), see Fig. S10 and Table S3.†ePd residue determined by SEM/EDX. (B) Synthetic process of the interfacial polymerization. (C1) Image of a BOC-PANI film. (C2) Image of a bent BOC-PANI film. (C3) Image of a DMF-swollen BOC-PANI film. (D) FT-IR spectra of linear and cross-linked BOC-PANI prepared via homogeneous and interfacial polymerization, respectively. | ||
When (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) (XPhos) was employed as a ligand, a PANI film with BOC protecting groups (BOC-PANI) was generated at a complete monomer conversion. After separating it from the reaction mixture, the BOC-PANI film was isolated in 81% yield (entry 5 in Fig. 1A and B). However, solvent reflux resulted in nonuniform distribution of the polymer film at the interface. Then, the reaction temperature was decreased to below the boiling points of both solvents (90 °C, entry 6). Consequently, the polymerization provided BOC-PANI as a thin layer of purple film in good isolated yield. While the thickness of BOC-PANI films (4–20 μm, entries 6–8, Table S1†) could be changed by simply regulating the monomer concentration under otherwise identical reaction conditions, the cross-linking densities (CLDs) were maintained at about 2% (Fig. S10 and Table S3†). Based on the simple operation, we believe that BOC-PANI films of different diameters and thicknesses could be prepared. After purification by washing with organic solvents, the Pd residues contained in films were less than 0.3 wt% as determined by elemental analysis (Fig. S11 and Table S4†). The BOC-PANI films could be bent without breaking (Fig. 1(C1) and (C2)) and be directly cut into different shapes without requiring operations such as mold pressing or spin coating, revealing the mechanical stability and ease of processing of materials obtained via the interfacial growth.
The BOC-PANI materials were analyzed by Fourier-transform infrared (FT-IR) spectroscopy. For linear BOC-PANI (Table S1, entry 2, Fig. S8†), a signal at 1510 cm−1, which belongs to the adsorption of the diphenylamine unit, was clearly observed (Fig. 1D). In comparison, for the cross-linked BOC-PANI film synthesized via the interfacial growth, a shoulder peak at 1475 cm−1 was observed due to the formation of cross-linking units of triphenyl amines as promoted by the chemoselective C–N bond formation between diphenyl amine embedded in the polymer backbone and terminal aryl-bromide group. The presence of triphenyl amine was also confirmed by comparison with a PANI material synthesized from a triphenyl amine derivative (Fig. S12 and S13†).
During the acid doping process, the pre-anchored BOC groups on the polymeric networks were utilized to generate PCL-PANI by releasing gas during deprotection (Fig. 2A and B). When a solvent-swollen BOC-PANI film was treated with hydrochloric acid under an air atmosphere, the purple film gradually turned into green (Fig. 2C) accompanied by the release of lots of CO2 bubbles. The appearance change indicated the successful doping of PANI from BOC-PANI to emeraldine salt (ES), transforming from the non-conductive form to the electroactive state.50 As shown in UV-vis (Fig. 2D), while a BOC-PANI film possessed a clear absorption peak at 320 nm wavelength attributed to the π → π* transition of the benzene ring, PCL-PANI exhibited an additional peak at 635 nm, which was due to the adsorption of the quinoid structure.35 In the FT-IR spectra, the absorption peaks that belong to the alkyl and carbonyl groups from BOC substituents (i.e., peaks at 2974, 1707 and 1506 cm−1, Fig. 2E) were clearly diminished, further confirming the successful deprotection.
 | ||
| Fig. 2 Synthesis and characterization of PCL-PANI films. (A) Acid doping of BOC-PANI. (B) Synthetic scheme of the deprotection process. (C) Image of a PCL-PANI film. (D) UV-vis spectra of BOC-PANI and PCL-PANI films. (E) FT-IR of BOC-PANI and PCL-PANI films. (F) Thermogravimetric analysis of BOC-PANI and PCL-PANI films. (G1 and G2) SEM images of BOC-PANI and PCL-PANI films, respectively. | ||
The thermal stability of PANI films was measured by thermogravimetric analysis (TGA, Fig. 2F). For BOC-PANI, the material underwent apparent weight loss at about 200 °C due to the decomposition of BOC groups under thermal conditions, the amount of which (36.3 wt%) was close to the theorical mass fraction (35.5 wt%) of BOC groups contained in the polymer. For PCL-PANI, the material only showed slow weight loss below 500 °C due to the release of HCl, and no detectable decomposition was observed below 200 °C, suggesting outstanding thermal stability of PCL-PANI. Next, solvent resistance was examined. After submerging BOC-PANI and PCL-PANI films in solvents for above 10 days, both materials showed no discernible decomposition against a variety of organic solvents (e.g., N,N-dimethylformamide, dimethyl sulfoxide, dichloromethane, tetrahydrofuran, Fig. 1(C3) and S15†) and water as a result of the inherently cross-linked networks via covalent bonds, revealing the high stability of obtained binder-free and self-standing films.
The morphology of conducting polymers plays a critical role especially in all-polymer supercapacitors, where electrodes are not soaked in liquid electrolytes to increase the electrode/electrolyte interfacial contact.17,51 The porous morphology of PCL-PANI was characterized using a scanning electron microscope (SEM). As shown in Fig. 2(G1), the planar surface without a porous structure was observed for the BOC-PANI film before deprotection. In sharp contrast, the morphology changed dramatically with plenty of pores being present throughout the film after releasing gas (Fig. 2(G2)), furnishing a number of open channels for charge storage and transportation.
The effect of thin-film thickness on the current response was investigated by cyclic voltammetry (CV) at a scan rate of 5 mV s−1. For PCL-PANI electrodes of three different thicknesses, their CV measurements afforded quasi–rectangular curves embedded with multiple couples of redox peaks corresponding to transitions between different oxidation states of PANI ranging from 0.0 to 1.2 V (Fig. 3A). Notably, all PCL-PANI materials exhibited larger enclosed areas of CV curves than electrodes based on commercial linear PANI, and the capacity scaled reversely with the thickness increasing from 4 to 20 μm for PCL-PANI electrodes. Subsequently, the surface areas of PCL-PANI films were calculated by measuring nitrogen adsorption and desorption isotherms using Brunauer–Emmett–Teller (BET) analysis (Fig. 3B). From 20 to 4 μm thickness, surface areas increased from 23.63 to 53.57 m2 g−1. These results suggested that the specific capacitance was markedly influenced by the thickness and surface area of PCL-PANI films, with PCL-PANI-1 (4 μm) providing the largest enclosed area and highest capacitance. It was also confirmed that the capacitance could be regulated by the thickness of films using the same deprotection strategy.
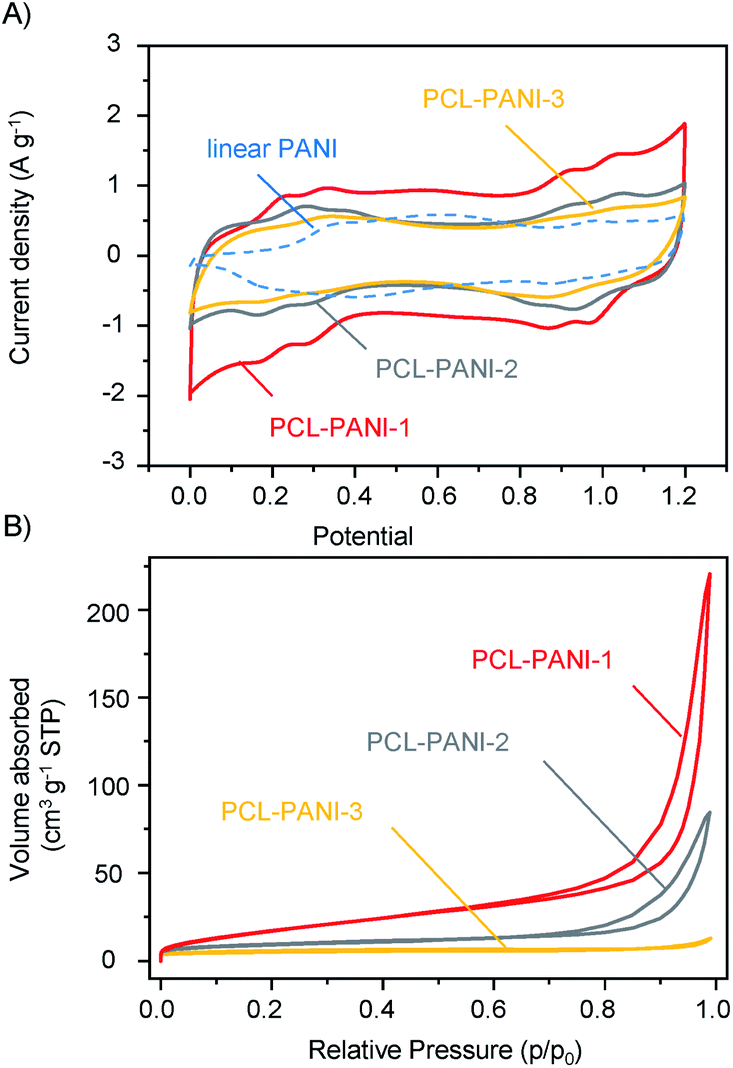 | ||
| Fig. 3 Comparison of PCL-PANI films of different thicknesses. (A) Cyclic voltammetry curves of PCL-PANI films of different film thicknesses and commercial linear PANI (scan rate = 5 mV s−1). (B) Adsorption–desorption curves of PCL-PANI films. Thicknesses of three films: PCL-PANI-1 = 4 ± 0.5 μm, PCL-PANI-2 = 8 ± 0.7 μm, PCL-PANI-3 = 20 ± 1.0 μm. | ||
Next, PCL-PANI films of 4 μm thickness were assembled into supercapacitors and employed to evaluate the electrochemical performance. Two pieces of PCL-PANI films were integrated as additive-free electrodes into a sandwich-like symmetric supercapacitor between two pieces of fluorine-doped tin oxide (FTO) conducting glass (Fig. S17†). As shown in Fig. 4A, the current densities of CV curves increased along with the scan rate while the redox peaks were retained when the scan rate reached up to 50 mV s−1, manifesting good reversibility of the PCL-PANI electrodes. PCL-PANI electrodes were further investigated by galvanostatic charge–discharge cycling (GCD) in a potential range of 1.2 V at varied current densities from 0.5 to 5 A g−1 to reveal the electrochemical behaviour of corresponding devices in repeated cycles. The GCD profiles (Fig. 4B) showed deviation from the linear pattern of capacitive charge and discharge, which demonstrated the contribution of pseudocapacitive properties of PANI films. Owing to the electronic conductivity of the PCL-PANI thin-films measured to be 21.78 S cm−1 (Table S5†), an internal resistance (IR) drop of 8 mV due to the internal resistance was observed at the onset of each discharge cycle.
 | ||
| Fig. 4 Comparative electronic performance of PCL-PANI and linear PANI as electrodes for supercapacitors. (A) CV curves of the PCL-PANI supercapacitor with different scan rates. (B) Galvanostatic charge and discharge curves of the PCL-PANI supercapacitor with different current densities. (C) Capacitance performance comparison between PCL-PANI and the linear PANI composite. (D) Ragone plots of gravimetric energy density versus gravimetric power density for the PCL-PANI supercapacitors and linear PANI composite supercapacitors. | ||
The specific capacitance of the supercapacitor can be estimated based on the discharge curves of the galvanostatic cycling (Fig. 4C and eqn (S1)). Notably, the specific capacitances of the PCL-PANI based devices were more than twice higher than those made of commercial linear PANI materials. Specifically, the highest capacitance was found to be 435 F g−1 with a current density of 0.5 A g−1, as opposed to 220 F g−1 of linear PANI (Fig. S20–S22†). It is worth pointing out that the gravimetric capacitances of these PCL-PANI electrodes are superior to the commercial activated carbon (AC) based electrodes (<200 F g−1),52 which are common inorganic materials for electrodes of supercapacitors. They are also comparable to or higher than some of the nanostructured carbon/metal oxide enhanced PANI electrodes.17,53,54
Furthermore, in conventional methods of electrode fabrication, binders and substrates are required to form integrated electrodes with the active materials. Hence, capacitances normalized to masses of active materials only are typically reported, and discussions were raised that it could cause overestimation of capacitance.55 Benefitting from the integration of polymer synthesis and formation of a cross-linked network in a one-step process, the resultant free-standing PCL-PANI electrodes eliminate the use of binders, additives or substrates, and thereby the gravimetric capacitances normalized to the electrode weight are identical to that normalized to PCL-PANI films, presenting much higher actual amounts of active materials for practical applications. Since substances other than conducing polymers have been almost always added to composed electrodes, very few studies have disclosed the gravimetric capacitance of the entire electrodes,25,41 and this work reports the highest value based on the entire electrode or PANI to our knowledge, especially given that this value was derived from a two-electrode cell setup, which typically yields more realistic but about half of the capacitance measured in a three-electrode cell setup.56,57 In addition, upon increasing the current density from 0.5 to 5 A g−1, the PCL-PANI supercapacitor still retains more than 70% of the specific capacitance, suggesting that the highly porous nature of the polymeric film could facilitate the charge transfer and ion diffusion of the supercapacitor.
The Ragone plots in relation to gravimetric power density and energy density of supercapacitors are fitted in Fig. 4D (eqn (S2) and (S3)). The PCL-PANI-based devices offered a maximum energy density of 23 W h kgelectrode−1 operating within the 1.2 V potential range, exceeding the best performance of devices made of linear PANI (10 W h kgelectrode−1) and commercial supercapacitors (5 W h kg−1)58 from low to high power densities. Notably, for the high power density region (above 10000 W kg−1), while linear PANI and commercial supercapacitors could barely deliver such superior rate capability, the PCL-PANI-based device provided a remarkable gravimetric power density of 16000 W kgelectrode−1. These results demonstrate that the additive-free and porous electrodes could dramatically improve the electrochemical capacitive behaviour of conducting polymer-based supercapacitors.
In order to demonstrate the long-term stability of the PCL-PANI electrodes, capacitance retention of supercapacitors based on the PCL-PANI film and commercial PANI material was compared at 1 A g−1 (Fig. 5A). The PCL-PANI system quickly stabilized after initial activating cycles owing to the partial oxidization by air for most conducting polymer-based pseudocapacitors.20 In subsequent charge–discharge cycling, the PCL-PANI supercapacitor displayed robust cycling and high coulombic efficiency (above 94.25%) without clear capacitance loss after 18000 cycles. On the other hand, the supercapacitor made of linear PANI experienced an abrupt decay of capacitance, which quickly reduced below 40% of the initial capacitance within 2000 cycles with rather unstable coulombic efficiency. It is postulated that the linear PANI composite electrode tends to pulverize when binders and active materials start dissolving in the electrolyte as a result of volume swelling and shrinkage during charge–discharge cycles. In sharp contrast, the PCL-PANI electrodes exhibited unchanged specific capacitance performance while delivering high gravimetric power and energy, which was so far the best cycling stability compared with other pure PANI and PANI-composite systems.14,17,47,53,54,59 Electrochemical impedance spectroscopy (EIS) was performed before and after long-time cycling to investigate the kinetics of current response and internal resistance change during the charge–discharge. As revealed by Fig. 5B, at high frequencies, the interface between the electrolyte and electrode was displayed as a semi-circle, which is typically found for supercapacitors and batteries. In the low frequency region, the high phase angle of the curve indicates good capacitive behavior of the PCL-PANI electrodes. It was observed that the electrolyte resistance and interface resistance changed from 36.8 to 49.8 Ω after 18000 cycles, which represented a small resistance growth, attributed to the structural integrity of the cross-linked structure as well as the good interfacial stability of the in situ formed film when subjected to long-term cycling.
 | ||
| Fig. 5 Cycling stability of the PCL-PANI supercapacitor. (A) Long-term cycling performance of the PCL-PANI-based supercapacitor versus the commercial linear PANI-based supercapacitor. (B) AC impedance spectra of PCL-PANI-based supercapacitors before and after cycling. (C) The specific capacitance and the resistance as a function of the floating time. | ||
To further assess the cycling performance of the PCL-PANI-based device, a more stringent floating test was adopted with unidirectional galvanostatic polarization. Specifically, the symmetric device was held at the highest charging potential (1.2 V) for 110 h. Subsequently, periodic sampling was performed every 12 h to keep track of capacitance and resistance change of the system. As evident in Fig. 5C, the capacitance of the PCL-PANI electrodes was retained at around 400 F gelectrode−1 over 110 h of charging, while the internal resistance experienced very limited change during the course, demonstrating the outstanding strain accommodating capability and mechanical stability of the free-standing conducting films.
Conclusions
In summary, we have developed a bottom-up synthetic approach to obtain PANI films that are cross-linked, highly porous and additive-free. These conducting materials can be readily assembled into all-polymer supercapacitors of excellent performance with gravimetric power capabilities of around 16000 W kgelectrode−1 and energy densities up to 23 W h kgelectrode−1. Importantly, owing to the covalently cross-linked structure and abundant open channels formed throughout the films, PCL-PANI-based electrodes effectively addressed the cycle-life issue of PANI electrodes, successfully extending the cycle life to above 18000 cycles without discernible capacitance deterioration. This tandem synthetic approach of interfacial polymerization/deprotection provides an unprecedented strategy for facile synthesis and material engineering of conducting polymers or other microporous polymers in one-pot, which might broaden opportunities to advance emerging energy storage technology in stretchable/wearable devices and microelectronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (no. 21971044, 52003231, and 22065037), the Program for Changjiang Scholars and Innovative Research Team in University (IRT17R94), and the State Key Laboratory of Molecular Engineering of Polymers.Notes and references
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581 CrossRef CAS PubMed.
- J. Janata and M. Josowicz, Nat. Mater., 2003, 2, 19 CrossRef CAS PubMed.
- A. Eftekhari, Nanostructured conductive polymers, Wiley, 2010 Search PubMed.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591 CrossRef CAS PubMed.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 22, 14225 CrossRef PubMed.
- T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS PubMed.
- Z. Sun, J. Zhang, F. Ye, W. Wang, G. Wang, Z. Zhang, S. Li, Y. Zhou and J. Cai, J. Power Sources, 2019, 443, 227246 CrossRef CAS.
- K. Gholami Laelabadi, R. Moradian and I. Manouchehri, ACS Appl. Energ. Mater., 2020, 3, 5301 CrossRef CAS.
- T. Li, X. Wang, P. Liu, B. Yang, S. Diao and Y. Gao, Synthetic Met, 2019, 258, 116194 CrossRef CAS.
- J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- Y. Wang, F. Chen, Z. Liu, Z. Tang, Q. Yang, Y. Zhao, S. Du, Q. Chen and C. Zhi, Angew. Chem., Int. Ed., 2019, 58, 15707 CrossRef CAS PubMed.
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., Int. Ed., 2016, 55, 9196 CrossRef CAS PubMed.
- M. Yu, Y. Zhang, Y. Zeng, M. S. Balogun, K. Mai, Z. Zhang, X. Lu and Y. Tong, Adv. Mater., 2014, 26, 4724 CrossRef CAS PubMed.
- K. Li, J. Liu, Y. Huang, F. Bu and Y. Xu, J. Mater. Chem. A, 2017, 5, 5466 RSC.
- M.-H. Bai, T.-Y. Liu, F. Luan, Y. Li and X.-X. Liu, J. Mater. Chem. A, 2014, 2, 10882 RSC.
- Y. G. Wang, H. Q. Li and Y. Y. Xia, Adv. Mater., 2006, 18, 2619 CrossRef CAS.
- A. Eftekhari, L. Li and Y. Yang, J. Power Sources, 2017, 347, 86 CrossRef CAS.
- Y. Liao, H. Wang, M. Zhu and A. Thomas, Adv. Mater., 2018, 30, e1705710 CrossRef PubMed.
- P. Jimenez, E. Levillain, O. Aleveque, D. Guyomard, B. Lestriez and J. Gaubicher, Angew. Chem., Int. Ed., 2017, 56, 1553 CrossRef CAS PubMed.
- H. Gao, L. Xue, S. Xin and J. B. Goodenough, Angew. Chem., Int. Ed., 2018, 57, 5449 CrossRef CAS PubMed.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- H. Y. Shi, Y. J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359 CrossRef CAS PubMed.
- Y. Wang, X. Yang, L. Qiu and D. Li, Energy Environ. Sci., 2013, 6, 477 RSC.
- S. R. Sivakkumar, W. J. Kim, J.-A. Choi, D. R. MacFarlane, M. Forsyth and D.-W. Kim, J. Power Sources, 2007, 171, 1062 CrossRef CAS.
- D. E. Stilwell and S. Park, J. Electrochem. Soc., 1989, 136, 688 CrossRef CAS.
- J. Huang and R. B. Kaner, Chem. Commun., 2006, 2006, 367 RSC.
- J. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851 CrossRef CAS PubMed.
- Y. Wei, R. Hariharan and S. A. Patel, Macromolecules, 1990, 23, 758 CrossRef CAS.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397 RSC.
- G. Ciric-Marjanovic, Synth. Met., 2013, 177, 1 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- J. P. Sadighi, R. A. Singer and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 4960 CrossRef CAS.
- X.-X. Zhang, J. P. Sadighi, T. W. Mackewitz and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 7606 CrossRef CAS.
- N. Spetseris, R. E. Ward and T. Y. Meyer, Macromolecules, 1998, 31, 3158 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27 RSC.
- W. Weng, P. Chen, S. He, X. Sun and H. Peng, Angew. Chem., Int. Ed., 2016, 55, 6140 CrossRef CAS PubMed.
- L. Pan, A. Chortos, G. Yu, Y. Wang, S. Isaacson, R. Allen, Y. Shi, R. Dauskardt and Z. Bao, Nat. Commun., 2014, 5, 3002 CrossRef PubMed.
- G. Shi, S. Jin, G. Xue and C. Li, Science, 1995, 267, 994 CrossRef CAS PubMed.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B. S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531 CrossRef CAS PubMed.
- C. Yu, X. Zhu, C. Wang, Y. Zhou, X. Jia, L. Jiang, X. Liu and G. G. Wallace, Nano Energy, 2018, 53, 475 CrossRef CAS.
- V. D. Thao, B. L. Giang and T. V. Thu, RSC Adv., 2019, 9, 5445 RSC.
- B. J. Waghmode, S. H. Patil, M. M. Jahagirdar, V. S. Patil, R. P. Waichal, D. D. Malkhede, S. D. Sathaye and K. R. Patil, Colloid Polym. Sci., 2014, 292, 1079 CrossRef CAS.
- M. F. Jimenez-Solomon, Q. Song, K. E. Jelfs, M. Munoz-Ibanez and A. G. Livingston, Nat. Mater., 2016, 15, 760 CrossRef CAS PubMed.
- M. Matsumoto, L. Valentino, G. M. Stiehl, H. B. Balch, A. R. Corcos, F. Wang, D. C. Ralph, B. J. Mariñas and W. R. Dichtel, Chem, 2018, 4, 308 CAS.
- W. Dai, F. Shao, J. Szczerbinski, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schluter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213 CrossRef CAS PubMed.
- S. H. Domingues, R. V. Salvatierra, M. M. Oliveira and A. J. Zarbin, Chem. Commun., 2011, 47, 2592 RSC.
- For other examples of all-polymer supercapacitors, see: (a) Y. Wang, F. Chen, Z. Liu, Z. Tang, Q. Yang, Y. Zhao, S. Du, Q. Chen and C. Zhi, Angew. Chem., Int. Ed., 2019, 58, 15707 CrossRef CAS PubMed; (b) M. Zhu, Y. Huang, Y. Huang, H. Li, Z. Wang, Z. Pei, Q. Xue, H. Geng and C. Zhi, Adv. Mater., 2017, 29, 1605137 CrossRef PubMed; (c) A. M. P. Hussain and A. Kumar, J. Power Sources, 2006, 161, 1486 CrossRef CAS.
- A. G. MacDiarmid and A. J. Epstein, Faraday Discuss. Chem. Soc., 1989, 88, 317 RSC.
- S. Grover, S. Goel, R. B. Marichi, V. Sahu, G. Singh and R. K. Sharma, Electrochim. Acta, 2016, 196, 131 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- Z. Niu, W. Zhou, X. Chen, J. Chen and S. Xie, Adv. Mater., 2015, 27, 6002 CrossRef CAS PubMed.
- L. Yuan, X. Xiao, T. Ding, J. Zhong, X. Zhang, Y. Shen, B. Hu, Y. Huang, J. Zhou and Z. L. Wang, Angew. Chem., Int. Ed., 2012, 124, 5018 CrossRef.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917 CrossRef CAS PubMed.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 RSC.
- V. Khomenko, E. Frackowiak and F. Béguin, Electrochim. Acta, 2005, 50, 2499 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- L. Liu, Z. Niu, L. Zhang, W. Zhou, X. Chen and S. Xie, Adv. Mater., 2014, 26, 4855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic methods and experimental results, Fig. S1–S19, Tables S1–S5, and eqn (S1)–(S9). See DOI: 10.1039/d0sc05061j |
| This journal is © The Royal Society of Chemistry 2021 |