
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA helical amplification system composed of artificial nucleic acids†
Hiromu
Kashida
 *a,
Keiji
Nishikawa
a,
Wenjing
Shi
a,
Toshiki
Miyagawa
a,
Hayato
Yamashita
b,
Masayuki
Abe
b and
Hiroyuki
Asanuma
*a
*a,
Keiji
Nishikawa
a,
Wenjing
Shi
a,
Toshiki
Miyagawa
a,
Hayato
Yamashita
b,
Masayuki
Abe
b and
Hiroyuki
Asanuma
*a
aDepartment of Biomolecular Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: kashida@chembio.nagoya-u.ac.jp; asanuma@chembio.nagoya-u.ac.jp
bGraduate School of Engineering Science, Osaka University, 1-3, Machikaneyama, Toyonaka, Osaka 560-8531, Japan
First published on 12th January 2021
Abstract
Herein we report an amplification system of helical excess triggered by nucleic acid hybridization for the first time. It is usually impossible to prepare achiral nanostructures composed of nucleic acids because of their intrinsic chirality. We used serinol nucleic acid (SNA) oligomers for the preparation of achiral nanowires because SNA oligomers with symmetrical sequences are achiral. Nanowire formation was confirmed by atomic force microscopy and size exclusion chromatography. When a chiral nucleic acid with a sequence complementary to SNA was added to the nanostructure, helicity was induced and a strong circular dichroism signal was observed. The SNA nanowire could amplify the helicity of chiral nucleic acids through nucleobase stacks. The SNA nanostructures have potential for use as platforms to detect chiral biomolecules under aqueous conditions because SNA can be readily functionalized and is water-soluble.
Introduction
Amplification of helicity has been paid great attention owing not only to its relevance to the origin of homochirality but also to its practical applications such as asymmetric catalysis and chiral sensors.1 Helical amplification that relies on non-covalent assembly is often described as a “sergeants and soldiers effect”, in which a small number of chiral units (the sergeants) control a large number of achiral units (the soldiers). The effect was originally proposed based on polymers by Green et al.2 and was later extended to supramolecular assemblies with the pioneering work of Meijer et al.3 Many systems have been reported for helical amplification that rely on self-assembly of small molecules mostly in organic solvents.4 In order to realize such amplification systems, molecular assemblies of achiral molecules have to be prepared. However, de novo design of such molecular assemblies is usually difficult, and development of a versatile platform for helical amplification in water has proven challenging.Nucleic acids are highly water-soluble and can form various nanostructures due to base-sequence-dependence of double-helix formation.5 However, it is impossible to prepare achiral nanostructures with natural nucleic acids because of their intrinsic chirality. Peptide nucleic acid (PNA) oligomers are achiral,6 and Nielsen et al. demonstrated that helicity of PNA can be induced by the covalent attachment of a chiral amino acid or nucleotide at its termini.7 Häner et al. reported a helical amplification system using non-covalent interaction between achiral and chiral oligomeric pyrene strands.8 However, helical amplification systems triggered by nucleic acid hybridization have not been reported so far. Here we report a helical amplification system of artificial nucleic acids for the first time. Such systems are desirable since they could be applied to detection of chiral biomolecules in aqueous solution.
Our group developed serinol nucleic acid (SNA), which is a nucleic acid analog.9 SNA is based on the serinol (2-amino-1,3-propanediol) moiety, and monomer units, which carry a natural base and are linked via a phosphodiester bond. An SNA oligomer with a symmetrical sequence is achiral and does not show circular dichroism (CD) signals (see Fig. S1 in the ESI†). In contrast, the helicity of SNA oligomers with asymmetric sequences can be inversed by reversing their sequences. It is a quite unique character of SNA and cannot be realized by other natural/artificial nucleic acids. SNA oligomers are highly water-soluble and can form stable duplexes with complementary SNA. Furthermore, SNA can recognize even natural DNA and RNA. Here we used SNA to prepare a helical amplification system. One of the advantages of using nucleic acid nanostructures is the ease of programmability.
Results and discussion
We first designed SNA oligomers that could form a one-dimensional SNA nanostructure through hybridization. The SNA decamers S1 and S2 (Fig. 1a) are partially complementary and can, therefore, form a one-dimensional nanowire. This strategy has been used for construction of a DNA nanowire.10 We reasoned that since the one-dimensional structure is formed from only achiral SNA strands, the structure will show no CD signals but that a CD signal will be induced when a strand composed of a chiral nucleic acid is incorporated into the nanowire (Fig. 1b). We selected acyclic D-threoninol nucleic acid (D-aTNA) and L-threoninol nucleic acid (L-aTNA) as chiral sources since they are intrinsically chiral and can form a stable duplex with SNA.11 Moreover, the D-aTNA/SNA duplex showed a CD spectrum similar to that of the D-aTNA/D-aTNA duplex,11b indicating that D-aTNA or L-aTNA can induce helicity of the SNA nanostructure efficiently. DT1 (D-aTNA) and LT1 (L-aTNA) have sequences identical to that of S1. S2F, which can form a fully complementary duplex with S1, was used as a control.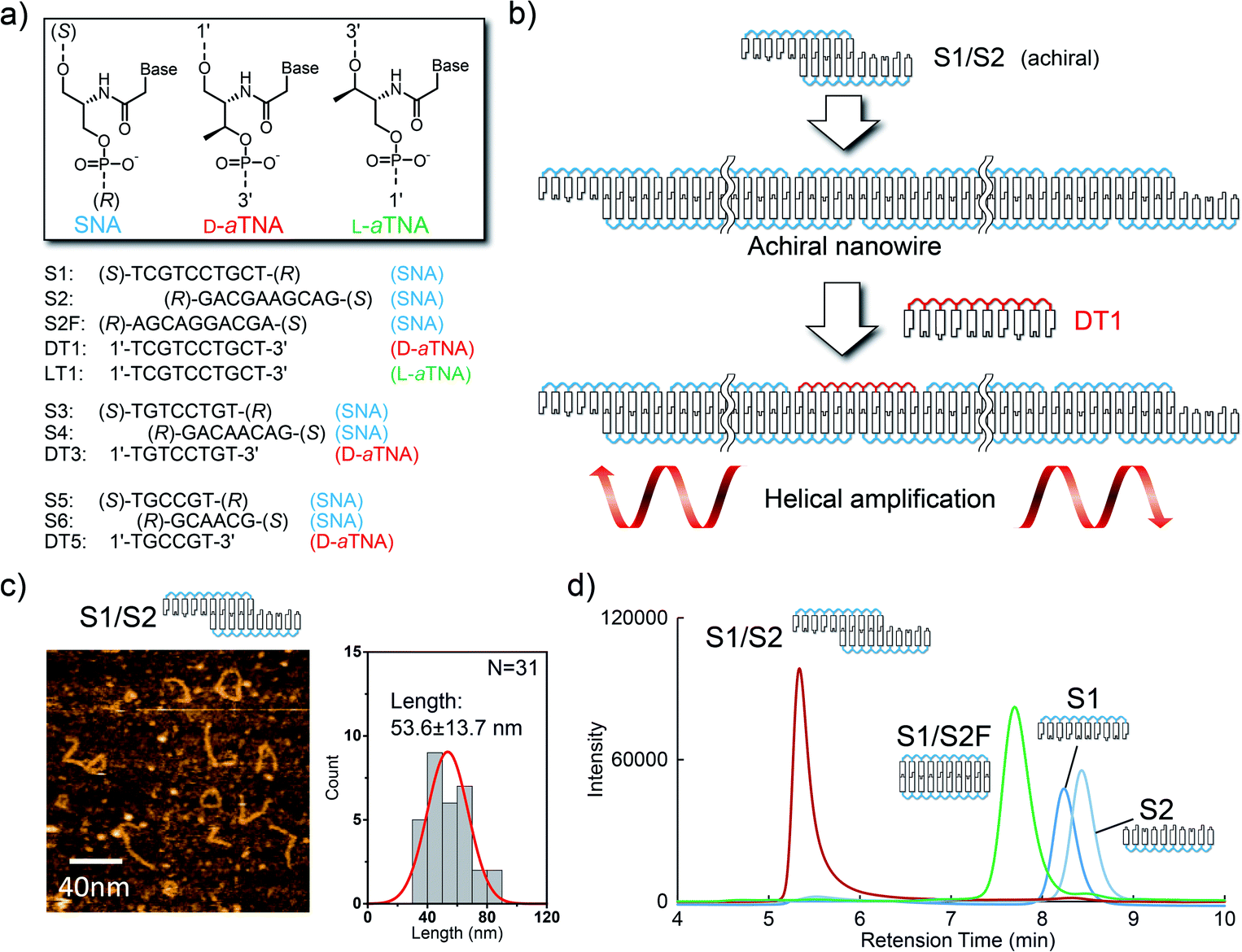 | ||
| Fig. 1 (a) Sequences of SNA, D-aTNA, and L-aTNA oligomers used in this study. (R) and (S) termini are named according to the chirality of the terminal residues. The chemical structures of S1 and S2 are shown in Fig. S2.† (b) Schematic illustration of helical amplification. S1 and S2 form an achiral nanowire. DT1 is efficiently incorporated into the nanostructure because DT1 can form a more stable duplex with S2 than with S1. Helical excess is amplified by inducing helicity in the SNA nanowire. (c) Atomic force microscopy images of S1/S2 in solution. The histogram of the lengths of observed structures is shown on the right. (d) Size-exclusion chromatograms of S1/S2 (red), S1/S2F (green), single-stranded S1 (dark blue) and single-stranded S2 (light blue). | ||
We first characterized the nanowire formed by S1 and S2. Atomic force microscopy imaging in solution indicated the formation of wire-like structures. The length approximately followed Gaussian distribution with an average length of 53.6 nm (Fig. 1c). Size-exclusion chromatography was also performed to analyze nanowire formation. The retention time for the peak observed when the solution of S1 and S2 was analyzed was much shorter than that for peaks corresponding to single strands (Fig. 1d). Moreover, the peak present in the S1/S2 solution had a much shorter retention time than did the S1/S2F duplex. These results clearly demonstrate that SNA strands S1 and S2 form a nanowire.
Melting analyses were then performed to assess the stability of the S1/S2 nanowire. Melting analysis of S1/S2 showed a melting temperature (Tm) of 46.7 °C, indicating that these strands formed a stable complex at room temperature (Fig. S3 and Table S1†). The Tm of DT1/S2 was 53.4 °C, and LT1/S2 had the same Tm as DT1/S2; this was expected since they are enantiomers. These results indicate that D- and L-aTNA/SNA duplexes are more stable than the SNA/SNA structure. In order to confirm incorporation of D-aTNA into the SNA nanostructure, thermodynamic parameters of fully complementary duplexes, S1/S2F and DT1/S2F, were determined from van't Hoff plots (Fig. S4 and Table S2†). The 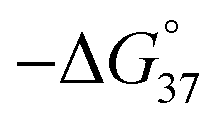 of formation of the DT1/S2F duplex was 4.3 kcal mol−1 higher than that of S1/S2F, showing that the D-aTNA/SNA duplex is much more stable than the SNA/SNA duplex. The binding constant of D-aTNA to SNA was 1000 times higher than that of SNA to SNA. These results indicate that D- and L-aTNA can be efficiently incorporated into the SNA achiral nanowires. We also determined the thermodynamic parameters of S1sh/S2sh (CTGCT/GACGA), which is the overlapping portion of the S1/S2 nanowire (Fig. S5 and Table S3†). The binding constant of S1sh/S2sh at 20 °C was only 1.1 × 106 M−1. It was reported that stacking interaction with neighboring strands drastically enhances the duplex formation.12 The Tm of S1/S2 (46.7 °C) was actually much higher than that of S1sh/S2sh (22.9 °C; Table S1†). These results clearly demonstrate that the stacking interaction with neighboring strands drastically enhanced the nanowire formation.
of formation of the DT1/S2F duplex was 4.3 kcal mol−1 higher than that of S1/S2F, showing that the D-aTNA/SNA duplex is much more stable than the SNA/SNA duplex. The binding constant of D-aTNA to SNA was 1000 times higher than that of SNA to SNA. These results indicate that D- and L-aTNA can be efficiently incorporated into the SNA achiral nanowires. We also determined the thermodynamic parameters of S1sh/S2sh (CTGCT/GACGA), which is the overlapping portion of the S1/S2 nanowire (Fig. S5 and Table S3†). The binding constant of S1sh/S2sh at 20 °C was only 1.1 × 106 M−1. It was reported that stacking interaction with neighboring strands drastically enhances the duplex formation.12 The Tm of S1/S2 (46.7 °C) was actually much higher than that of S1sh/S2sh (22.9 °C; Table S1†). These results clearly demonstrate that the stacking interaction with neighboring strands drastically enhanced the nanowire formation.
We then investigated helical amplification by SNA nanowires through CD measurements performed at 0 °C. The fully complementary duplex, S1/S2F, had no distinct CD signals since it forms an achiral duplex like “meso helix” (Fig. 2a).13 In sharp contrast, a strong CD signal at about 260 nm was induced when DT1 was added. The signal increased with the number of equivalents of DT1 added. In the absence of DT1, there was no CD signal from the S1/S2 nanowire as the nanowire is achiral. However, there was significant signal intensity at around 260 nm even upon the addition of 0.01 equivalents of DT1 (Fig. 2b). The intensity increased as the number of equivalents of DT1 increased, but it was almost saturated after 0.2 equivalents. A plot of the magnitude of the CD couplet at 260 nm (ΔCD) versus DT1 equivalents revealed a linear relationship between S1/S2F (Fig. 2c). ΔCD instead of CD was used for the quantification of helicity in order to reduce the influence of baseline noise (Fig. 2a). That this plot was linear indicated that, although a duplex was formed, there was no helical amplification. In contrast, the CD signal of S1/S2 significantly increased dramatically and non-linearly in the presence of equivalents of up to 0.2 of DT1. Such non-linearity strongly indicates that the helicity of D-aTNA transferred to the achiral SNA nanostructure. The degree of helical amplification was evaluated from CD intensities at 0 °C to avoid complexity due to insufficient hybridization. Plots of ΔCD at 20 °C (Fig. S6†) revealed the same behavior as observed at 0 °C. The helical excess of S1/S2 upon the addition of DT1 was also calculated and similar behavior with the ΔCD plot was observed (Fig. S7†). Although S1/S2 and S1/S2F have identical base compositions, the amplitude of helical amplification was drastically different in the presence of a small amount of DT1. The observed difference clearly shows that a nanostructure is required for helical amplification. Each CD spectrum was measured after annealing at 80 °C in order to avoid complexity caused by slow kinetics. CD change of S1/S2 upon the addition of DT1 at 20 or 30 °C was also measured (Fig. S8†). The CD spectra after incubation with sufficient time showed almost the same intensity as those after incubation. These results clearly demonstrate that helical amplification can occur without annealing.
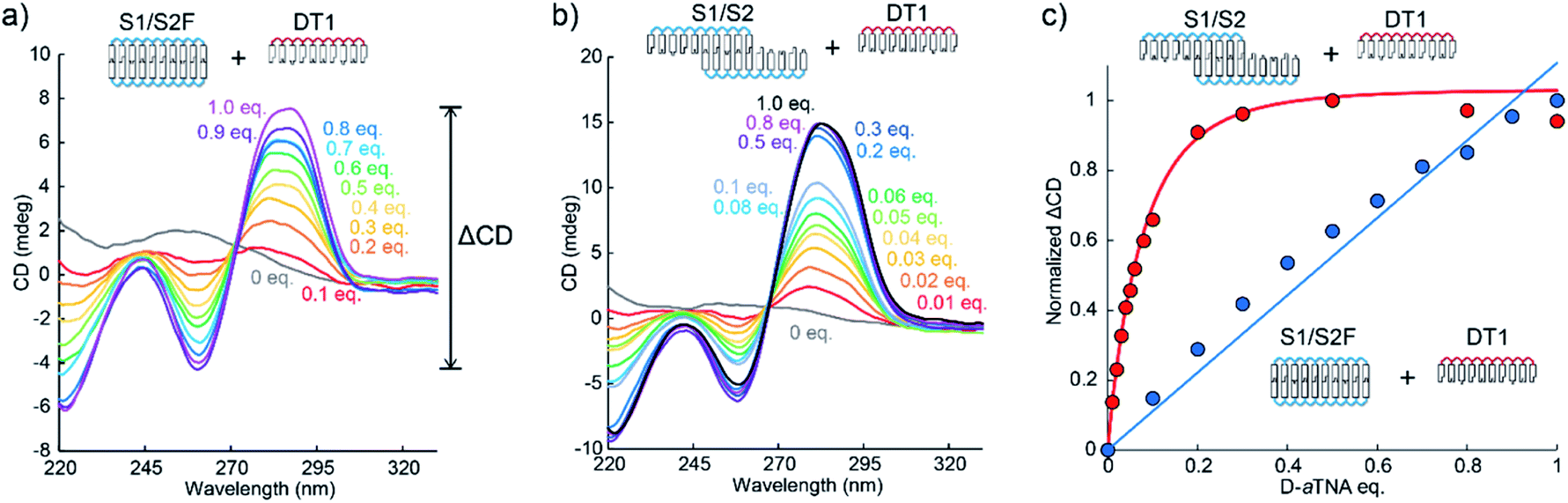 | ||
| Fig. 2 (a) CD spectra of the S1/S2F duplex in the presence of indicated equivalents of DT1. ΔCD was calculated by subtracting the minimum CD from the maximum CD at around 260 nm. (b) CD spectra of the S1/S2 nanowire in the presence of indicated equivalents of DT1. (c) Plots of change in ΔCD of S1/S2 and S1/S2F versus number of equivalents of DT1. Solution conditions were 4.0 μM each SNA strand, 100 mM NaCl, 10 mM phosphate buffer (pH 7.0), 10 mm path length, 0 °C. | ||
The induced helicity was inverted by adding L-aTNA, an enantiomer of D-aTNA, to the SNA nanowire. The CD spectrum of S1/S2 with 1.0 equivalent of DT1 had a negative peak at about 260 nm and a positive peak at about 285 nm, whereas the spectrum with 1 equivalent of LT1 had a positive peak at 260 nm and a negative peak at 285 nm (Fig. 3). The helicity of L-aTNA could also be amplified by the SNA nanostructure as revealed by the plot between ΔCD and the number of equivalents of LT1 (Fig. S9†).
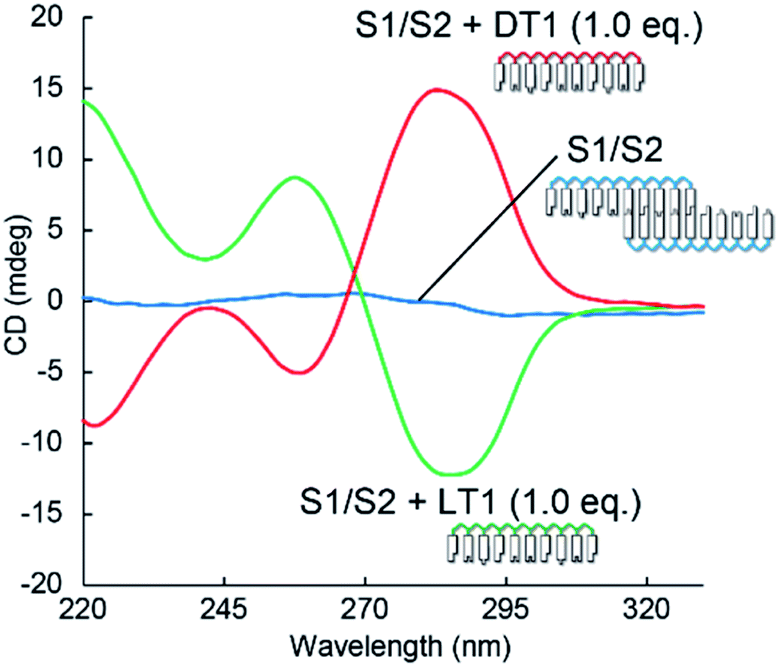 | ||
| Fig. 3 Comparison of CD spectra of S1/S2 alone or with DT1 or LT1. Solution conditions were 4.0 μM each strand, 100 mM NaCl, 10 mM phosphate buffer (pH 7.0), 10 mm path length, 0 °C. | ||
Effects of mismatches were assessed to evaluate the significance of stacking interactions in helical amplification. When a T–T or a C–T mismatch was incorporated at the ends of SNA strands, the degree of helical amplification severely decreased (Fig. 4a). Although the ΔCD was non-linear, it was not saturated even with 0.8 equivalents of D-aTNA. The degree of amplification further decreased when the nanowire contained gaps, yielding almost linear behavior (Fig. 4a). These results clearly demonstrated the importance of stacking interactions in helical amplification. In other words, stacking interaction between nucleobases drove the induction of helicity. The dependence of helical amplification on the strand length was investigated. Non-linear curves were observed with SNA nanowires composed of 8-mer and 6-mer strands, and the degrees of amplification were similar to that of the nanowire composed of 10-mer SNAs (Fig. 4b). These results strongly indicate that the degree of amplification is governed not by the length of the nanowire but by the number of nicks, suggesting that the flexibility of the nicked duplex14 might induce the helical inversion. The ΔCD of S1/S2 with 0.01 equivalents of DT1 was about 14 times higher than the value estimated from the linear increment (Fig. 2c). This result indicates that one D-aTNA can induce helicity of seven S1/S2 duplexes. Although the detailed structure of the SNA/SNA duplex has not been reported, seven S1/S2 duplexes correspond to about 25 nm by assuming 3.4 Å bp−1. In contrast, the average length of the SNA nanowire was determined to be 53.6 nm by AFM imaging (Fig. 1c). These rough estimates also indicate that helical inversion occurs within the SNA nanowire.
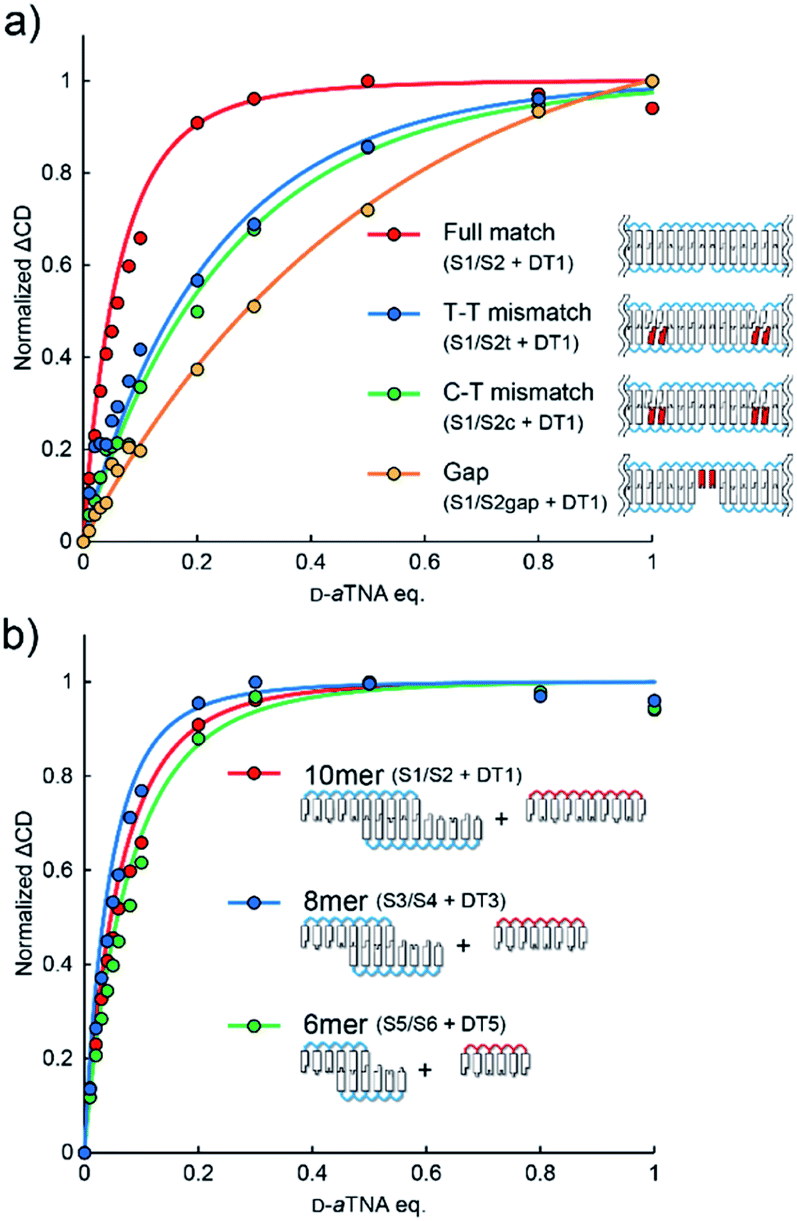 | ||
| Fig. 4 (a) Effects of mismatches and gaps on helical amplification. ΔCD was calculated from the spectra shown in Fig. S10.† (b) Effects of strand length on helical amplification. ΔCD was calculated from the spectra shown in Fig. S11.† | ||
Conclusions
In conclusion, a helicity amplification system composed of nucleic acids was successfully prepared by using the SNA nanostructure. The helicity of threoninol nucleic acids was amplified through base stacks of the SNA nanowire. Since our system utilizes duplex formation as a trigger, various helical circuits could be prepared by preparing more complex SNA nanostructures. In this study, artificial nucleic acid was used as a chiral source; however, the SNA-based nanowires will be useful for the detection of other types of biomolecules. For example, the system could be used to detect DNA or RNA as SNA can recognize natural nucleic acids. Since SNA can be readily functionalized, CD at the desired wavelength or even circularly polarized luminescence (CPL) could be induced by attaching chromophores to SNA. Furthermore, other biomolecules, such as proteins and sugars, could be detected if these molecules could induce helicity in SNA nanostructures. SNA nanostructures would be a versatile platform to convert chiral information of biomolecules to chiroptical signals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant numbers JP18H03933 (H. A.), JP16H05925 (H. K.), JP17H05150 (H. K.), JP19K22250 (H. K.), JP20H02858 (H. K.), JP20H03223 (H. Y.) and JP19H05789 (M. A.), and by AMED under grant number 19am0401007 (H. A.). Support from the Asahi Glass Foundation (H. K.) is gratefully acknowledged.Notes and references
- (a) A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS; (b) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS; (c) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- (a) L. Brunsveld, B. G. G. Lohmeijer, J. A. J. M. Vekemans and E. W. Meijer, Chem. Commun., 2000, 2305–2306 RSC; (b) L. J. Prins, P. Timmerman and D. N. Reinhoudt, J. Am. Chem. Soc., 2001, 123, 10153–10163 CrossRef CAS; (c) K. Toyofuku, M. A. Alam, A. Tsuda, N. Fujita, S. Sakamoto, K. Yamaguchi and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 6476–6480 CrossRef CAS; (d) A. Lohr and F. Würthner, Chem. Commun., 2008, 2227–2229 RSC; (e) M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS; (f) F. Helmich, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 16753–16755 CrossRef CAS; (g) H. Ito, M. Ikeda, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2011, 133, 3419–3432 CrossRef CAS; (h) F. Helmich, M. M. J. Smulders, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 12238–12246 CrossRef CAS; (i) D. J. van Dijken, J. M. Beierle, M. C. A. Stuart, W. Szymański, W. R. Browne and B. L. Feringa, Angew. Chem., Int. Ed., 2014, 53, 5073–5077 CAS; (j) T. Kim, T. Mori, T. Aida and D. Miyajima, Chem. Sci., 2016, 7, 6689–6694 RSC.
- (a) N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS; (b) P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS; (c) N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS.
- (a) P. Wittung, M. Eriksson, R. Lyng, P. E. Nielsen and B. Norden, J. Am. Chem. Soc., 1995, 117, 10167–10173 CrossRef CAS; (b) I. A. Kozlov, L. E. Orgel and P. E. Nielsen, Angew. Chem., Int. Ed., 2000, 39, 4292–4295 CrossRef CAS.
- A. L. Nussbaumer, D. Studer, V. L. Malinovskii and R. Häner, Angew. Chem., Int. Ed., 2011, 50, 5490–5494 CrossRef CAS.
- H. Kashida, K. Murayama, T. Toda and H. Asanuma, Angew. Chem., Int. Ed., 2011, 50, 1285–1288 CrossRef CAS.
- (a) Y. Ohya, H. Noro, M. Komatsu and T. Ouchi, Chem. Lett., 1996, 25, 447–448 CrossRef; (b) T. Akasaka, K. Matsuura and K. Kobayashi, Bioconjugate Chem., 2001, 12, 776–785 CrossRef CAS.
- (a) H. Asanuma, T. Toda, K. Murayama, X. Liang and H. Kashida, J. Am. Chem. Soc., 2010, 132, 14702–14703 CrossRef CAS; (b) K. Murayama, Y. Tanaka, T. Toda, H. Kashida and H. Asanuma, Chem.–Eur. J., 2013, 19, 14151–14158 CrossRef CAS; (c) K. Murayama, H. Kashida and H. Asanuma, Chem. Commun., 2015, 51, 6500–6503 RSC.
- M. J. Lane, T. Paner, I. Kashin, B. D. Faldasz, B. Li, F. J. Gallo and A. S. Benight, Nucleic Acids Res., 1997, 25, 611–617 CrossRef CAS.
- R. Wechsel, J. Raftery, D. Cavagnat, G. Guichard and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 9657–9661 CrossRef CAS.
- H. Kashida, A. Kurihara, H. Kawai and H. Asanuma, Nucleic Acids Res., 2017, 45, e105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05245k |
| This journal is © The Royal Society of Chemistry 2021 |