
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of chiral porphyrin macrocyclic hosts and kinetic enantiorecognition of viologen guests†
Pieter J.
Gilissen‡
 a,
Annemiek D.
Slootbeek‡
a,
Jiangkun
Ouyang
a,
Nicolas
Vanthuyne
b,
Rob
Bakker
a,
Johannes A. A. W.
Elemans
*a and
Roeland J. M.
Nolte
*a
a,
Annemiek D.
Slootbeek‡
a,
Jiangkun
Ouyang
a,
Nicolas
Vanthuyne
b,
Rob
Bakker
a,
Johannes A. A. W.
Elemans
*a and
Roeland J. M.
Nolte
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: r.nolte@science.ru.nl; j.elemans@science.ru.nl
bAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France
First published on 13th January 2021
Abstract
The construction of macromolecular hosts that are able to thread chiral guests in a stereoselective fashion is a big challenge. We herein describe the asymmetric synthesis of two enantiomeric C2-symmetric porphyrin macrocyclic hosts that thread and bind different viologen guests. Time-resolved fluorescence studies show that these hosts display a factor 3 kinetic preference (ΔΔG‡on = 3 kJ mol−1) for threading onto the different enantiomers of a viologen guest appended with bulky chiral 1-phenylethoxy termini. A smaller kinetic selectivity (ΔΔG‡on = 1 kJ mol−1) is observed for viologens equipped with small chiral sec-butoxy termini. Kinetic selectivity is absent when the C2-symmetric hosts are threaded onto chiral viologens appended with chiral tails in which the chiral moieties are located in the centers of the chains, rather than at the chain termini. The reason is that the termini of the latter guests, which engage in the initial stages of the threading process (entron effect), cannot discriminate because they are achiral, in contrast to the chiral termini of the former guests. Finally, our experiments show that the threading and de-threading rates are balanced in such a way that the observed binding constants are highly similar for all the investigated host–guest complexes, i.e. there is no thermodynamic selectivity.
Introduction
Chiral recognition and selection of substrate (guest) molecules are well-established features of enzymes and natural receptors.1,2 Inspired by these naturally occurring chiral hosts, chemists have developed a variety of synthetic chiral macromolecular architectures that function as receptors. Examples include macrocyclic arenes,3–8 cyclodextrins,9 and metal–organic cages.10–13 Calixarenes have been reported that display enantiorecognition towards chiral carboxylates3 and chiral amines.4–6 Amino acid recognition has been achieved with calixarenes,4 cyclodextrins,9 and metal–organic cages.10 Chiral metal–organic cages have also been used for the enantioseparation of small alcohols and carboxylic acids,11,12 and for the separation of atropisomeric compounds, such as 1,1′-bi-2-naphthol (BINOL) derivatives.11 Triptycene-based hosts have been developed for the recognition of chiral trimethylammonium compounds.14 The reported receptors showed different degrees of thermodynamic enantiorecognition (up to 12-fold difference in binding constant Kassoc for the binding of the two enantiomers). Studies on kinetic enantiorecognition, such as differences in threading rate constants (kon-values), however, have rarely been reported.15Our research aims at encoding information into synthetic polymers using a macrocyclic porphyrin catalyst based on the glycoluril framework, i.e.Mn1 (Fig. 1) that can thread onto a polymer containing alkene double bonds, e.g. a polybutadiene chain, and processively epoxidize these double bonds.16–18 Since we intend to write a binary code on said polymer in the form of chiral epoxides ((R,R)-epoxide = digit 0, (S,S)-epoxide = digit 1), our desired writing system requires a chiral variant of Mn1 that is capable of performing sequential processive threading and catalysis. Two important requirements for sequential processive catalysis are spatiotemporal control and unidirectionality,19 both of which are determined by kinetic factors. We hypothesized that unidirectional threading of polymer chain through a catalytic machine might be accomplished by aligning chiral structural information in both the catalyst and the polymer. Therefore, it is crucial that the catalytic machine displays a kinetic preference for one of the enantiomers of a chiral (polymeric) guest. In earlier work, we reported on the post-modification of the achiral porphyrin cage H2120 by providing it with a nitro function, yielding a racemic mixture of planar chiral nitro-functionalized porphyrin cage H22.21 The enantiomers of H22 could be resolved by chiral HPLC22 and by installing a chiral auxiliary on the corresponding amino-functionalized host.23 The enantiopure hosts investigated earlier were shown to have small differences in affinity for chiral viologen guests (up to 3-fold difference in binding constant Kassoc).22,23 In this paper we report an alternative approach to synthesize a chiral C2-symmetric porphyrin cage compound, i.e. a host in which the porphyrin macrocycle is linked to the glycoluril framework via chiral linkers (H23, Fig. 1). Furthermore, we show that the enantiomers of H23 display a kinetic preference for the threading and binding of viologen guests equipped with a chiral head group.
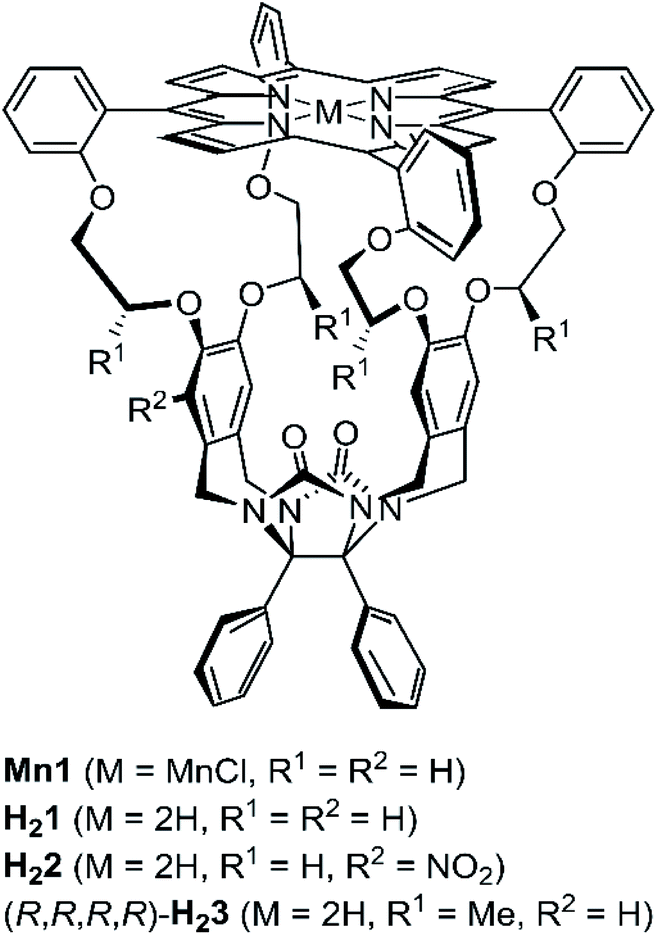 | ||
| Fig. 1 Chemical structures of porphyrin macrocycles. | ||
Results and discussion
Synthesis and characterization
The synthetic route to the novel porphyrin macrocyclic hosts (R,R,R,R)-H23 and (S,S,S,S)-H23 is depicted in Fig. 2. The key synthesis step was the introduction of the chiral centers in intermediate 7. Burkard and Effenberger reported the synthesis of (R,R)-7 in one step from pyrocatechol and the very reactive triflate ester of ethyl (S)-lactate under basic conditions.24 According to their communicated procedure, the resulting diester (R,R)-7 was obtained diastereomerically pure. In our hands, however, this procedure was not scalable and it resulted in an inseparable 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 diastereomeric mixture of (R,R)-7 and the meso-compound (R,S)-7 (via epimerization of one of the chiral centers). To prevent epimerization, we resided to the Mitsunobu reaction. Initially, we attempted a double Mitsunobu reaction on pyrocatechol with ethyl (S)-lactate to afford diester (R,R)-7 in a single step, which however failed. In contrast to the lack of literature procedures for Mitsunobu reactions on pyrocatechol, there are successful examples of Mitsunobu reactions on 2-alkoxyphenols employing enantiopure lactates.25–27
:6 diastereomeric mixture of (R,R)-7 and the meso-compound (R,S)-7 (via epimerization of one of the chiral centers). To prevent epimerization, we resided to the Mitsunobu reaction. Initially, we attempted a double Mitsunobu reaction on pyrocatechol with ethyl (S)-lactate to afford diester (R,R)-7 in a single step, which however failed. In contrast to the lack of literature procedures for Mitsunobu reactions on pyrocatechol, there are successful examples of Mitsunobu reactions on 2-alkoxyphenols employing enantiopure lactates.25–27
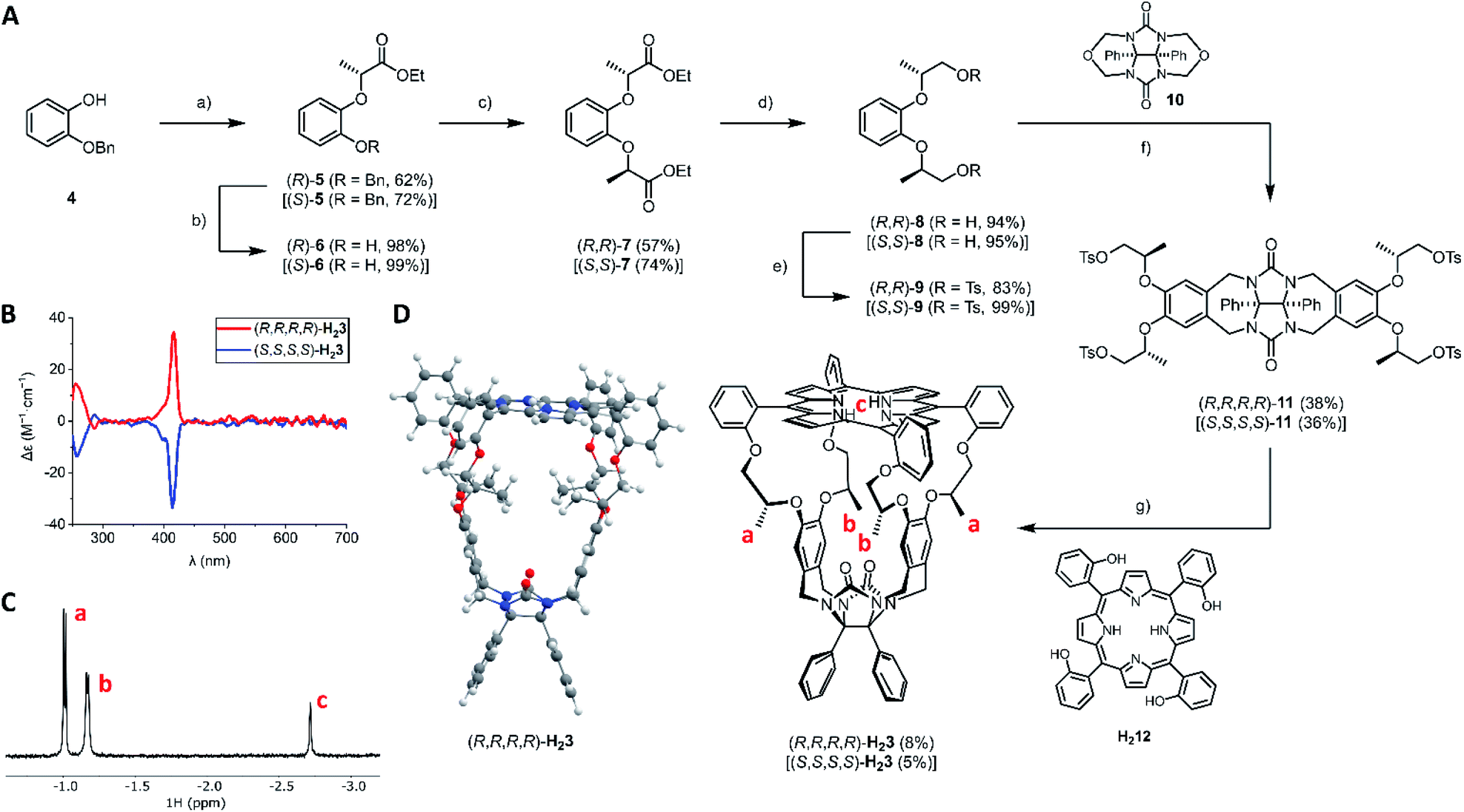 | ||
| Fig. 2 Synthesis and characterization of H23. (A) Synthesis of H23; reagents and conditions: (a) ethyl (S)-lactate (for (R)-5) or ethyl (R)-lactate (for (S)-5), DIAD, PPh3, toluene, 0 → 20 °C; (b) H2, Pd/C, EtOAc, 20 °C; (c) ethyl (S)-lactate (for (R,R)-7) or ethyl (R)-lactate (for (S,S)-7), DIAD, PPh3, toluene, 0 → 20 °C; (d) LiAlH4, THF, 0 → 20 °C; (e) TsCl, pyridine, CH2Cl2, 0 → 20 °C; (f) ZnCl2, SOCl2, CH2Cl2, 20 °C; (g) K2CO3, CH3CN, reflux. For clarity, the synthetic scheme only displays the structures of all (R)-derivatives; yields of all (S)-derivatives are indicated in square brackets. (B) ECD spectra of (R,R,R,R)-H23 and (S,S,S,S)-H23 (c = 3 × 10−5 M in CHCl3/CH3CN, 1:1, v/v). (C) Negative chemical shift region of the 1H NMR spectrum of (R,R,R,R)-H23 (400 MHz, c = 10−3 M in CDCl3/CD3CN, 1:1, v/v) with assignment of protons a–c. (D) Front view of the DFT-optimized structure of (R,R,R,R)-H23 at the B3LYP/6-311+G(d) level. Color code: white = hydrogen, grey = carbon, blue = nitrogen, red = oxygen. | ||
Hence, we designed a stepwise protocol for the introduction of the chiral centers. The Mitsunobu reaction of 2-benzyloxyphenol with ethyl (S)-lactate or ethyl (R)-lactate afforded esters (R)-5 and (S)-5 in 62% and 72% yield, respectively. After quantitative removal of the benzyl protecting group, the other chiral center was installed with a second Mitsunobu reaction. In this way, diesters (R,R)-7 and (S,S)-7 were obtained in reasonable yields (57% and 74%, respectively) and with excellent diastereomeric ratios (>98:2). Both diesters 7 were reduced with lithium aluminum hydride, to afford the corresponding primary alcohols 8 in excellent yields. The primary alcohols were then transformed into tosylate leaving groups, affording (R,R)-9 (83%) and (S,S)-9 (99%). According to the established procedure for the synthesis of achiral cage compound H21,28 the N,O-acetals of compound 10 were activated with zinc chloride in the presence of thionyl chloride, and the resulting N-acyliminium intermediates were reacted with electron-rich arenes (R,R)-9 and (S,S)-9 to afford chiral clip molecules (R,R,R,R)-11 (38%) and (S,S,S,S)-11 (36%). The syntheses of the two enantiomers of H23 were completed by reacting chiral clips 11 under highly dilute basic conditions with 1 equivalent of porphyrin tetraol H212. After two chromatographic purification steps (alumina followed by 60H silica), the enantiopure cage compounds (R,R,R,R)-H23 (8%) and (S,S,S,S)-H23 (5%) were obtained as purple solids. Electronic circular dichroism (ECD) measurements (Fig. 2B) showed that the chirality of the flexible spacers is clearly transferred to the porphyrin, as opposite signs of the CD signals were observed for the Soret bands (λmax = 416 nm) of the enantiomeric hosts (R,R,R,R)-H23 and (S,S,S,S)-H23. 1H NMR analysis (Fig. 2C) revealed that the methyl groups of the C2-symmetric macrocycles are in uncommon – highly shielded – chemical environments, as their protons resonate at negative chemical shift (assigned as protons a and b). This effect can be explained by the close proximity of the shielding area of the aromatic porphyrin ring, indicating that the methyl groups are pointing into the cavity of the cage. 2D ROESY experiments indicated the close proximity of the methyl groups a and b, as we observed mutual NOE contacts. DFT calculations (Fig. 2D) confirmed that the minimum energy structure of H23 is the one with the methyl groups pointing into the cavity of the porphyrin macrocyclic host.
Host–guest binding and threading studies
We reported earlier that H21 is an excellent host for the binding of viologen derivatives.20 Hence, we investigated the effect of the chiral spacers in the C2-symmetric hosts H23 on the binding thermodynamics and the threading kinetics (Table 1) of various achiral and chiral viologen guests (Fig. 3). We studied threading by using approach-to-equilibrium fluorescence quenching spectroscopy, in which we follow the fluorescence intensity of the host as a function of time, as it is quenched upon binding the viologen moiety (Fig. 4, for details see the ESI, Fig. S8–S55†). The initial stages of this process follow second-order kinetics.29,30 Subsequently, de-threading of the viologen guests from the hosts was studied by dilution-induced fluorescence recovery, for which the initial stages follow first-order kinetics (see the ESI, Fig. S56–S103†).29,30 Binding constants were obtained by dividing the threading kon-values by the de-threading koff-values. For the smallest possible viologen, i.e. methyl viologen 13, the kinetics of threading and de-threading were too fast to measure, and therefore the binding constants of this guest in the enantiomers of host H23 were obtained by fluorescence titrations (see the ESI, Tables S4–S6†). The obtained binding constants with guest 13 were slightly higher than the binding constant between 13 and the parent porphyrin macrocyclic host H21 (Table 1, entries 1–3).20 This result implies that the methyl groups, which are inside the cavity, do not have a negative effect on the thermodynamics of the binding process. In fact, a 1H NMR titration of (S,S,S,S)-H23 with viologen 13 showed a remarkably large effect on the methyl protons a and b of the chiral spacers (see the ESI, Fig. S104†). Upon binding the guest, the signals of these protons were deshielded by ∼2 ppm units, indicating that the methyl groups orient themselves out of the cavity, thereby facilitating binding of the guest inside the cavity (see the ESI, Fig. S105†). Apparently, this structural reorganization of the host has no negative effect on the binding strength of 13. An explanation for the slightly enhanced binding may be the presence of stabilizing van der Waals interactions between the bound guest and the spacer methyl groups of the host.:1, v/v) at 298 K
| Entry | Guest | Host | k on (M−1 s−1) | ΔG‡on (kJ mol−1) | ΔΔG‡on (kJ mol−1) | k off (s−1) | ΔG‡off (kJ mol−1) | K assoc (M−1) | ΔGassoc (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Values taken from ref. 20. b Determined by fluorescence titration. c Values taken from ref. 30. d Estimated error 20%. e K assoc = kon/koff, unless stated otherwise, estimated error 30%. See the ESI for details. | |||||||||
| 1a | 13 | H21 | — | — | — | — | — | 6.0 × 105 | −33.0 |
| 2b | 13 | (R,R,R,R)-H23 | — | — | — | — | — | 1.9 × 106 | −35.8 |
| 3b | 13 | (S,S,S,S)-H23 | — | — | — | — | — | 1.6 × 106 | −35.4 |
| 4c | 14 | H21 | 4.0 × 104 | 46.7 | — | 1.4 × 10−3 | 89.3 | 2.9 × 107 | −42.5 |
| 5 | 14 | (R,R,R,R)-H23 | 2.0 × 103 | 54.1 | +0.0 | 1.3 × 10−4 | 95.2 | 1.5 × 107 | −41.0 |
| 6 | 14 | (S,S,S,S)-H23 | 2.0 × 103 | 54.1 | 1.2 × 10−4 | 95.3 | 1.7 × 107 | −41.2 | |
| 7b | (R,R)-15 | H21 | — | — | — | — | — | 8.6 × 106 | −39.6 |
| 8 | (R,R)-15 | (R,R,R,R)-H23 | 4.2 × 104 | 46.6 | +0.1 | 4.0 × 10−3 | 86.7 | 1.1 × 107 | −40.1 |
| 9 | (R,R)-15 | (S,S,S,S)-H23 | 4.4 × 104 | 46.5 | 3.9 × 10−3 | 86.7 | 1.1 × 107 | −40.2 | |
| 10b | (S,S)-15 | H21 | — | — | — | — | — | 1.1 × 107 | −40.1 |
| 11 | (S,S)-15 | (R,R,R,R)-H23 | 4.4 × 104 | 46.5 | +0.1 | 3.7 × 10−3 | 86.9 | 1.2 × 107 | −40.4 |
| 12 | (S,S)-15 | (S,S,S,S)-H23 | 4.5 × 104 | 46.4 | 4.1 × 10−3 | 86.6 | 1.1 × 107 | −40.2 | |
| 13 | (R,R)-16 | H21 | 3.6 × 104 | 47.0 | — | 2.3 × 10−3 | 88.0 | 1.5 × 107 | −41.0 |
| 14 | (R,R)-16 | (R,R,R,R)-H23 | 7.8 × 103 | 50.8 | −1.1 | 4.7 × 10−4 | 92.0 | 1.7 × 107 | −41.2 |
| 15 | (R,R)-16 | (S,S,S,S)-H23 | 4.9 × 103 | 51.9 | 3.7 × 10−4 | 92.6 | 1.3 × 107 | −40.6 | |
| 16 | (S,S)-16 | H21 | 3.7 × 104 | 46.9 | — | 2.4 × 10−3 | 87.9 | 1.5 × 107 | −41.0 |
| 17 | (S,S)-16 | (R,R,R,R)-H23 | 4.7 × 103 | 52.0 | +1.0 | 3.4 × 10−4 | 92.8 | 1.4 × 107 | −40.8 |
| 18 | (S,S)-16 | (S,S,S,S)-H23 | 7.0 × 103 | 51.0 | 4.4 × 10−4 | 92.1 | 1.6 × 107 | −41.1 | |
| 19 | (R,R)-17 | H21 | 1.8 × 103 | 54.4 | — | 1.5 × 10−4 | 94.9 | 1.2 × 107 | −40.4 |
| 20 | (R,R)-17 | (R,R,R,R)-H23 | 7.2 × 101 | 62.4 | +0.0 | 3.1 × 10−5 | 98.7 | 2.3 × 106 | −36.3 |
| 21 | (R,R)-17 | (S,S,S,S)-H23 | 7.1 × 101 | 62.4 | 2.9 × 10−5 | 98.9 | 2.5 × 106 | −36.5 | |
| 22 | (S,S)-17 | H21 | 1.9 × 103 | 54.3 | — | 1.4 × 10−4 | 94.9 | 1.3 × 107 | −40.6 |
| 23 | (S,S)-17 | (R,R,R,R)-H23 | 7.1 × 101 | 62.4 | +0.0 | 2.8 × 10−5 | 99.0 | 2.5 × 106 | −36.5 |
| 24 | (S,S)-17 | (S,S,S,S)-H23 | 7.2 × 101 | 62.4 | 3.0 × 10−5 | 98.8 | 2.4 × 106 | −36.4 | |
| 25 | (R,R)-18 | H21 | 4.5 × 103 | 52.1 | — | 2.6 × 10−4 | 93.4 | 1.7 × 107 | −41.3 |
| 26 | (R,R)-18 | (R,R,R,R)-H23 | 7.2 × 101 | 62.4 | +3.0 | 1.1 × 10−5 | 101.3 | 6.6 × 106 | −38.9 |
| 27 | (R,R)-18 | (S,S,S,S)-H23 | 2.5 × 102 | 59.3 | 2.3 × 10−5 | 99.4 | 1.1 × 107 | −40.1 | |
| 28 | (S,S)-18 | H21 | 5.1 × 103 | 51.8 | — | 2.4 × 10−4 | 93.6 | 2.1 × 107 | −41.8 |
| 29 | (S,S)-18 | (R,R,R,R)-H23 | 2.2 × 102 | 59.6 | −2.9 | 2.4 × 10−5 | 99.3 | 9.2 × 106 | −39.7 |
| 30 | (S,S)-18 | (S,S,S,S)-H23 | 6.7 × 101 | 62.6 | 1.4 × 10−5 | 100.7 | 4.8 × 106 | −38.1 | |
 | ||
| Fig. 3 Chemical structures of viologen guests 13–18. | ||
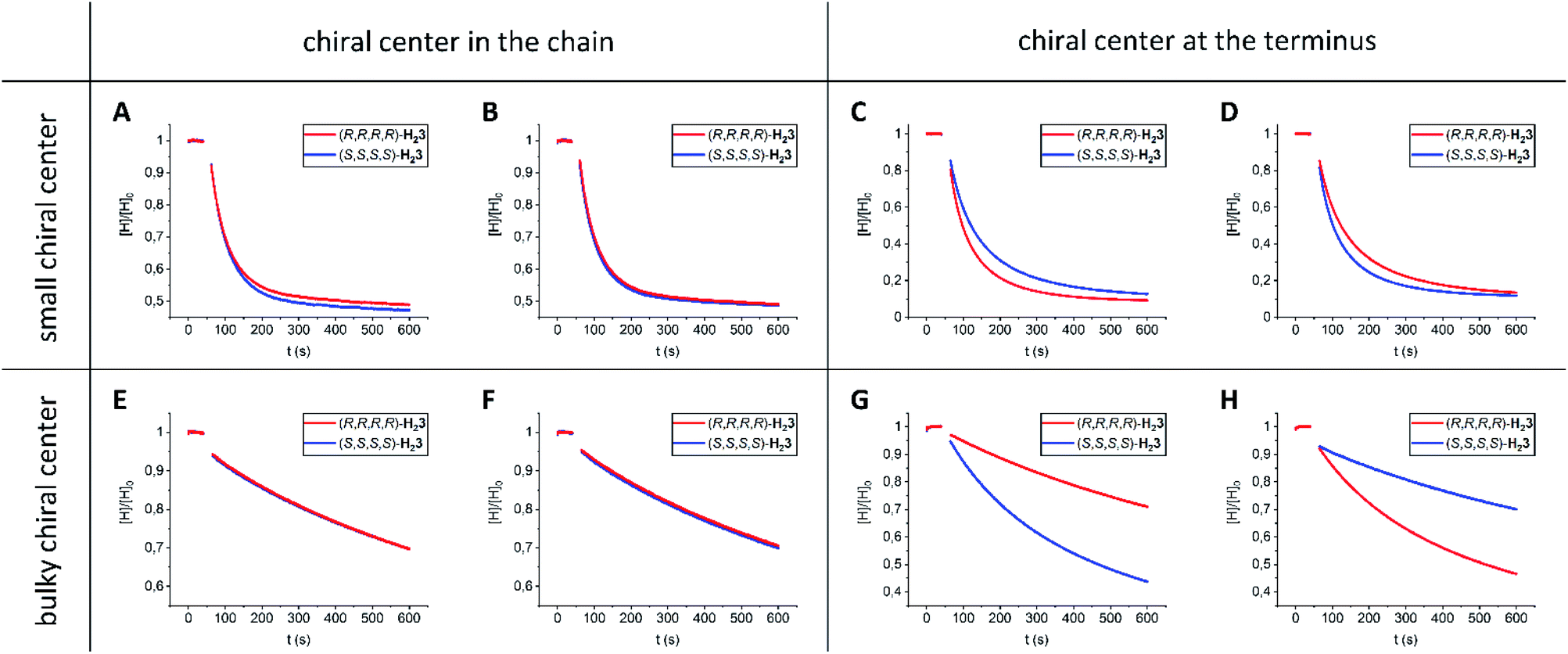 | ||
| Fig. 4 Threading of porphyrin macrocyclic hosts H23 onto chiral viologens 15–18 in CHCl3/CH3CN (1:1, v/v) at 298 K. Normalized fluorescence intensity of hosts H23 as a function of time after the addition (t = 50 s) of 1 equivalent of guest (A) (R,R)-15 (c = 2.5 × 10−7 M), (B) (S,S)-15 (c = 2.5 × 10−7 M), (C) (R,R)-16 (c = 3 × 10−6 M), (D) (S,S)-16 (c = 3 × 10−6 M), (E) (R,R)-17 (c = 10−5 M), (F) (S,S)-17 (c = 10−5 M), (G) (R,R)-18 (c = 10−5 M), and (H) (S,S)-18 (c = 10−5 M). | ||
We then studied the threading of achiral polymer 14 through macrocyclic hosts H2130 and H23 (Table 1, entries 4–6). The kon-value for the threading of polymer 14 through H21 was 20 times higher than that for the threading through the two enantiomers of H23. In addition, the koff-value for the de-threading of polymer 14 from H21 was 14 times higher than that for the de-threading of 14 from the enantiomers of H23. Even though the kinetic processes of both threading through and de-threading from H21 are a lot faster compared to those determined for H23, the on- and off-rates are balanced, resulting in similar Kassoc values. The NMR spectra of the host–guest complexes of H23 with 14 again showed deshielding of methyl protons a and b by ∼2 ppm units (see the ESI, Fig. S106†).
To investigate whether enantiomeric viologens with chiral N-substituents would exhibit stereoselective threading behavior through the cavities of the enantiomers of H23, we examined the threading and de-threading processes of chiral viologens (R,R)-15 and (S,S)-15 (Table 1, entries 7–12).23 The threading experiments (Fig. 4A and B) revealed that the enantiomers of H23 display no kinetic preference for the binding of either (R,R)-15 or (S,S)-15. Also the de-threading (see the ESI, Fig. S60–S67†) of these guests from both enantiomers of H23 occurred at the same rate. As a consequence, there is no thermodynamic preference for the binding of the chiral guests. The binding constants of all complexes H23·15 were verified independently by fluorescence titrations and the values were similar to those of H21·15 (see the ESI, Tables S7–S13†). The 1H NMR spectra of all combinations of host–guest complexes H23·15 were nearly fully superimposable (see the ESI, Fig. S107†). The spectra again showed deshielding of the methyl groups of the chiral spacers of the hosts, as we also observed for the host–guest complexes of H23 with achiral guests. We propose that the lack of kinetic selectivity of chiral hosts H23 for threading onto chiral guests 15 arises from the absence of chirality at the termini of the viologen substituents, i.e. the part of the guest molecule that has to initiate the threading process. Previously, we have shown that the threading of polymeric viologens through H21 involves a kinetically favorable ‘entron’ effect, which involves an internal filling of the cavity of the porphyrin macrocyclic host by the first 5–8 atoms of the polymer chain.30 Starting from the chain termini, the stereogenic centers of viologens 15 only appear at the 6th chain atom. We hypothesize that the remote location of the chirality in the chain inhibits the kinetic stereoselectivity of the threading process, and that the initial entron effect is only governed by the threading of the achiral isopropyl termini of the chains of 15, which are identical for both enantiomers. Alternatively, the lack of stereoselectivity could be due to the lack of steric bulk near the chiral centers of guest 15.
To investigate these hypotheses further, we designed and synthesized chiral guest molecules 16–18 (see the ESI, Scheme S1†). Guest 16 contains small sec-butoxy chiral groups at the chain termini and guest 17 contains bulky 1-(4-octylphenyl)ethoxy chiral groups in the center of the chains. Guest 18 contains a viologen binding station appended at the chain termini with chiral 1-phenylethoxy moieties.
We found that guests 16 (Table 1, entries 13–18) display a kinetic preference of a factor 1.6 (ΔΔG‡on = 1 kJ mol−1) for threading through the enantiomers of H23, i.e. (R,R)-16 prefers (R,R,R,R)-H23 over (S,S,S,S)-H23 and (S,S)-16 prefers (S,S,S,S)-H23 over (R,R,R,R)-H23 (Fig. 4C and D). The small selectivity is attributed to the location of the small chiral moieties at the termini of the guests.
Then, we investigated guests 17 (Table 1, entries 19–24), which contain bulky chiral moieties in locations remote from the chain termini. We found that threading of hosts H23 onto guests 17 proceeded very slowly, i.e. 1–3 orders of magnitude slower than measured for the other investigated guests. This deceleration is caused by the steric bulk in the chains of the guests. The threading experiments (Fig. 4E and F) also indicated that guests 17 do not display kinetic selectivity, which is line with the experiments involving guests 15. Hence, for such selectivity the location of the chiral moiety at the terminus of the guest is pivotal.
In a final set of experiments, we investigated the threading of guests 18 (Table 1, entries 25–30), in which the bulky chiral moieties are positioned at the chain termini. The threading rates of guests 17 and 18 were similar, which is expected based on the similar degree of steric bulk. More interestingly, (R,R)-18 showed a factor 3 (ΔΔG‡on = 3 kJ mol−1) kinetic preference for threading host (S,S,S,S)-H23 over (R,R,R,R)-H23 (Fig. 4G). The opposite kinetic selectivity was observed when guest (S,S)-18 was threaded (Fig. 4H). The results indicate that the chiral environment of the chain termini attached to the viologen moiety determines the extent of kinetic selectivity. Interestingly, de-threading of guests 16–18 from hosts H23 (see the ESI, Fig. S68–S103†) followed the same trends as observed for the corresponding threading processes, i.e. a kinetic preference in the threading process is associated with a kinetic preference in the de-threading process. As a result, the thermodynamic selectivity for all diastereomeric complexes of guests 16–18 with hosts H23 is low, i.e. all binding constants are similar. Moreover, all the host–guest complexes H23·16, H23·17, and H23·18 display similar 1H NMR spectra (see the ESI, Fig. S108–S110†). The methyl protons a and b are again deshielded by ∼2 ppm units. In addition, the aromatic viologen protons are shielded by 2–4 ppm units. The complexation induced shifts (CIS-values) for the protons of the macrocyclic hosts H23 and the protons of the guests 16–18 near the viologen binding station are nearly identical for all host–guest complexes H23·16, H23·17, and H23·18 (see the ESI, Tables S65–S67†). Finally, for all the investigated guests 16–18, the kinetic events of threading and de-threading involving H21 occur faster than those involving H23, but the resulting thermodynamic equilibria remain mostly unaffected.
Conclusions
We have successfully synthesized chiral C2-symmetric porphyrin macrocyclic hosts (R,R,R,R)-H23 and (S,S,S,S)-H23 in an enantioselective fashion from the naturally abundant compounds ethyl (S)-lactate and ethyl (R)-lactate, respectively. The chiral information present in the flexible spacers of (R,R,R,R)-H23 and (S,S,S,S)-H23 is effectively transferred throughout the entire macrocycle up to the porphyrin, as was evidenced by CD spectroscopy and confirmed by DFT calculations. The macrocyclic hosts display the characteristic features of the achiral macrocycle H21, such as binding and threading of viologen guests. While the thermodynamics of binding achiral guests in H21 and H23 are very similar, the underlying kinetics are completely different, since threading of the guests through H23 is significantly slower than threading through H21. This deceleration allows for a more controlled threading process. As we aim to store data on polymers via catalytic reactions on a polymer chain, gaining spatio-temporal control is highly desired. Another step in this direction is the realization of diastereoselectivity in the threading process. Chiral viologen guests 16 and 18, equipped with chiral sec-butoxy and 1-phenylethoxy termini, respectively, display a significant kinetic preference (ΔΔG‡on = 1–3 kJ mol−1) for binding in one enantiomer of H23 over the other. In contrast, dihydrocitronellyl-appended viologens 15 and 1-(4-octylphenyl)ethoxy-appended viologens 17 do not display such a kinetic preference. The reason for this difference in kinetic stereoselectivity is that the chirality of guests 16 and 18 is located at the termini of their chain substituents, which are involved in the initial interaction with the chiral cavities of hosts H23. The rate of the subsequent threading process is dictated by the relative stereochemistry of host and guest. The termini of guests 15 and 17, however, are achiral, and therefore the initial interactions of these termini with the cavities of the enantiomers of hosts H23 are similar. Therefore, we conclude that chirality at the chain terminus of the viologen guest is a prerequisite for realizing kinetic stereoselectivity. This conclusion fits with the previously observed ‘entron’ effect, which states that an initial binding event between the chain terminus of the guest inside the cavity of the host dictates the rate of the threading process.30 A bulky chiral moiety at the chain terminus further enhances the kinetic stereoselectivity, either by steric interactions or by favourable supramolecular interactions of the phenyl substituent. We are currently investigating manganese(III) derivatives of (R,R,R,R)-H23 and (S,S,S,S)-H23, which may potentially be used as processive catalysts for the stereoselective epoxidation of double bond-containing chiral polymer chains.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council (ERC Advanced Grant No. 74092 to R. J. M. N.) and by the Dutch Ministry of Education, Culture, and Science (Gravitation program 024.001.035). We thank Prof. Floris P. J. T. Rutjes for discussions and his interest in this work.References
- A. Stank, D. B. Kokh, J. C. Fuller and R. C. Wade, Acc. Chem. Res., 2016, 49, 809–815 CrossRef CAS.
- M. K. Gilson and H.-X. Zhou, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 21–42 CrossRef CAS.
- Y.-S. Zheng and C. Zhang, Org. Lett., 2004, 6, 1189–1192 CrossRef CAS.
- L. Mutihac, J. H. Lee, J. S. Kim and J. Vicens, Chem. Soc. Rev., 2011, 40, 2777–2796 RSC.
- J. Luo, Q.-Y. Zheng, C.-F. Chen and Z.-T. Huang, Tetrahedron, 2005, 61, 8517–8528 CrossRef CAS.
- Y. Kubo, S. Maeda, S. Tokita and M. Kubo, Nature, 1996, 382, 522–524 CrossRef CAS.
- M. Tlustý, P. Slavík, M. Kohout, V. Eigner and P. Lhoták, Org. Lett., 2017, 19, 2933–2936 CrossRef.
- S. J. Nemat, H. Jędrzejewska, A. Prescimone, A. Szumna and K. Tiefenbacher, Org. Lett., 2020, 22, 5506–5510 CrossRef CAS.
- Q. Huang, L. Jiang, W. Liang, J. Gui, D. Xu, W. Wu, Y. Nakai, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Org. Chem., 2016, 81, 3430–3434 CrossRef CAS.
- W. Xuan, M. Zhang, Y. Liu, Z. Chen and Y. Cui, J. Am. Chem. Soc., 2012, 134, 6904–6907 CrossRef CAS.
- K. Wu, K. Li, Y.-J. Hou, M. Pan, L.-Y. Zhang, L. Chen and C.-Y. Su, Nat. Commun., 2016, 7, 10487 CrossRef CAS.
- T. Liu, Y. Liu, W. Xuan and Y. Cui, Angew. Chem., Int. Ed., 2010, 49, 4121–4124 CrossRef CAS.
- L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC.
- G.-W. Zhang, P.-F. Li, Z. Meng, H.-X. Wang, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2016, 55, 5304–5308 CrossRef CAS.
- B.-Y. Wang, S. Stojanović, D. A. Turner, T. L. Young, C. M. Hadad and J. D. Badjić, Chem.–Eur. J., 2013, 19, 4767–4775 CrossRef CAS.
- S. Varghese, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Sci., 2015, 6, 6050–6058 RSC.
- M. G. T. A. Rutten, F. W. Vaandrager, J. A. A. W. Elemans and R. J. M. Nolte, Nat. Rev. Chem., 2018, 2, 365–381 CrossRef.
- J. A. A. W. Elemans and R. J. M. Nolte, Chem. Commun., 2019, 55, 9590–9605 RSC.
- X. Wang, Q. Gan, B. Wicher, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2019, 58, 4205–4209 CrossRef CAS.
- J. A. A. W. Elemans, M. B. Claase, P. P. M. Aarts, A. E. Rowan, A. P. H. J. Schenning and R. J. M. Nolte, J. Org. Chem., 1999, 64, 7009–7016 CrossRef CAS.
- S. Varghese, B. Spierenburg, A. Swartjes, P. B. White, P. Tinnemans, J. A. A. W. Elemans and R. J. M. Nolte, Org. Lett., 2018, 20, 3719–3722 CrossRef CAS.
- J. Ouyang, A. Swartjes, M. Geerts, P. J. Gilissen, D. Wang, P. C. P. Teeuwen, P. Tinnemans, N. Vanthuyne, S. Chentouf, F. P. J. T. Rutjes, J.-V. Naubron, J. Crassous, J. A. A. W. Elemans and R. J. M. Nolte, Nat. Commun., 2020, 11, 4776 CrossRef CAS.
- S. Varghese, B. Spierenburg, J. P. J. Bruekers, A. Swartjes, P. B. White, J. A. A. W. Elemans and R. J. M. Nolte, Eur. J. Org. Chem., 2019, 3525–3533 CrossRef CAS.
- U. Burkard and F. Effenberger, Chem. Ber., 1986, 119, 1594–1612 CrossRef CAS.
- S. Müller, C. M. Afraz, R. De Gelder, G. J. A. Ariaans, B. Kaptein, Q. B. Broxterman and A. Bruggink, Eur. J. Org. Chem., 2005, 1082–1096 CrossRef.
- Y. Nishiyama, Y. Han-ya, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2014, 136, 6598–6601 CrossRef CAS.
- S. Chacko, H. I. M. Boshoff, V. Singh, D. M. Ferraris, D. R. Gollapalli, M. Zhang, A. P. Lawson, M. J. Pepi, A. Joachimiak, M. Rizzi, V. Mizrahi, G. D. Cuny and L. Hedstrom, J. Med. Chem., 2018, 61, 4739–4756 CrossRef CAS.
- P. J. Gilissen, A. Swartjes, B. Spierenburg, J. P. J. Bruekers, P. Tinnemans, P. B. White, F. P. J. T. Rutjes, R. J. M. Nolte and J. A. A. W. Elemans, Tetrahedron, 2019, 75, 4640–4647 CrossRef CAS.
- R. G. E. Coumans, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19647–19651 CrossRef CAS.
- A. B. C. Deutman, C. Monnereau, J. A. A. W. Elemans, G. Ercolani, R. J. M. Nolte and A. E. Rowan, Science, 2008, 322, 1668–1671 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, kinetic data, DFT calculations, and copies of NMR spectra of all new compounds. See DOI: 10.1039/d0sc05233g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |