
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic resolution of racemic allylic alcohols via iridium-catalyzed asymmetric hydrogenation: scope, synthetic applications and insight into the origin of selectivity†
Haibo
Wu
 a,
Cristiana
Margarita
a,
Jira
Jongcharoenkamol‡
a,
Mark D.
Nolan
a,
Thishana
Singh
b and
Pher G.
Andersson
*ab
a,
Cristiana
Margarita
a,
Jira
Jongcharoenkamol‡
a,
Mark D.
Nolan
a,
Thishana
Singh
b and
Pher G.
Andersson
*ab
aDepartment of Organic Chemistry, Stockholm University, Arrhenius Laboratory, 106 91, Stockholm, Sweden. E-mail: pher.andersson@su.se
bSchool of Chemistry and Physics, University of Kwazulu-Natal, Private Bag X54001, Durban, 4000, South Africa
First published on 8th December 2020
Abstract
Asymmetric hydrogenation is one of the most commonly used tools in organic synthesis, whereas, kinetic resolution via asymmetric hydrogenation is less developed. Herein, we describe the first iridium catalyzed kinetic resolution of a wide range of trisubstituted secondary and tertiary allylic alcohols. Large selectivity factors were observed in most cases (s up to 211), providing the unreacted starting materials in good yield with high levels of enantiopurity (ee up to >99%). The utility of this method is highlighted in the enantioselective formal synthesis of some bioactive natural products including pumiliotoxin A, inthomycin A and B. DFT studies and a selectivity model concerning the origin of selectivity are presented.
Introduction
The demand for enantiomerically pure alcohols is steadily increasing. In particular, chiral allylic alcohols have tremendous synthetic relevance to natural products, pharmaceuticals, agricultural chemicals and specialty materials.1 Kinetic resolution (KR) is a useful and direct approach to access such compounds, in a manner where they are obtained in high enantiopurity, from inexpensive racemic starting material.1c Notably, KR is also one of the most common methods that is used to prepare optically active alcohols on an industrial scale.2Transition-metal catalyzed asymmetric hydrogenation3 is one of the most efficient and well-established transformations in asymmetric catalysis. Owing to its perfect atom economy and excellent enantioselectivity, this process was frequently used in the preparation of enantiomericially enriched compounds in both academia and industry. However, when compared to other fundamental transformations such as epoxidation4 and acylation,5 its application in kinetic resolution is still a far less explored and challenging task. Pioneering work in this field was described by Noyori et al. in 1988 using the Ru–BINAP catalyst system (Fig. 1a), which was initially found to be efficient for few aliphatic cyclic substrates.6 In 2015, Zhou et al. reported an impressive KR of saturated aliphatic alcohols via iridium catalyzed asymmetric hydrogenation of ester (Fig. 1b),7 giving excellent selectivity and high efficiency without the need to convert the OH group to other functionalities. By using rhodium catalysts, Vidal-Ferran et al. used rhodium catalysts to develop the KR of racemic vinyl sulfoxides and vinyl phosphane oxides via hydrogenation of the terminal olefin (Fig. 1c).8 Despite this progress, there is still no general hydrogenation protocol available for KR of a wide range of allylic alcohols.
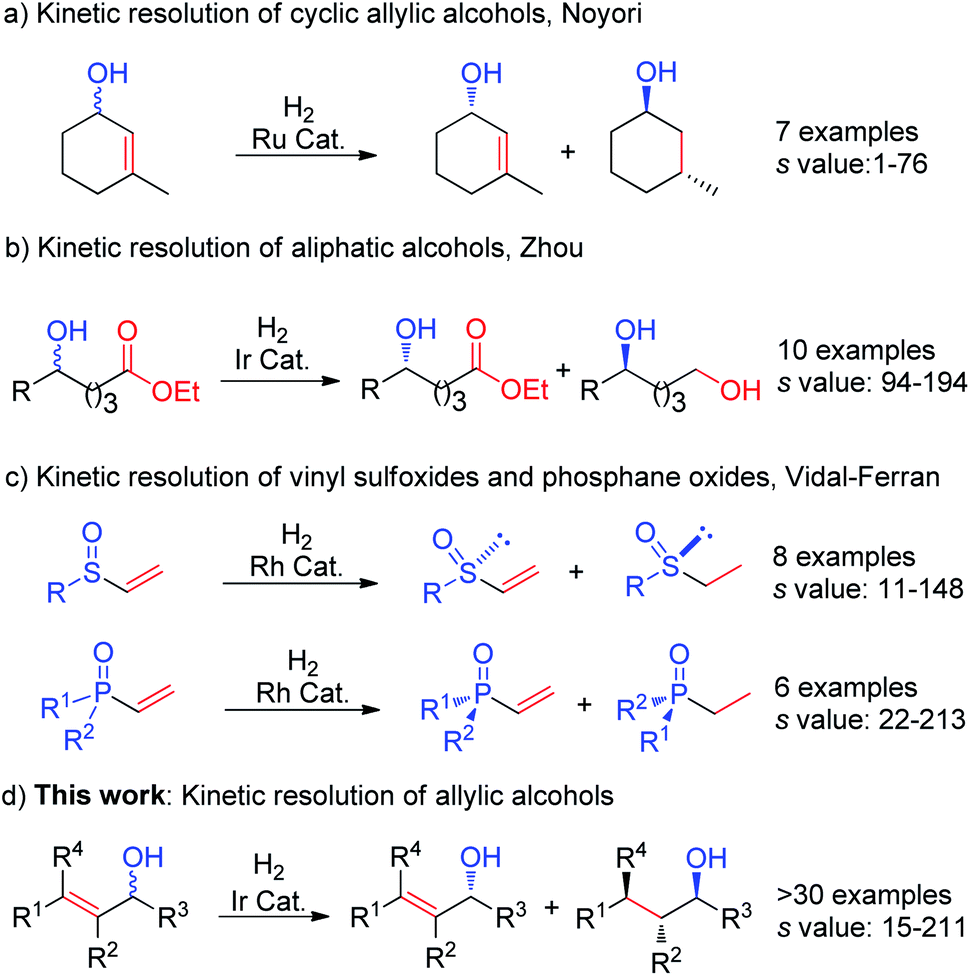 | ||
| Fig. 1 Kinetic resolution via asymmetric hydrogenation. | ||
Over the past two decades, our group has developed a variety of iridium N,P complexes, that are successfully employed in the asymmetric hydrogenation of different types of olefins with different functionalities.9 As a continuous interest, further application of asymmetric hydrogenation in kinetic resolution of allylic substrates is highly desired. Iridium catalyzed asymmetric hydrogenation of primary allylic alcohols has been well developed.10 The secondary or tertiary allylic alcohols are relatively acid-senstive, and are challenging substrates10 for iridium catalyzed asymmetric hydrogenation due to the Lewis or Brønsted acidity of these transition-metal–hydride complexes.11 There are several known competing transformations such as allylic substitution,12 elimination,13 and isomerization14 that are likely to occur when the iridium hydride species is involved. Additionally, the selective hydrogenation of one olefin in the presence of another olefin is still a generally unsolved but useful task.15 Herein, we disclose the first iridium-catalyzed KR of racemic secondary and tertiary allylic alcohols via asymmetric hydrogenation.
Results and discussion
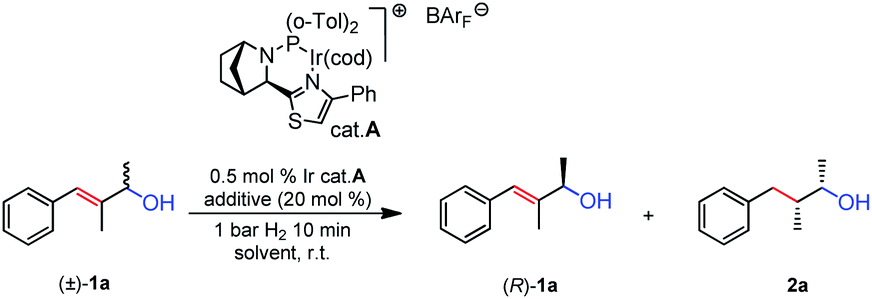
We began our investigation using racemic allylic alcohol 1a and the Ir-N,P thiazole-based catalyst A in the screening (Table 1). Preliminary results without an additive (entry 1) showed the allylic alcohol was hydrogenated with high conversion (89%) under 1 bar of H2 and 0.5% catalyst loading. The remaining starting material had very low ee (11%). When AcOH was used as the additive (entry 2), the conversion was similar (91%), and the remaining alcohol 1a did not show any enantiomeric enrichment. These results indicated that, even in the absence of an acid, the acidity of the Ir-N,P catalyst under hydrogenation conditions was sufficient to enable the process of carbocation formation. To our delight, with the addition of a small amount of base, the reaction proceeded cleanly, we observed only the hydrogenated product and the remaining starting material in the reaction mixture. A number of different bases were screened, either at 10 or 20 mol% loading. The use of K3PO4 (entry 3) provided 43% conversion and 63% ee of 1a, which corresponded to kinetic resolution with a good level of selectivity (Table 1). Then KOAc was evaluated (entry 4) and even at a lower loading, it afforded a slightly better result than the previous one, increasing the selectivity of the reaction to s = 24. The use of K2CO3 had a significant effect and enhanced the KR performance (entry 5). With 20 mol% base a conversion slightly higher than ideal (55%) was observed, and 1a was resolved to an excellent ee. This corresponded to the highest selectivity factor for this substrate (s = 26). Two other bases were screened (entries 6 and 7) and good results were obtained with regard to conversion and the s factor, however they did not outperform K2CO3. With the best basic additive chosen, a screening of a few different solvents was carried out. When the reaction was run in benzene (entry 8) it gave a comparable outcome to that in toluene but the overall selectivity was not improved. The use of CH2Cl2 was completely detrimental to the process (entry 9): even when the reaction was stopped at 55% conversion, the remaining alcohol 1a was present only in 38% ee. When α,α,α-trifluorotoluene was tested (entry 10), the system gave good selectivity (s = 21), but with lower conversion and enantiopurity of 1a (79% ee). Eventually, the reaction was carried out on a preparative scale (0.2 mmol) with 10 mol% K2CO3 (entry 11), and gave similar selectivity to that of the smaller scale (entry 5).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Additive | Conv.b (%) | ee of 1ac (%) | s |
| a Reaction conditions: (±)-1a (0.05 mol), 0.5 mol% catalyst and 20 mol% additive in the solvent (1.0 mL) under 1 bar H2 at room temperature for 10 min, unless otherwise specified. b Conversion was determined by 1H NMR spectroscopy, the combined recovery ratio of 1a and 2a >99%, unless specified in parentheses. c Enantiomeric excesses were determined by SFC analysis. d The selectivity factors: s = ln[(1 − conv.)(1 − ee)]/ln[(1 − conv.)(1 + ee)]. e 3 min reaction time. f 10 mol% additive. g 0.2 mmol scale reaction. | |||||
| 1 | Toluene | None | 89(81) | 11.0 | 1.1 |
| 2e,f | Toluene | HOAc | 91(78) | 0.5 | 1.0 |
| 3 | Toluene | K3PO4 | 43 | 62.5 | 20.5 |
| 4f | Toluene | KOAc | 44 | 65.7 | 23.6 |
| 5 | Toluene | K2CO3 | 56 | 94.6 | 25.9 |
| 6 | Toluene | KHCO3 | 54 | 85.5 | 17.2 |
| 7 | Toluene | Na2CO3 | 54 | 89.7 | 21.4 |
| 8 | Benzene | K2CO3 | 58 | 97.7 | 24.6 |
| 9e | CH2Cl2 | K2CO3 | 55(75) | 37.9 | 2.6 |
| 10 | PhCF3 | K2CO3 | 50 | 79.0 | 21.4 |
| 11f,g | Toluene | K2CO3 | 60 | 99.0 | 24.2 |
We then evaluated the allylic alcohol scope (Table 2), with a systematic study on the substitution covering a range of different functionalities. The substrates that have different substituents on the allylic position were found to be suitable candidates for the Ir-catalyzed hydrogenation/KR, and excellent ees and high s values were obtained in most cases (1b–f).
| a Reaction conditions: (±)-substrate 1 (0.2 mmol), 0.2–1.0 mol% catalyst A and 10 mol% K2CO3 in toluene (1.0 mL) under 1–3 bar H2 at room temperature for 10 min to 1 h, unless otherwise specified in the ESI. Isolated yield. Conversion was determined by 1H NMR spectroscopy. The selectivity factors: s = ln[(1 − conv.)(1 − ee)]/ln[(1 − conv.)(1 + ee)]. Enantiomeric excesses were determined by SFC or GC analysis. b Isolated as a mixture with hydrogenated product. c Catalyst B was used. |
|---|
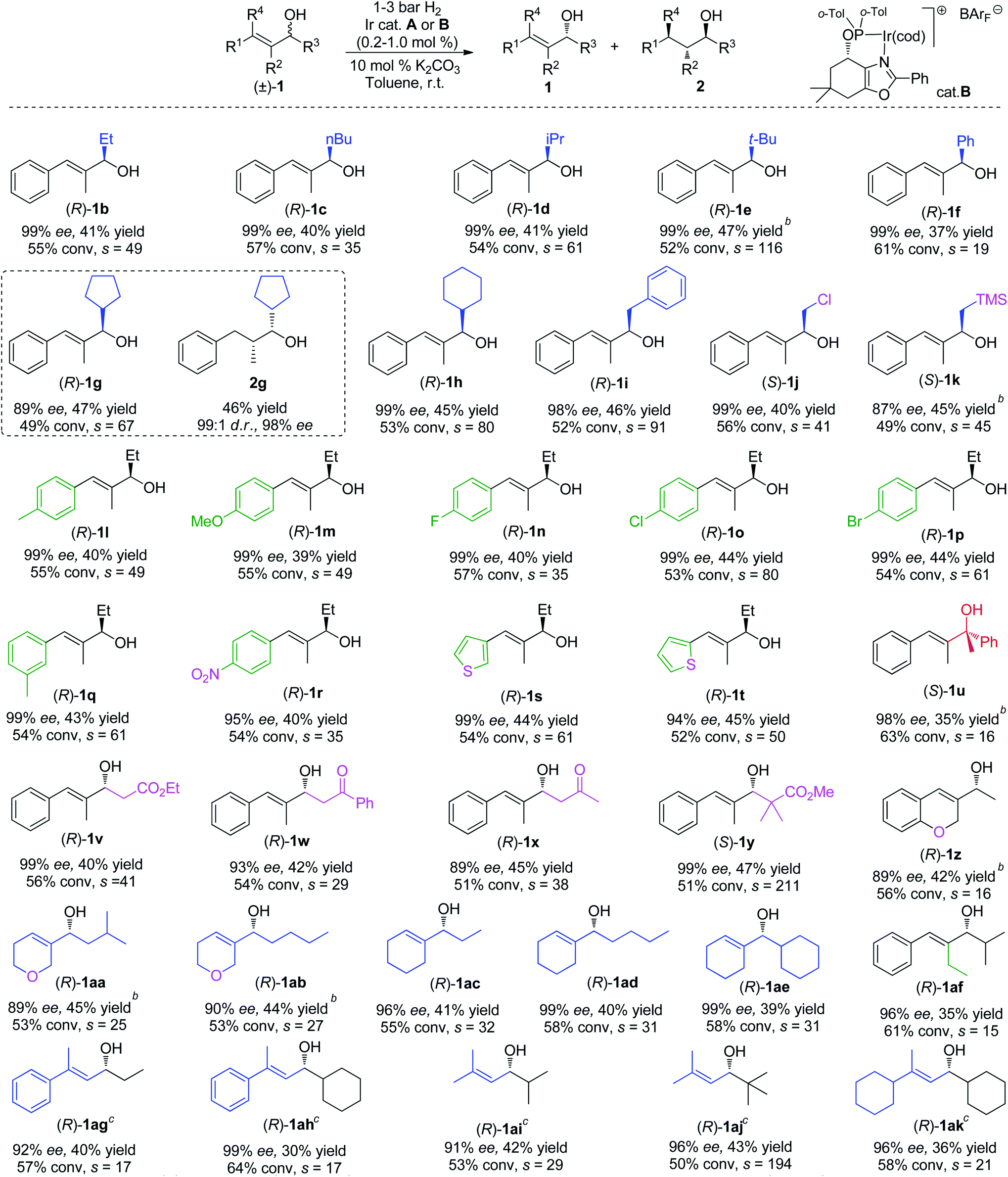
|
These results also indicate how the large steric bulk influences the catalyst in discriminating between the two enantiomers of the alcohol. The current method could be also applied in the synthesis of saturated alcohols with excellent diastereo- and enantio-selectivity. As an illustrative example, substrate 1g was resolved with slightly lower conversion affording the hydrogenated product 2g in 46% isolated yield with excellent d.r. (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and ee (98%). From the measurement of the optical rotation of an isolated sample of resolved compound 1b and by comparison with literature values,16 the assigned absolute configuration for the alcohol was determined to be (R). The stereochemistry of the other resolved alcohols was assigned by analogy. The investigation continued with the evaluation of different functionalities (1j, 1k) such as –CH2Cl, –CH2TMS,17 which were well tolerated by the system and gave good selectivity.
:1) and ee (98%). From the measurement of the optical rotation of an isolated sample of resolved compound 1b and by comparison with literature values,16 the assigned absolute configuration for the alcohol was determined to be (R). The stereochemistry of the other resolved alcohols was assigned by analogy. The investigation continued with the evaluation of different functionalities (1j, 1k) such as –CH2Cl, –CH2TMS,17 which were well tolerated by the system and gave good selectivity.
Substrates (1l–r) bearing an electron-withdrawing group or electron-donating group at the para or meta position on the phenyl ring were successfully resolved, affording excellent ee for the remaining starting material. However, the ortho-substituted analogue of 1l cannot be kinetically resolved, probably due to the unfavorable steric effect. Good results were also achieved for the heteroaromatic compounds 1s and 1t containing a thiophene ring. Surprisingly, the challenging tertiary alcohol 1u, which is a sensitive substrate under hydrogenation conditions, was resolved (98% ee) at an higher conversion (63%).
Next, a set of allylic alcohols with a β-carbonyl functionality were also evaluated in our KR system, given their high synthetic utility. The first substrate in this class was ethyl ester 1v. The Ir-N,P catalyst accomplished efficient kinetic resolution (99% ee) at 56% conversion, corresponding to a high selectivity (s = 41). Good results were also observed in the KR of ketones 1w and 1x, with relatively high s factors. When the bulky alcohol 1y was applied in this hydrogenation/KR strategy, excellent selectivity (s = 211) was obtained. Cyclic substrate 1z with a methyl group on the allylic position also proceeded with lower selectivity (s = 16). We encountered lower selectivity again when manipulating the substitution on the olefin (1af). Several fully aliphatic allylic alcohols (1aa–ae) were tested using the method as well. To our delight, good selectivity was achieved for these substrates and good to excellent ee was attained when the reaction conversion was controlled to a range of 55% to 60%. Finally, a few β,β-substituted allylic alcohols (1ag–1ak) including both aromatic and pure aliphatic substrates were tested using the oxazole-type catalyst B. High to excellent selectivities (s factor up to 194) were obtained.
Different stereoisomers of a biologically active molecule can have completely different effects in biological systems.18 It is well known that the inherent problem of the KR process is that only a 50% maximum isolated yield is permitted. The excellent enantio-discriminating ability of our catalytic system in the hydrogenation of racemic allylic alcohols, used all the starting materials. Hence, we continued to explore this double stereo-differentiation potential, to provide both enantiomers of saturated secondary alcohol 2i bearing two contiguous chiral centers (Scheme 1). Resolved alcohol (R)-1i was subjected to secondary hydrogenation using catalyst ent-A without an additive under 3 bar of hydrogen for 10 minutes. This afforded saturated alcohol (2R,3S)-2i with excellent diastereo- and enantioselectivity (>99:1 d.r., 99% ee). Beginning with racemic allylic alcohol 1i, the combined hydrogenations afforded clean reactions with 96% overall yield of the two separated enantiomers.
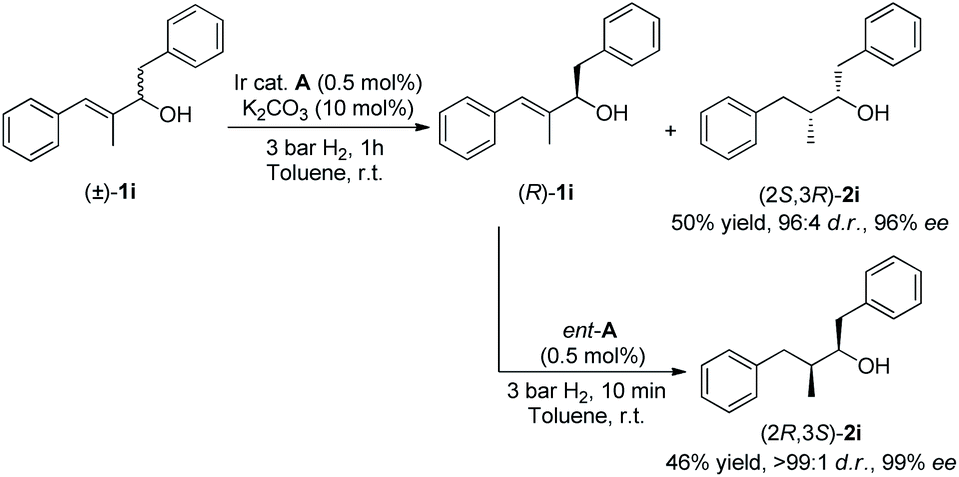 | ||
| Scheme 1 Double stereo-differentiation. | ||
In order to demonstrate the utility of this approach, we have performed a gram-scale kinetic resolution of allylic alcohol 1b with 0.2 mol% catalyst loading under 1 bar hydrogen pressure for 30 minutes, and the resolved allylic alcohol was obtained with 99% ee at 54% conversion (Scheme 2). The crude product was benzylated and underwent ozonolysis to yield methyl ketone 3, which was previously synthesized in 7 steps from glycidol.19a This is a key intermediate for the total synthesis of pharmacologically active dendrobatid alkaloid pumiliotoxin A.19b
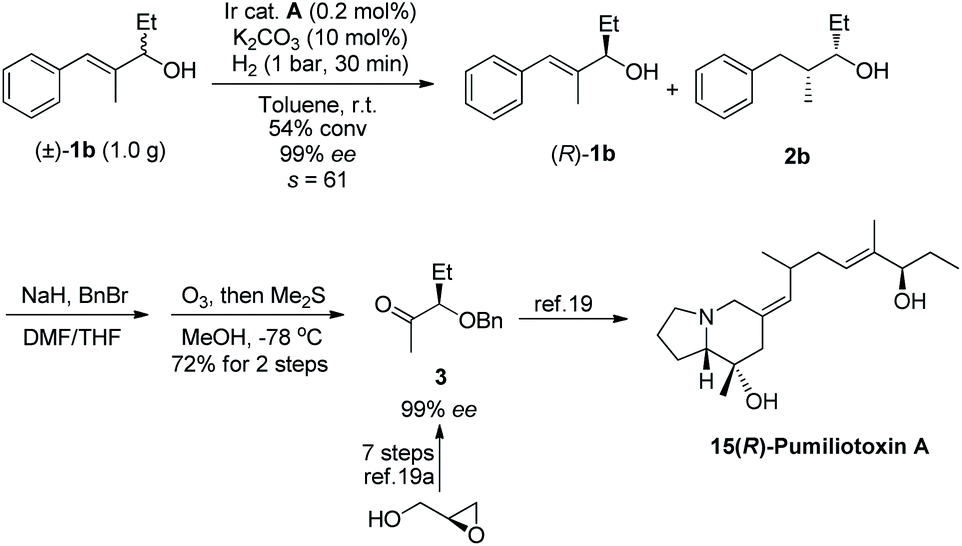 | ||
| Scheme 2 Gram-scale kinetic resolution and concise synthesis of the chiral building block of 15(R)-pumiliotoxin A. | ||
Encouraged by the above success, we then turned our attention to the synthesis of the natural products inthomycin A and B with our new method as a key step (Scheme 3). The inthomycins, a small family of polyene natural products were first isolated from Streptomyces sp.20 have attracted considerable attention from synthetic chemists21 due to their unique structural features and interesting bioactivities.22 Our synthesis began with KR of racemic alcohol 1y, which has already showed excellent selectivity on a small scale affording the resolved alcohol with over 99% ee at 52% conversion (s > 100). Subsequent TBS protection and ozonolysis delivered the enantioenriched ketone 4 in 38% overall yield. Surprisingly, the Horner–Wadsworth–Emmons reaction of methyl ketone 4 with dimethyl 3-trimethylsilylpropynyl phosphonate23 followed by a deprotection of TMS afforded the Z-enyne 5 in 81% yield with an unprecedented exclusive stereoselectivity.
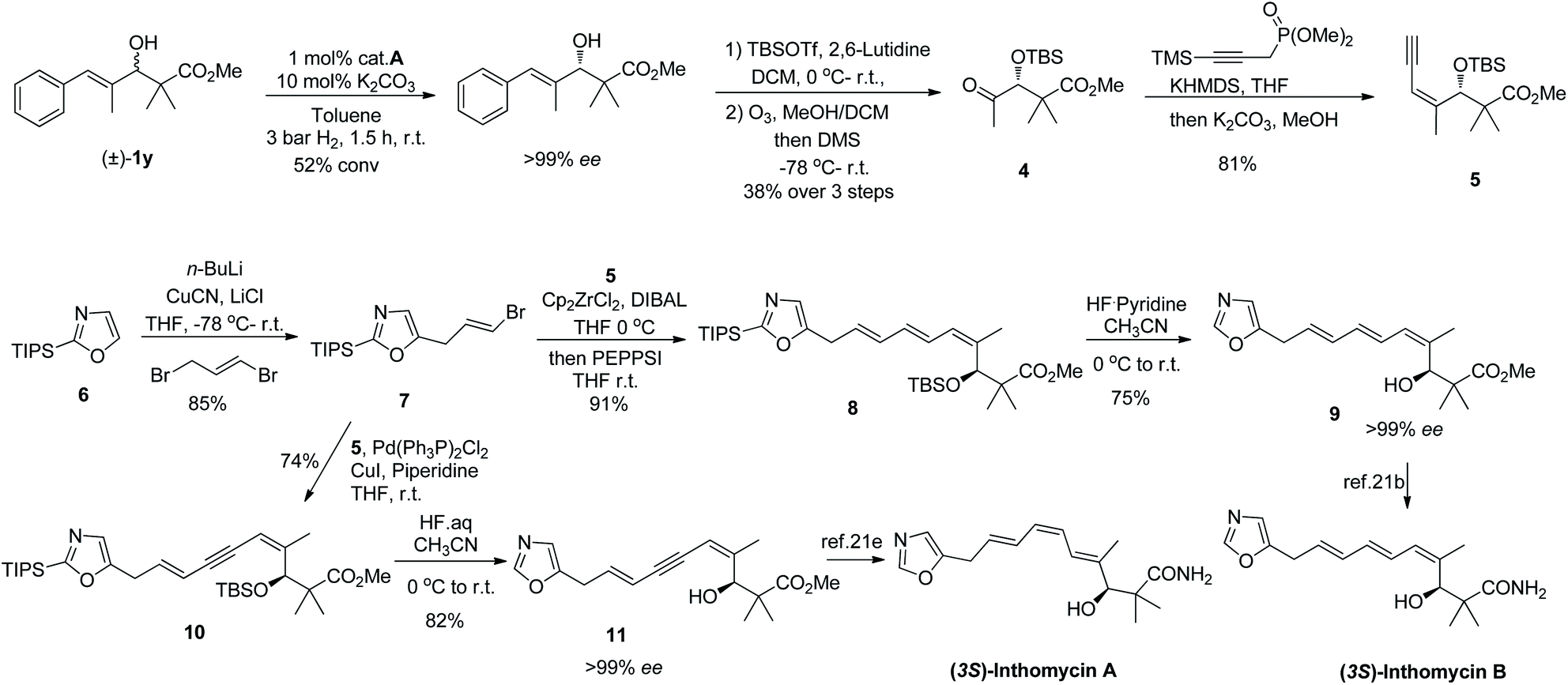 | ||
| Scheme 3 Enantioselective formal total synthesis of inthomycin A and B. | ||
Separately, lithiation of TIPS protected oxazole 621g followed by allylation at the C-5 position to give vinyl bromide 7 in high yield. With 5 and 7 in hand, we then focused on coupling the two fragments corresponding to the geometry of the target inthomycins. Firstly, using a modified version of Negishi's method,24 enyne 5 was subjected to hydrozirconation and then coupled with 7 to give the triene 8 in good yield with excellent stereoselectivity. After desilylation of 8, we obtained the reported alcohol 921b,c with over 99% ee, thus achieving the formal total synthesis of (3S)-inthomycin B. It should be noted that, fragments 5 and 7 was also connected by Sonogashira cross-coupling to afford intermediate 10, which was further converted to alcohol 1121e after deprotection. Over 99% ee was also achieved for alcohol 11 and this constitutes the formal enantioselective synthesis of inthomycin A. This synthetic sequence provides one of the most efficient routes to the inthomycins, with remarkable selectivity.
To rationalize the origin of the enantio-discrimination, a quadrant model analysis correlated with preliminary density functional theory (DFT) studies were performed (Fig. 2).10,25 According to a previously developed quadrant model, the iridium coordination sphere was divided into four planar quadrants based on the calculated catalyst structure. When the olefin substrate was fitted into this model (Fig. 2b), the smallest substituent (H atom) always occupies the most hindered quadrant, resulting in a fixed conformation for the transition state. As shown in the three-dimensional quadrant model (Fig. 2c), there is possible hydrogen bonding between the hydroxyl group and the axial iridium hydride which may account for the enantio-discrimination, since the corresponding calculated distance is around 2.5 Å for both enantiomers.
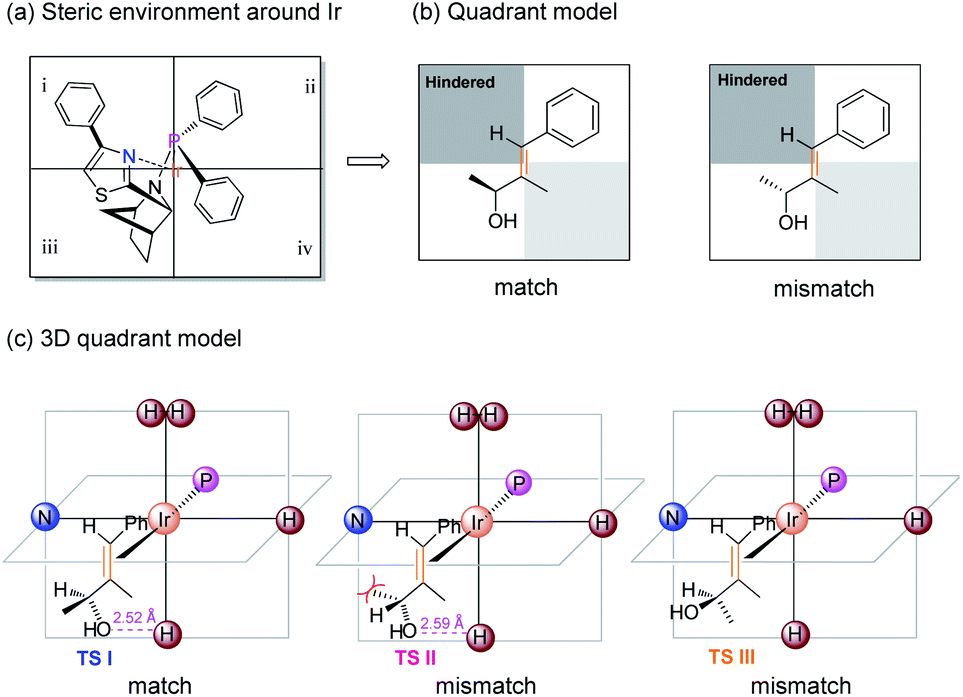 | ||
| Fig. 2 Origin of selectivity depicting: (a) steric environment around Ir. (b) Quadrant model illustrating the matched and mismatched allylic alcohol. (c) 3D quadrant model. | ||
Further relative energy calculations for the possible transition states were conducted (Fig. 3). For transition state TS I of the matched enantiomer (S)-1a, hydrogen bonding leads to a conformation such that the methyl group at the carbinol points away from the ligand backbone. In contrast, hydrogen bonding in transition state TS II of the matched enantiomer (R)-1a results in the methyl group pointing forward to the ligand backbone, which is not favored for steric reasons. The cost of this steric clash resulted in an energy difference of 3.0 kcal mol−1 for the two transition states. This result also indicates that substrates with a bulky substituent would produce better selectivity, which is consistent with the experimental finding. Another possible transition state TS III for the mismatched enantiomer, adopts a less sterically hindered mode of coordination with the catalyst. Here the hydrogen bond breaks, resulting in an even higher energy barrier. Additionally, the absolute configurations obtained for the recovered allylic alcohols are in agreement with the theoretical prediction.
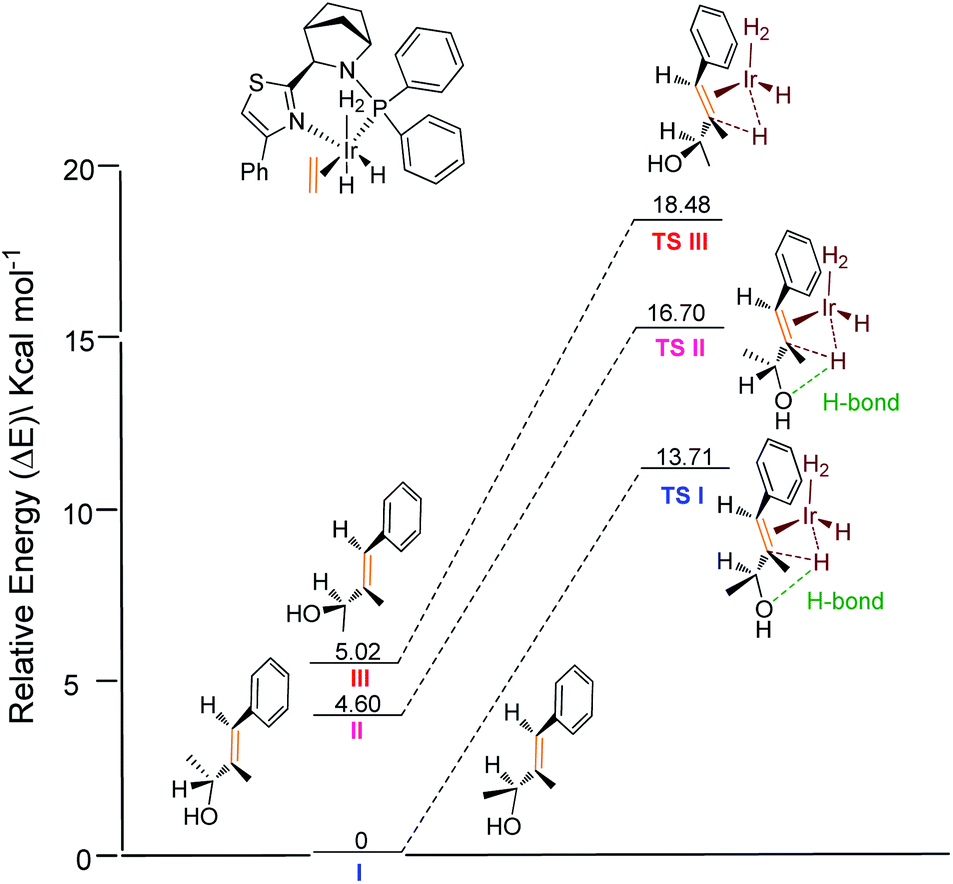 | ||
| Fig. 3 Calculated relative energy profile. | ||
Conclusions
To summarize, we have developed an efficient kinetic resolution protocol for a variety of trisubstituted allylic alcohols via Ir-N,P-catalyzed asymmetric hydrogenation. High selectivity factors were observed using this methodology. These compare well with those reported for other KR systems, especially taking into consideration the mild reaction conditions, short reaction times and operational simplicity. The utility of this strategy is illustrated by the concise formal synthesis of bioactive 15(R)-pumiliotoxin A, (3R)-inthomycin A and B. DFT calculations and the quadrant model analysis indicated that a medium-to-strong hydrogen bond between the alcohol and the iridium center is responsible for the selectivity. This kinetic resolution of diverse allylic alcohols via asymmetric hydrogenation provides new and exciting opportunities for enantioselective synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Swedish Research Council (VR), the Knut och Alice Wallenberg Stiftelsen for funding this research. J. J. thanks the Chulabhorn Graduate Institute, Chulabhorn Royal Academy, Thailand for an exchange scholarship to Stockholm University. T. S. acknowledges the NRF South Africa for the Post-PhD Track Thuthuka Grant. All computations were carried out using the computational cluster resources at the National Supercomputer Centre based at Linkoping University, Sweden and at the Center for High Performance Computing, South Africa.Notes and references
- For recent reviews, see: (a) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394 CrossRef CAS; (b) H. Jiang, N. Holub and K. Anker Jørgensen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20630 CrossRef CAS; (c) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef CAS.
- For reviews, see: (a) T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Chem. Rec., 2016, 16, 2754 CrossRef; (b) X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272 CrossRef CAS; (c) C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346 CrossRef CAS; (d) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS; (e) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS; (f) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS; (g) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS.
- For a review, see (a) M. M. Heravi, T. B. Lashaki and N. Poorahmad, Tetrahedron: Asymmetry, 2015, 26, 405 CrossRef CAS. For some recent synthetic examples, see: (b) H. Li, S.-J. Shen, C.-L. Zhu and H. Xu, J. Am. Chem. Soc., 2018, 140, 10619 CrossRef CAS; (c) D. Katsumi, K. Nakasone, N. Terayama, E. Yasui, M. Mizukami, M. Miyashita and S. Nagumo, J. Org. Chem., 2019, 84, 1553 CrossRef CAS; (d) L. Jeanne-Julien, G. Masson, E. Astier, G. Genta-Jouve, V. Servajean, J.-M. Beau, S. Norsikian and E. Roulland, Org. Lett., 2017, 19, 4006 CrossRef CAS; (e) M. Kanto and M. Sasaki, Org. Lett., 2016, 18, 112 CrossRef CAS; (f) B. Seetharamsingh, P. R. Rajamohanan and D. S. Reddy, Org. Lett., 2015, 17, 1652 CrossRef CAS; (g) S. Tang, Y.-L. Deng, J. Li, W.-X. Wang, G.-L. Ding, M.-W. Wang, Z.-P. Xiao, Y.-C. Wang and R.-L. Sheng, J. Org. Chem., 2015, 80, 12599 CrossRef CAS; (h) Y.-G. Wang, R. Takeyama and Y. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 3320 CrossRef CAS.
- (a) J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492 CrossRef CAS; (b) E. Vedejs and X. Chen, J. Am. Chem. Soc., 1996, 118, 1809 CrossRef CAS; (c) S. Bellemin-Laponnaz, J. Tweddell, J. C. Ruble, F. M. Breitling and G. C. Fu, Chem. Commun., 2000, 12, 1009 RSC.
- M. Kitamura, I. Kasahara, K. Manabe, R. Noyori and H. Takaya, J. Org. Chem., 1988, 53, 708 CrossRef CAS.
- X.-H. Yang, K. Wang, S.-F. Zhu, J.-H. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2014, 136, 17426 CrossRef CAS.
- (a) H. Fernández-Pérez and A. Vidal-Ferran, Org. Lett., 2019, 21, 7019 CrossRef; (b) J. R. Lao, H. Fernández-Pérez and A. Vidal-Ferran, Org. Lett., 2015, 17, 4114 CrossRef CAS.
- For selected examples, see: (a) K. Källström, C. Hedberg, P. Brandt, A. Bayer and P. G. Andersson, J. Am. Chem. Soc., 2004, 126, 14308 CrossRef; (b) P. Cheruku, J. Diesen and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 5595 CrossRef CAS; (c) A. Paptchikhine, P. Cheruku, M. Engman and P. G. Andersson, Chem. Commun., 2009, 5996 RSC; (d) J. J. Verendel, T. Zhou, J.-Q. Li, A. Paptchikhine, O. Lebedev and P. G. Andersson, J. Am. Chem. Soc., 2010, 132, 8880 CrossRef CAS; (e) B. K. Peters, J. Liu, C. Margarita, W. Rabten, S. Kerdphon, A. Orebom, T. Morsch and P. G. Andersson, J. Am. Chem. Soc., 2016, 138, 11930 CrossRef CAS; (f) J. Liu, S. Krajangsri, J. Yang, J.-Q. Li and P. G. Andersson, Nat. Catal., 2018, 1, 438 CrossRef CAS; (g) S. Ponra, W. Rabten, J. Yang, H. Wu, S. Kerdphon and P. G. Andersson, J. Am. Chem. Soc., 2018, 140, 13878 CrossRef CAS; (h) S. Kerdphon, S. Ponra, J. Yang, H. Wu, L. Eriksson and P. G. Andersson, ACS Catal., 2019, 9, 6169 CrossRef CAS.
- J.-Q. Li, J. Liu, S. Krajangsri, N. Chumnanvej, T. Singh and P. G. Andersson, ACS Catal., 2016, 6, 8342 CrossRef CAS.
- (a) R. G. Pearson, Chem. Rev., 1985, 85, 41 CrossRef CAS; (b) G. Jia and C.-P. Lau, Coord. Chem. Rev., 1999, 190–192, 83 CrossRef CAS; (c) Y. Zhu, Y. Fan and K. Burgess, J. Am. Chem. Soc., 2010, 132, 6249 CrossRef CAS; (d) Y. Zhu and K. Burgess, Adv. Synth. Catal., 2008, 350, 979 CrossRef CAS; (e) Y. Zhu and K. Burgess, RSC Adv., 2012, 2, 4728 RSC.
- For some reviews, see: (a) J. F. Hartwig and M. J. Pouy, in Iridium Catalysis, ed. P. G. Andersson, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 169–208 Search PubMed; (b) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539 CrossRef CAS; (c) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855 CrossRef CAS.
- H. Li, D. Fiorito and C. Mazet, ACS Catal., 2017, 7, 1554 CrossRef CAS.
- (a) E. Erbing, A. Vázquez-Romero, A. Bermejo Gómez, A. E. Platero-Prats, F. Carson, X. Zou, P. Tolstoy and B. Martín-Matute, Chem.–Eur. J., 2016, 22, 15659 CrossRef CAS; (b) H. Li and C. Mazet, Acc. Chem. Res., 2016, 49, 1232 CrossRef CAS; (c) J.-Q. Li, B. Peters and P. G. Andersson, Chem.–Eur. J., 2011, 17, 11143 CrossRef CAS.
- (a) J. Liu, S. Krajangsri, T. Singh, G. De Seriis, N. Chumnanvej, H. Wu and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 14470 CrossRef CAS; (b) C. Margarita, W. Rabten and P. G. Andersson, Chem.–Eur. J., 2018, 24, 8022 CrossRef CAS.
- (a) S. Duce, M. Jorge, I. Alonso, J. L. G. Ruano and M. B. Cid, Eur. J. Org. Chem., 2013, 7067 CrossRef CAS; (b) L. Pisani, S. Superchi, A. D'Elia, P. Scafato and C. Rosini, Tetrahedron, 2012, 68, 5779 CrossRef CAS.
- S. Krajangsri, H. Wu, J. Liu, W. Rabten, T. Singh and P. G. Andersson, Chem. Sci., 2019, 10, 3649 RSC.
- S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353 CrossRef CAS.
- (a) S. Aoyagi, S. Hirashima, K. Saito and C. Kibayashi, J. Org. Chem., 2002, 67, 5517 CrossRef CAS; (b) N.-H. Lin, L. E. Overman, M. H. Rabinowitz, L. A. Robinson, M. J. Sharp and J. Zablocki, J. Am. Chem. Soc., 1996, 118, 9062 CrossRef CAS.
- T. Henkel and A. Zeeck, Liebigs Ann. Chem., 1991, 1991, 367 CrossRef.
- Two racemic and two enantioselective reported routes for synthesis of inthomycin A, three reported enantioselective routes for synthesis of inthomycin B, for details see: (a) N. Hénaff and A. Whiting, Org. Lett., 1999, 1, 1137 CrossRef; (b) M. Yoshino, K. Eto, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Biomol. Chem., 2012, 10, 8164 RSC; (c) M. R. Webb, C. Donald and R. J. K. Taylor, Tetrahedron Lett., 2006, 47, 549 CrossRef CAS; (d) M. R. Webb, M. S. Addie, C. M. Crawforth, J. W. Dale, X. Franci, M. Pizzonero, C. Donald and R. J. K. Taylor, Tetrahedron, 2008, 64, 4778 CrossRef CAS; (e) M. Kumar, L. Bromhead, Z. Anderson, A. Overy and J. W. Burton, Chem.–Eur. J., 2018, 24, 16753 CrossRef CAS; (f) B. K. Senapati, L. Gao, S. I. Lee, G.-S. Hwang and D. H. Ryu, Org. Lett., 2010, 12, 5088 CrossRef CAS; (g) S. Balcells, M. B. Haughey, J. C. L. Walker, L. Josa-Culleré, C. Towers and T. J. Donohoe, Org. Lett., 2018, 20, 3583 CrossRef CAS; (h) K. J. Hale, M. Grabski, S. Manaviazar and M. Maczka, Org. Lett., 2014, 16, 1164 CrossRef CAS.
- (a) Y. Tanaka, I. Kanaya, Y. Takahashi, M. Shinose, H. Tanaka and S. Omura, J. Antibiot., 1993, 46, 1208 CrossRef CAS; (b) S. Omura, Gene, 1992, 115, 141 CrossRef CAS; (c) M. Kawada, H. Inoue, I. Usami and D. Ikeda, Cancer Sci., 2009, 100, 150 CrossRef CAS; (d) M. Kawada, Y. Yoshimoto, K. Minamiguchi, H. Kumagai, T. Someno, T. Masuda, M. Ishizuka and D. Ikeda, Anticancer Res., 2004, 24, 1561 CAS.
- A. W. Gibson, G. R. Humphrey, D. J. Kennedy and S. H. B. Wright, Synthesis, 1991, 1991, 414 CrossRef.
- G. Wang, S. Mohan and E.-i. Negishi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11344 CrossRef CAS.
- T. L. Church, T. Rasmussen and P. G. Andersson, Organometallics, 2010, 29, 6769 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05276k |
| ‡ Present address: Chulabhorn Graduate Institute, Chulabhorn Royal Academy, 54 Kamphaeng Phet 6 Road, Laksi, Bangkok 10210, Thailand. |
| This journal is © The Royal Society of Chemistry 2021 |