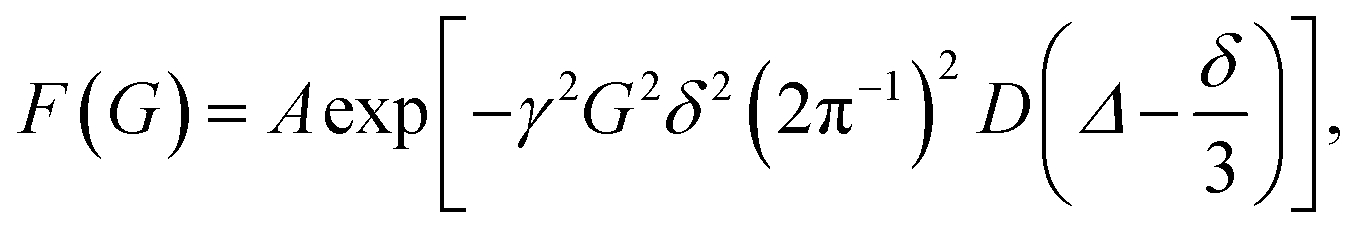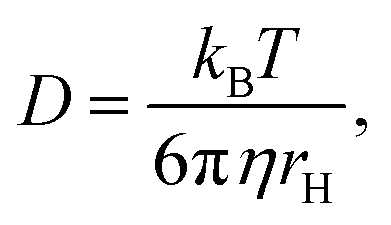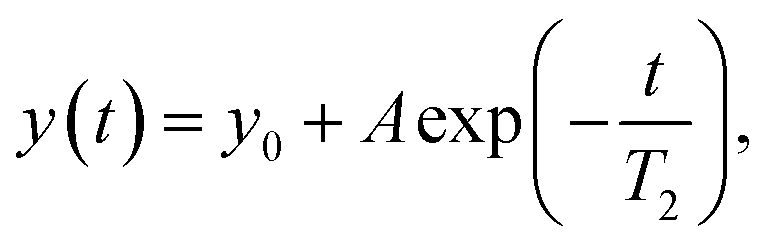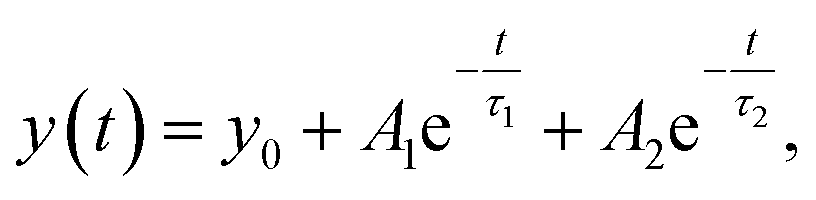DOI:
10.1039/D0SC05299J
(Edge Article)
Chem. Sci., 2021,
12, 412-426
Template-assisted design of monomeric polyQ models to unravel the unique role of glutamine side chains in disease-related aggregation†
Received
24th September 2020
, Accepted 28th October 2020
First published on 28th October 2020
Abstract
Expanded polyglutamine (polyQ) sequences cause numerous neurodegenerative diseases which are accompanied by the formation of polyQ fibrils. The unique role of glutamines in the aggregation onset is undoubtedly accepted and a lot structural data of the fibrils have been acquired, however side-chain specific structural dynamics inducing oligomerization are not well understood yet. To analyze spectroscopically the nucleation process, we designed various template-assisted glutamine-rich β-hairpin monomers mimicking the structural motif of a polyQ fibril. In a top-down strategy, we use a template which forms a well-defined stable hairpin in solution, insert polyQ-rich sequences into each strand and monitor the effects of individual glutamines by NMR, CD and IR spectroscopic approaches. The design was further advanced by alternating glutamines with other amino acids (T, W, E, K), thereby enhancing the solubility and increasing the number of cross-strand interacting glutamine side chains. Our spectroscopic studies reveal a decreasing hairpin stability with increased glutamine content and demonstrate the enormous impact of only a few glutamines – far below the disease threshold – to destabilize structure. Furthermore, we could access sub-ms conformational dynamics of monomeric polyQ-rich peptides by laser-excited temperature-jump IR spectroscopy. Both, the increased number of interacting glutamines and higher concentrations are key parameters to induce oligomerization. Concentration-dependent time-resolved IR measurements indicate an additional slower kinetic phase upon oligomer formation. The here presented peptide models enable spectroscopic molecular analyses to distinguish between monomer and oligomer dynamics in the early steps of polyQ fibril formation and in a side-chain specific manner.
Introduction
The implication of the mutational expansion of polyglutamine (polyQ) repeats in proteins became obvious by numerous neurodegenerative polyQ diseases, including Huntington's disease. The affected proteins show no sequence homology except for the abnormally expanded polyQ tracts.1,2 The pathogenic threshold is in the range of 35–40 consecutive glutamines, varying somewhat among the diseases. There is a consensus that polyQ aggregates favor a fibrillar structure, and several studies indicate features of amyloids3 and antiparallel β-sheet-rich secondary structures.4–7 It is beyond all question that the side chains of the glutamine tracts are the determinants for the aggregation onset in polyQ diseases.8,9 Nevertheless, a detailed understanding is still missing. PolyQ repeats appear within larger proteins in vivo, but analyzing full length polyQ proteins is often impractical. To track solely the glutamines, isolated polyQ peptides were established in various experimental and theoretical studies.8,10–18 Those artificially isolated polyQ sequences may aggregate slightly differently as they would in full-length proteins,4,6,7,11,17 however model sequences enable a molecular understanding of the exceptional properties of glutamine repeats causing aggregation in all polyQ diseases. A β-hairpin monomer has been suggested as aggregation nucleus,4,19 whereas other works indicated that polyQ monomers populate a β-hairpin as the toxic conformation in polyQ diseases.20,21
Peptide models have been used in the past to analyze the structural composition of polyQ fibrils and aggregates. PolyQ chains were described as polar zippers forming β-sheets where hydrogen bonds are formed between both the glutamine side-chain and backbone amides.22 The steric zipper model suggests that the reorientation of intrasheet glutamine hydrogen bonds facilitates a close van der Waals contact of the glutamine methylene groups.23,24 Other studies demonstrated the importance of side-chain hydrogen bonds for the stabilization of polyQ fibrils.8,9 A recent study reported that the amyloid core of huntingtin exon 1 fibrils features β-sheets that interact via interdigitation of glutamine side chains.25 The monomeric structural motif of a polyglutamine amyloid fibril was shown to consist of an antiparallel β-hairpin structure.7 Thus many static structural details on fibrils and aggregates were acquired so far, but not much is known about the formation process, in particular about the dynamics of glutamine side chains during protein misfolding.
A systematic design of polyQ peptide models is desirable to understand side-chain specific characteristics of glutamines in polyQ fibril formation processes. The simplest design of polyQ peptides contains sequences of consecutive glutamines. One major challenge is the low solubility of glutamines in aqueous solution, making it highly difficult to reach the pathologic repeat length using such models. The solubility of polyQ sequences can be increased by introducing charged residues at the N- and C-terminus. Such a peptide design has been used in previous studies with different glutamine sequence lengths.4,7,14,22,26,27 Another design strategy to enhance the solubility is the interruption of polyQ sequences with other residues, for example with histidines28,29 or prolines.30 Furthermore, there is a substantial history on generating peptide model systems with β-hairpin motifs.30–32 In these bottom-up designs, one or several sequence motifs are chosen to favor a hairpin structure, for example DPro–Gly turns, disulfide cross-links, hydrophobic and/or Coulomb interactions.
In this study, we introduce a novel top-down design strategy by using a template which forms a well-defined and stable β-hairpin monomer in solution. We incorporated glutamine-rich sequences into the strands of the template hairpin and analyzed the effects on structure and dynamics. As template we chose the peptide Trpzip2 introduced by Cochran and coworkers.33 We and others have extensively studied the dynamics of Trpzip2 variants by laser-excited temperature-jump (T-jump) spectroscopy,34–42 including the effect of hydrophobic interactions43 and the β-turn region44 on the folding dynamics and stability. Thus, Trpzip2 is very well suited as β-hairpin template. We designed various template-assisted glutamine-rich peptides with the aim to maximize the number of cross-strand interacting glutamine side chains while keeping the β-hairpin structure and solubility. An extensive spectroscopic analysis of all peptide designs was performed using NMR-, CD- and IR-spectroscopic techniques. T-jump IR spectroscopy enabled us to access the sub-ms time domain and to address conformational dynamics of polyQ monomers and oligomers. These states precede the much slower fibril elongation and aggregation kinetics occurring on a min-to-h timescale. Although the repeat lengths in our peptide models are much below the disease threshold, these studies are physiological relevant since they reveal the significant impact of individual glutamines on structure and dynamics of polyQ repeats.
Methods
Peptide synthesis and purification
Peptides were synthesized by peptides&elephants GmbH (Hennigsdorf, Germany) using FMOC-based-solid-phase-peptide-synthesis. The purity (96–99%) of the peptides was confirmed by HPLC-MS (ESI†). To eliminate spectral interference from trifluoroacetate (TFA) counterions remaining from the peptide synthesis, peptides were dissolved in 0.1 M DCl and lyophilized three times.
CD spectroscopy
CD samples were prepared at 0.1 mg mL−1 in H2O and 1 mm quartz cells were used. CD measurements were performed with a J815 spectrometer (JASCO, USA). Data were recorded between 300 nm and 180 nm with a scanning speed of 100 nm min−1 and a digital integration time of 1 s. The bandwidth was 1 nm. Final spectra were recorded as an average of 5 scans, smoothed and the water background spectrum was subtracted. Thermal variation of CD spectra was carried out in a temperature range of 5–95 °C in steps of ΔT = 5 °C. The heating rate was 1 °C per minute. The temperature was controlled by a Peltier sample holder with a regulated flow from a water bath (FL300, Julabo, Germany). Ellipticity was converted to molar ellipticity using the solution concentration determined by UV absorbance at 280 nm. The unit is given on a per molecule basis by dividing through the number of amino acids in the sequence.
High-resolution NMR spectroscopy
All NMR spectra of peptide samples were measured in 95% H2O and 5% D2O at various concentrations (0.1–3 mM) as indicated in the text. The pH values were between 2.7–4.0. All one-dimensional 1H as well as two-dimensional 1H–15N HSQC spectra were measured either with a 600 MHz Bruker Avance III or 800 MHz Avance NEO spectrometer equipped with a TCI cryogenically cooled probe at T = 298 K sample temperature. Additional TOCSY and NOESY spectra were recorded for the assignment of resonance signals comprising the template Trpzip2. The data were processed using MestReNova software. Fitting procedures of experimentally obtained data were carried out using Origin 2019b software.
Pulse-field gradient NMR diffusion experiments
Diffusion experiments of the Trpzip-Qn peptides were performed with pulse-field gradient NMR spectroscopy. NMR data were recorded for 21 different gradient strengths ranging between 0.06–0.6 Tm−1 2π−1. The diffusion time was set to 60 ms and a gradient duration of 3 ms was used.
The diffusion coefficients were determined with a nonlinear curve function:
| | 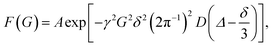 | (1) |
where
γ is the gyromagnetic ratio for protons,
G is the gradient strength,
δ is the duration of the gradient,
Δ is the diffusion time and
D is the diffusion coefficient. The measured value of the diffusion coefficient can be converted into the hydrodynamic radius
rH by the Stokes–Einstein equation:
| | 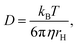 | (2) |
where
kB is the Boltzmann constant,
η is the viscosity of the solvent, and
T is the temperature.
The hydrodynamic radii of different proteins have been studied before using pulse field gradient NMR experiments. An empirical relationship between the measured hydrodynamic radius rH and the number of amino acids N in the sequence was established for natively folded proteins:45
Thus, the theoretically expected rH can be compared to rH obtained from our experimental data.
NMR relaxation experiments
Experiments to determine the transversal relaxation time T2 were performed using a CPMG-based approach applying relaxation delays ranging between 8 ms and 600 ms. The transverse relaxation time was determined with a non-linear function:| | 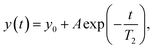 | (4) |
where y0 is an offset, and A is the pre-exponential factor.
FTIR spectroscopy
IR samples were prepared in D2O with a concentration of 10 mg mL−1 at acidic pH (after lyophilization from DCl) and were placed in home-made demountable cells consisting of CaF2 windows separated by a Teflon spacer with 100 μm optical pathlength. Spectra were acquired with an Equinox 55 FTIR spectrometer (Bruker, Germany). For each FTIR spectrum, 128 scans with a spectral resolution of 4 cm−1 were averaged at each temperature. Thermal variation of the IR spectra was carried out in a temperature range of 5–95 °C in steps of ΔT = 5 °C. The temperature of the sample holder was controlled by a water bath (Lauda Ecoline E300, Germany), and the sample cell temperature recorded with a Pt100 sensor. A home-built software-controlled shuttle device was used to measure sample and reference signals successively for each temperature allowing the subtraction of the solvent signal.46 Transition temperatures were determined as described in ESI† using Origin 2019b software.
Time-resolved IR-detected temperature-jump dynamics
T-jump IR measurements were performed using the quantum cascade laser-based spectrometer that has been described in detail previously.38,41,44,46 Relaxation dynamics of the peptides were probed at selected wavenumbers with a quantum cascade laser (QCL), installed in a MIRcat-QT laser system (Daylight Solutions Inc., USA). The continuous-wave (cw) QCL used (M2062-PCX) has a tuning range from 1730 to 1480 cm−1. The rapid temperature jump is initiated by a 10 ns pulse at 2090 nm of a Ho:YAG laser (IPG Photonics Corporation, USA), exciting a solvent (D2O) vibration, thereby rapidly increasing the peptide temperature. The pump pulse is split into two counter-propagating parts; one of the pump pulses is delayed by 5 ns to produce a homogenous heating profile in the sample cell preventing cavitation effects. The initial temperature of the sample was controlled by a water bath connected to the sample holder. The temperature jump was calculated by referencing the measured absorbance change of the solvent to the corresponding temperature-dependent FITR spectra of D2O.44 For the experiments, a T-jump magnitude of ∼5–8 °C was used. About 2000 transients were averaged. To exclude distorted transients caused by cavitation effects,47 a self-developed software filter (MATLAB2010, The MathWorks, USA) was applied to save the most reliable data after collection. To account for solvent kinetics, both solvent and peptide samples were measured sequentially. The solvent-only signal was scaled appropriately and subtracted from the peptide sample signal.46 The resulting transients were subjected to a quasi-logarithmic averaging procedure so that an equal number of points were distributed in each time decade (20 points per decade) leading to a significant reduction of noise and distortion signals.46 Measurements of the polyQ-rich peptides were performed at the same concentration 10 mg mL−1 as for the equilibrium FTIR measurements. The samples were heated to 70 °C for ∼10 min in an oven to dissolve possible aggregates before measurements. The relaxation times were evaluated in a time interval from 300 ns up to 1.2 ms using a mono-exponential decay at a final temperature of ∼25 °C. For a concentration study of Trpzip-Q6, samples were prepared to final concentrations of 5–20 mg mL−1. The relaxation times for Trpzip-Q6 were fitted with a bi-exponential decay function at a final temperature of ∼25 °C:| | 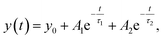 | (5) |
where y0 is an offset, A1 and A2 are pre-exponential factors for the corresponding relaxation times, τ1 and τ2. Fitting procedures were performed with Origin 2019b software.
Results and discussion
Design of model peptides
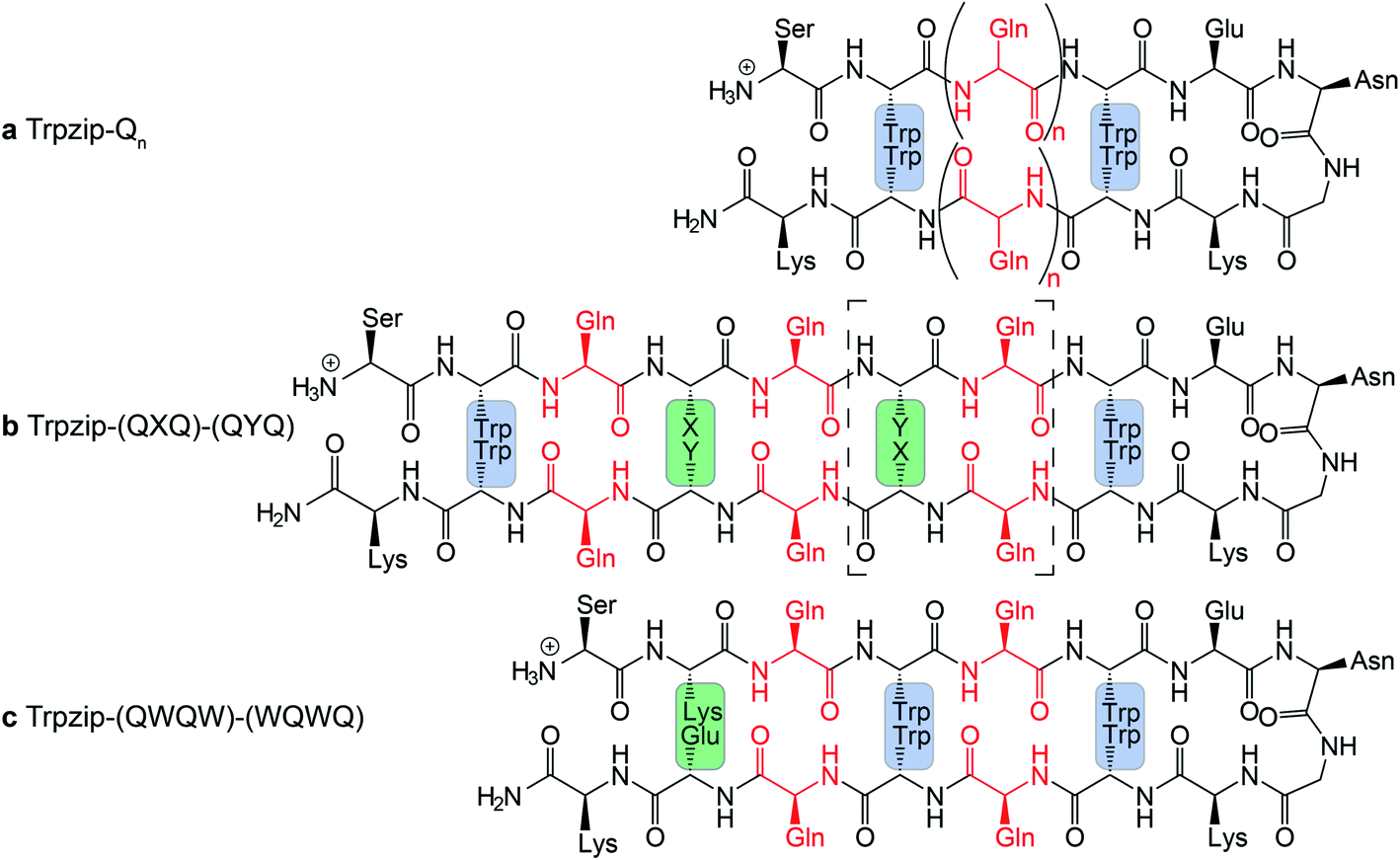
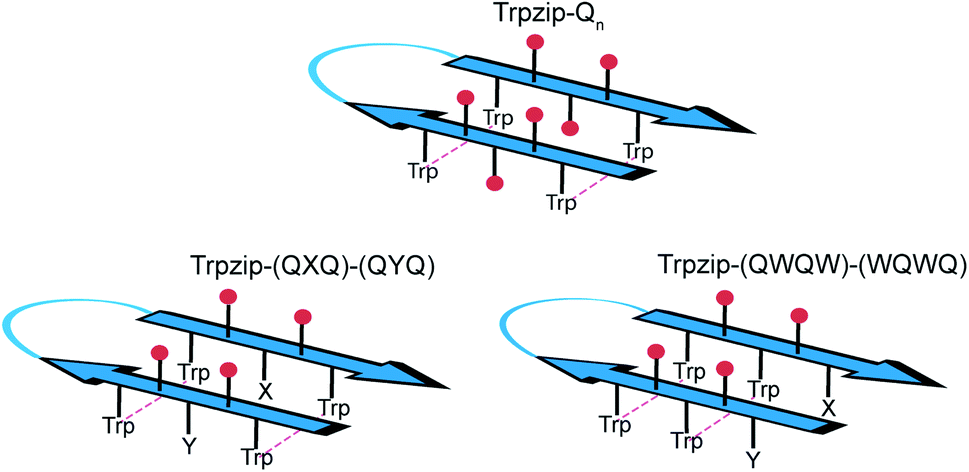
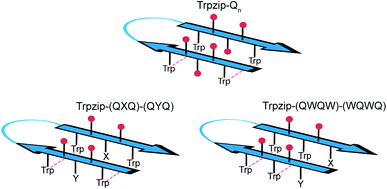
The template-assisted designs are shown in Fig. 1. The original Trpzip2 sequence contains 12 amino acids and is stabilized by two cross-strand tryptophan pairs (on positions 2–11 and 4–9), which are orientated in an edge-to-face geometry. The tryptophan hydrophobic cross-strand interactions are important to maintain a hairpin structure. Threonines (on positions 3 and 10) are enveloped by two tryptophans on each strand and were substituted with glutamine repeats in one variant (Fig. 1a). We call this design Trpzip-Qn (n is the total number of incorporated glutamines), for example Trpzip-Q6, the number of glutamines is six with three consecutive glutamines per strand. The number of incorporated glutamines determines the number of side chains pointing up and those pointing down with respect to the hairpin backbone plane, and thus the number of side-chain interactions. For Trpzip-Q6, two side chains per strand point up and one points down, thus resulting in three cross-strand interactions, two above and one below the hairpin plane (Fig. 2). The second design strategy focused on the increase of glutamine side-chain interactions while preserving the solubility of the monomeric hairpin motif. This design model is called Trpzip-(QXQ)-(QYQ) and contains polyQ repeats interrupted by other amino acids X and Y, so that all glutamine side chains are pointing to the same side of the hairpin (Fig. 1b). (QXQ) and (QYQ) indicate the interrupted polyQ repeats on the hairpin strands. The interrupted polyQ repeats start and end with tryptophans so that Trp–Trp cross-strand interactions enclose the polyQ-rich sequences to stabilize the hairpin structure. An increase from two to three glutamines per strand (Fig. 1b shown in dashed brackets) belongs to the same design variant and is called Trpzip-(QXQYQ)2 (Table 1). Threonines were used for X and Y like in the original template Trpzip2, and as alternative a glutamic acid for X and a lysine for Y. In the third design Trpzip-(QWQW)-(WQWQ) (Fig. 1c), the glutamines alternate with tryptophans, thus the same Trp–Trp positions are maintained as in the original Trpzip2. In addition, a Lys–Glu salt bridge is introduced close to the hairpin ends to enhance the solubility. All glutamine side chains are pointing to the same side of the hairpin.
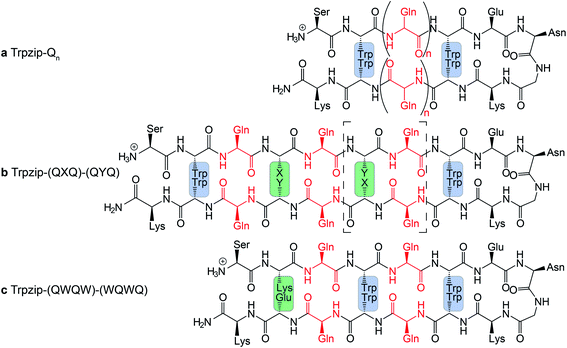 |
| | Fig. 1 PolyQ peptide designs based on the template Trpzip2 (SWTWENGKWTWK-NH2). All peptides are stabilized by aromatic cross-strand interactions of the tryptophan residues (blue boxes). (a) In Trpzip-Qn threonines were substituted by n/2 consecutive glutamines inserted in each strand. (b) In Trpzip-(QXQ)-(QYQ), glutamine repeats are interrupted by other amino acids X or Y (highlighted in green) so that glutamine side chains are orientated on the same side of the hairpin plane. X and Y were substituted with threonines similar to the Trpzip2 template. As alternative and for solubility enhancement, glutamic acid and lysine were used to insert salt bridge pairs. The dashed brackets show the potential elongation of the glutamine sequence, called Trpzip-(QXQYQ)2. (c) In Trpzip-(QWQW)-(WQWQ) the original arrangement of the tryptophans is maintained as in the template Trpzip2. The orientations of the glutamine side chains are like in the previous design. An additional salt bridge enhances the solubility (highlighted in green). | |
 |
| | Fig. 2 Schematic view of cross-strand interacting glutamine side chains (red) shown for examples representing each of the three design variants as outlined in Table 1. In Trpzip-Qn, the orientation of the adjacent glutamine side chains is alternating with respect to the hairpin plane (up/down). In Trpzip-(QXQ)-(QYQ) and Trpzip-(QWQW)-(WQWQ), glutamine sequences are interrupted by other amino acids (X, Y or W) so that glutamine side chains are orientated on the same side of the hairpin. | |
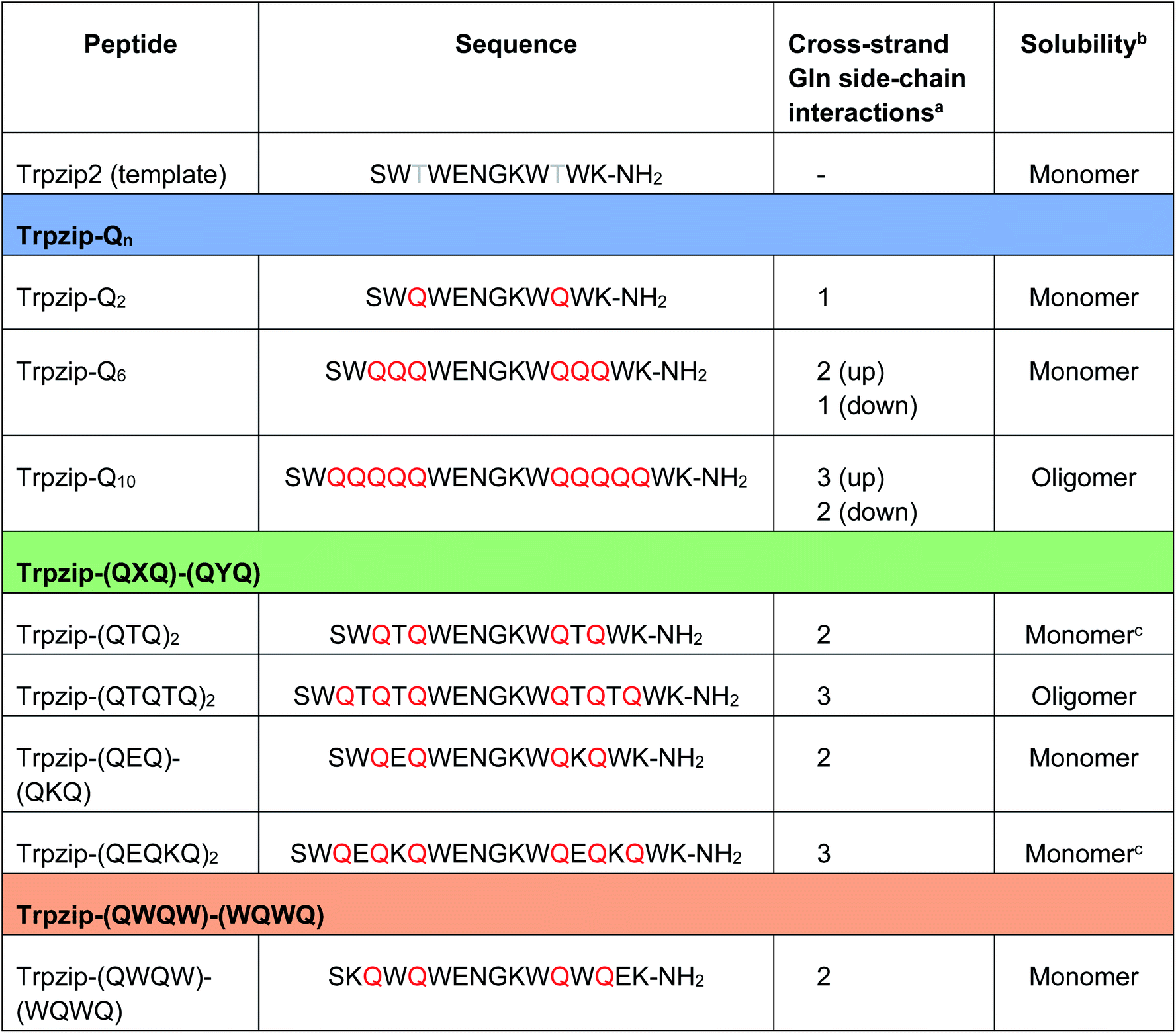
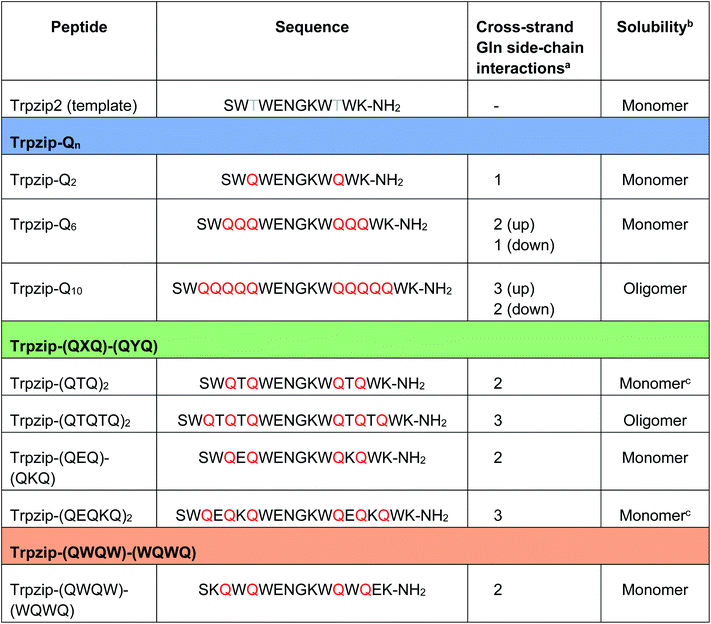
Table 1 Sequences of the template Trpzip2 and the three template-designed polyQ peptide models. Trpzip-Qn: n/2 is the number of consecutive glutamines per strand. Trpzip-(QXQ)-(QYQ): glutamine sequences are interrupted by other amino acids X and Y. Trpzip-(QWQW)-(WQWQ): the tryptophan pairs do not envelope the glutamine sequence as in the other designs, but alternate with the glutamines. Glutamines are highlighted in red
|
The number of cross-strand interacting glutamine side chains are counted for each side (up/down) of the β-hairpin plane.
The observation of the solubility is based on FTIR data measured for peptide concentrations of 10 mg mL−1 (Fig. S9–S13, ESI).
Peptides were converted into monomeric hairpin structures after heating and recooling (Fig. S11a, b, S12b, c and S16a, b, ESI).
|
|
The sequences of the template Trpzip2 and all designed model peptides, categorized as Trpzip-Qn, Trpzip-(QXQ)-(QYQ) and Trpzip-(QWQW)-(WQWQ), are listed in Table 1. In addition, the number of cross-strand Gln–Gln side-chain interactions are given for each variant as the maximization of interacting side chains was the focus of the peptide design. There are also Gln–Gln side-chain interactions within one strand, however adjacent glutamine side chains are pointing to different sites of the hairpin plane, thus the interaction of side chains separated by one amino acid is likely to be weaker as compared to cross-strand interactions, and were not explicitly listed here. The solubility of each variant is given in Table 1 based on FTIR measurements distinguishing between a monomeric and oligomeric state of the peptide. The first design Trpzip-Qn contains n/2 consecutive glutamines per strand. Trpzip-Q2 is monomeric and consists of one glutamine per strand resulting in one pair of cross-strand interacting side chains. Trpzip-Q6 exhibits three cross-strand Gln pairs. Both variants form monomers. Increasing the number of consecutive glutamines leads very quickly to the formation of oligomers as shown for Trpzip-Q10. The second design Trpzip-(QXQ)-(QYQ) aims to generate a monomeric peptide with an increased number of cross-strand interacting glutamine side chains. In the first attempt, threonines were used for X and Y, resulting in Trpzip-(QTQ)2 with two pairs of interacting glutamine side chains. Elongation of the polyQ-rich sequence to Trpzip-(QTQTQ)2 leads to oligomeric structures. Another attempt of this design uses charged amino acids, glutamic acid for X and lysine for Y, which can form a salt bridge pair and enhance the solubility of the peptide. By contrast to the threonine substitutions in Trpzip-(QTQTQ)2, Trpzip-(QEQKQ)2 with three cross-strand glutamine side-chain interactions stays in a monomeric state. In the third design Trpzip-(QWQW)-(WQWQ), the original Trp–Trp templating of Trpzip2 was retained. This strategy leads to a monomer with two interacting side chains on one side of the hairpin (Fig. 2).
Structural changes in hairpin conformation by insertion of polyQ sequences
Two-dimensional heteronuclear 1H–15N HSQC experiments were performed for Trpzip2 and the Trpzip-Qn templates to evaluate potential changes in the peptide fold upon insertion of glutamine residues (Fig. 3). In principle, each nitrogen bound proton in the peptide exhibits one cross signal in the spectra, thus backbone amide protons as well as side-chain bound nitrogen protons are monitored. Some side-chain signals and the C-terminal carboxamide group (resulting from the peptide synthesis) are well separated from the backbone amide protons, as well as the side-chain indole NH of tryptophans (black frame), the side-chain amide protons of asparagine and glutamines and the C-terminal carboxamide group of lysine (orange frame). The reference spectrum of the template Trpzip2 is in good agreement with the literature.33
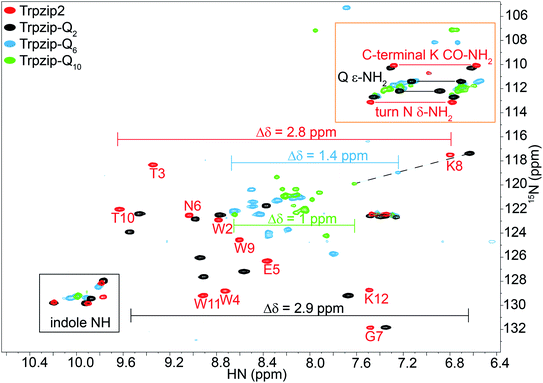 |
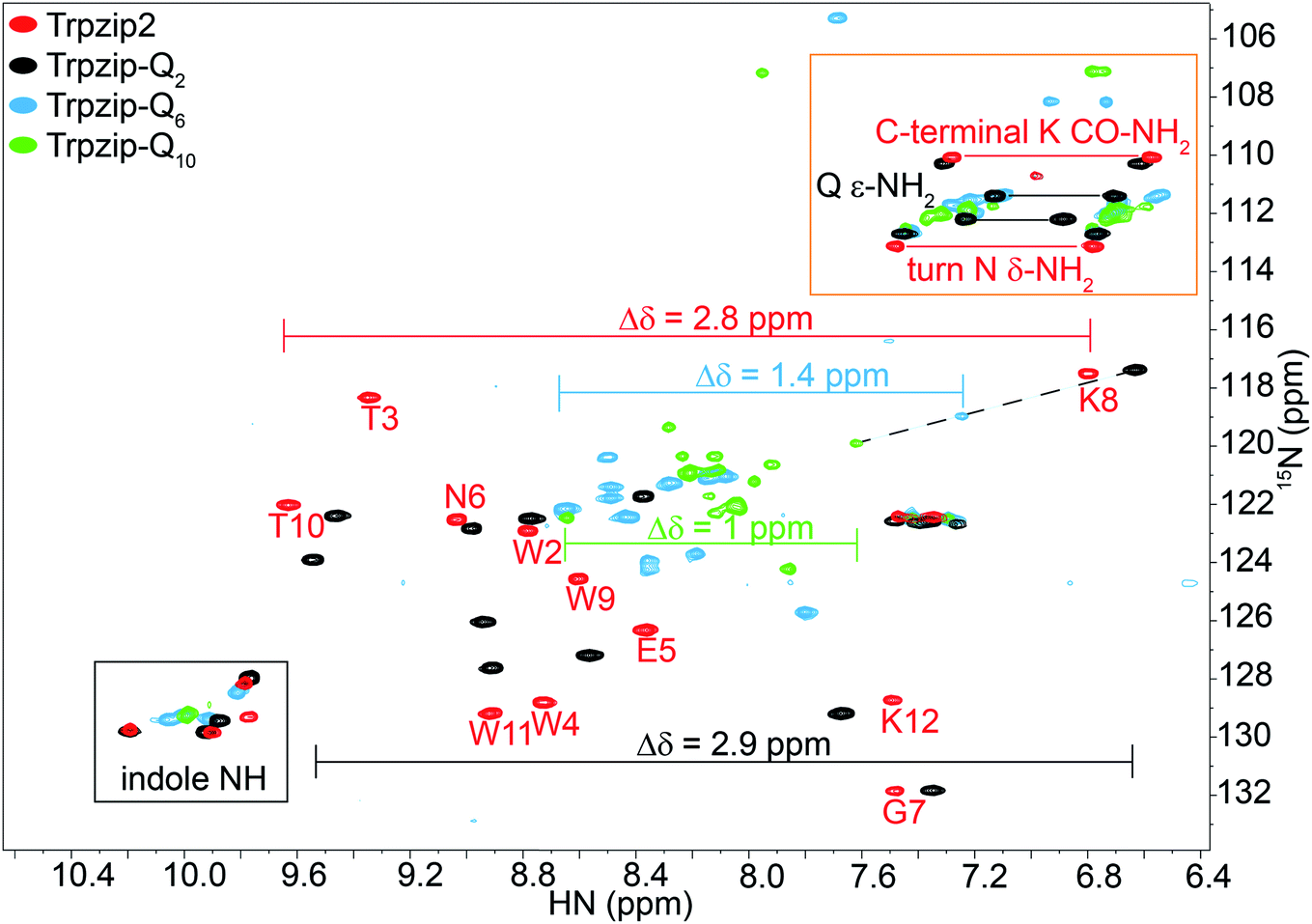
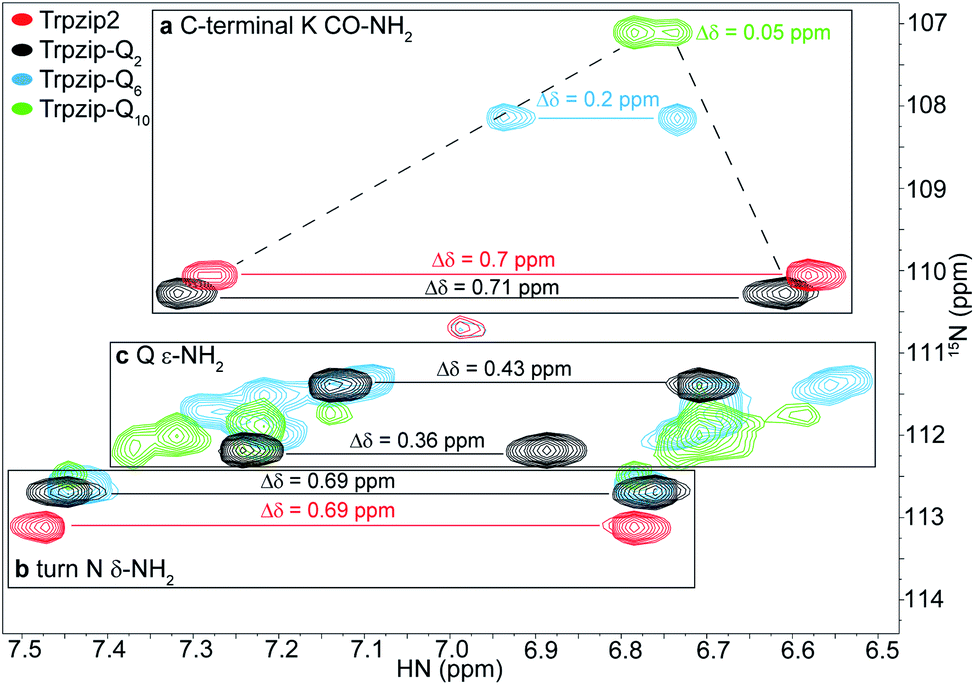
| | Fig. 3 Overlay of two-dimensional 1H–15N HSQC spectra acquired for Trpzip2 (red), Trpzip-Q2 (black), Trpzip-Q6 (blue) and Trpzip-Q10 (green) at T = 298 K. The assignment of backbone cross signals of Trpzip2 is compared with Trpzip-Qn models. The dispersion of chemical shifts comprising backbone amide protons, Δδ, decrease the more glutamines are inserted (Δδ = 2.9 ppm for Trpzip-Q2, Δδ = 1.4 ppm for Trpzip-Q6 and Δδ = 1 ppm for Trpzip-Q10). The NH cross signals of the tryptophan side-chain indole groups (black frame) indicate the distortion of the hairpin-stabilizing Trp–Trp cross-strand interactions (with a side-chain edge-to-face orientation for Trpzip2). The C-terminal signals of lysines (K CO–NH2) as well as the side-chain signals of asparagines (N δ-NH2) and glutamines (Q ε-NH2) are well separated (orange frame) and shown enlarged in Fig. 4. The C-terminal lysine and turn asparagine are present in all peptide sequences resulting in two pairs of signals (linked by red lines for Trpzip2). Each inserted glutamine exhibits one pair of signals (linked by black lines for Trpzip-Q2). Further details are in the text. The comparison of each variant with Trpzip2 is shown separately (Fig. S1a–c, ESI†). | |
The comparison of the signal dispersions with the Trpzip2 template is well suited to draw conclusions about potential structural changes due to the insertion of glutamines. We focused on the dispersion of chemical shifts for amide backbone protons to monitor global conformational changes in the hairpin backbone. Additional indicators for distortions in the hairpin structure are the four indole protons of the tryptophan side chains because the template hairpin Trpzip2 is mainly stabilized by the hydrophobic Trp–Trp cross-strand interactions. Furthermore, each model sequence has two lysines and one asparagine conserved in position, one lysine is located close to the Asn–Gly (NG) turn and one at the C-terminus. The lysines contribute with proton signals of the backbone amide (K8 and K12 in the case of Trpzip2) and the C-terminal carboxamide group. In particular, the backbone amide proton of K8 close to the turn can be very well identified for all variants due to no spectral overlap with other signals (visualized by the dashed black line in Fig. 3). The spectral region of carboxamide protons of the C-terminal lysine (K CO–NH2), the side-chain amide protons of the turn asparagine (N δ-NH2) and the glutamines (Q ε-NH2) is enlarged in Fig. 4. In Trpzip2, the C-terminal lysine and the asparagine each exhibit a pair of well dispersed signals which can be used to evaluate local structural changes at the end of one strand (C-terminal K CO–NH2) and in the turn (turn N δ-NH2) when glutamines are inserted. The polyQ variants exhibit additional signals of the glutamine side chains (Fig. 4c) and two pairs of well dispersed signals are observed for Trpzip-Q2 (Fig. 4c, linked by black lines). Trpzip-Q6 and Trpzip-Q10 naturally show more side-chain amide proton signals, which partially overlap (Fig. 4c, blue and green). Another peptide variant Trpzip-(QWQW)-(WQWQ) with four glutamine side-chain amides (Fig. S1d, ESI†) shows well separated and dispersed signals.
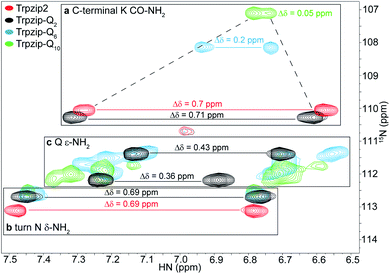 |
| | Fig. 4 Enlarged section of two-dimensional 1H–15N HSQC spectra acquired for Trpzip2 (red), Trpzip-Q2 (black), Trpzip-Q6 (blue) and Trpzip-Q10 (green) at T = 298 K. The spectral range shown is subdivided into three parts. (a) C-terminal lysine CO–NH2 cross signals show a decreasing dispersion of chemical shifts, Δδ, with an increasing number of inserted glutamines (highlighted by dashed lines). (b) Cross signals of turn asparagine δ-NH2 show similar dispersions of chemical shifts (Δδ = 0.69 ppm) for each variant. (c) Glutamine side-chain signals (Q ε-NH2) tend to overlap the more glutamines are introduced into the Trpzip2 template. | |
The 1H–15N HSQC spectrum of Trpzip2 represents the well-defined hairpin fold of the template and shows a broad dispersion of the peptide backbone amide proton chemical shifts (Δδ = 2.8 ppm). For Trpzip-Q2 (Fig. 3, 4, and S1a, ESI†), cross signals of all amide protons (black) are slightly shifted compared to Trpzip2 (red) showing that the insertion of only one glutamine per hairpin strand already has an impact on the backbone conformation. However, the dispersion of the backbone amide protons remains broad for Trpzip-Q2 (Δδ = 2.9 ppm) and hints that the overall fold of Trpzip-Q2 is very similar to the template. This is also supported by the indole NHs (Fig. 3 black frame) where the cross signals are only slightly shifted indicating that the tryptophan geometry is maintained to stabilize the hairpin structure. The dispersion of the lysine carboxamide CO–NH2 resonances remains basically the same (Fig. 4a) and two new and well dispersed signals of the glutamine side chains ε-NH2 show up (Fig. 4c). In Trpzip-Q6 (Fig. 3, 4, and S1b, ESI†), the dispersion of the backbone amide proton chemical shifts is about half compared to the template (Δδ = 1.4 ppm, blue) indicating significant conformational changes caused by the insertion of the glutamines. The cross signals of the indole NH resonances of the tryptophans partially overlap, but remain dispersed, supporting that the Trp–Trp cross-strand interactions are still effective enough to keep the peptide sequence in a (distorted) hairpin structure. Interestingly, the lysine close to the turn shows a significant shift of the backbone signal (Fig. 3), whereas the side-chain signals of the turn asparagine don't change (Fig. 4b) and we speculate that the side-chain movements are constrained by the turn. In contrast, the carboxamide of the C-terminal lysine shows a remarkable less dispersed set of cross signals (Δδ = 0.2 ppm, Fig. 4a) indicating a more disordered C-terminal strand. To get further insights into the backbone conformation of the inserted glutamine tracts we performed temperature-dependent 13C NMR spectroscopy and relaxation experiments of 13C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O isotopically labeled Trpzip-Q6 (Fig. S2 and Table S1, ESI†). The experimental data reveal that the backbone of the glutamines possesses a quite ordered structure as seen by dispersed chemical shifts and diverse transversal relaxation times. For Trpzip-Q10 (Fig. 3, 4, and S1c, ESI†), the dispersion of chemical shifts for all backbone amide protons is only about a third of Trpzip2 (Δδ = 1 ppm) indicating pronounced structural changes of the peptide fold. Only two NH signals are resolved for the indole groups in contrast to the template Trpzip2 with four dispersed signals caused by the cross-strand interacting tryptophans with edge-to-face geometry of the side chains. This suggests that the indole protons of Trpzip-Q10 have similar chemical environments and that the well-defined orientations of the Trp side chains, stabilizing the hairpin structure, are significantly lost. The overlaid signal of the C-terminal lysine supports an even more disordered C-terminus compared to Trpzip-Q6 (Fig. 4a). In addition, the side-chain signals observed for glutamines overlap more strongly (Fig. 4c). We conclude that Trpzip-Q10 adopts a predominantly disordered structure with only weak hairpin features maintained by the Asn–Gly turn and very weak remaining Trp–Trp interactions.
O isotopically labeled Trpzip-Q6 (Fig. S2 and Table S1, ESI†). The experimental data reveal that the backbone of the glutamines possesses a quite ordered structure as seen by dispersed chemical shifts and diverse transversal relaxation times. For Trpzip-Q10 (Fig. 3, 4, and S1c, ESI†), the dispersion of chemical shifts for all backbone amide protons is only about a third of Trpzip2 (Δδ = 1 ppm) indicating pronounced structural changes of the peptide fold. Only two NH signals are resolved for the indole groups in contrast to the template Trpzip2 with four dispersed signals caused by the cross-strand interacting tryptophans with edge-to-face geometry of the side chains. This suggests that the indole protons of Trpzip-Q10 have similar chemical environments and that the well-defined orientations of the Trp side chains, stabilizing the hairpin structure, are significantly lost. The overlaid signal of the C-terminal lysine supports an even more disordered C-terminus compared to Trpzip-Q6 (Fig. 4a). In addition, the side-chain signals observed for glutamines overlap more strongly (Fig. 4c). We conclude that Trpzip-Q10 adopts a predominantly disordered structure with only weak hairpin features maintained by the Asn–Gly turn and very weak remaining Trp–Trp interactions.
Trp–Trp interactions as structural indicator
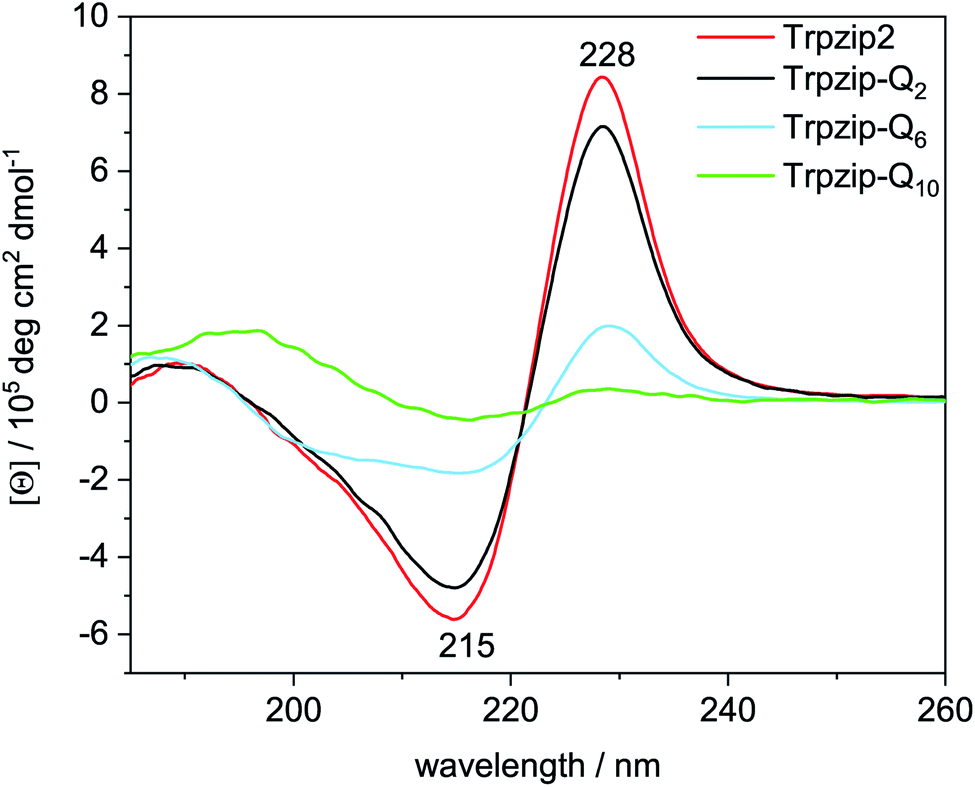
The two tryptophan pairs of the template Trpzip2 exhibit strong CD bands at 215 nm and 228 nm due to the π–π* transition of the exciton-coupled aromatic groups of the Trp side chains.48,49 The impact of the glutamine repeats on the hairpin structure could nicely be monitored by these bands since the glutamines are enveloped by the tryptophans and directly influence the cross-strand Trp–Trp interactions33 (Fig. 5). The bands show a slight decrease in intensities for Trpzip-Q2, which becomes significant for Trpzip-Q6 indicating the loss of the Trp–Trp cross-strand interactions. For Trpzip-Q10, no interactions between the Trp residues exist anymore. It should be noted that Trpzip-Q10 was heated before the experiment to dissolve oligomeric β-sheet structures (Fig. S7, ESI†).
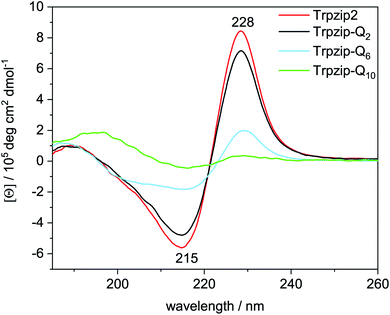 |
| | Fig. 5 Comparison of CD spectra acquired for Trpzip2 (red), Trpzip-Q2 (black), Trpzip-Q6 (blue) and Trpzip-Q10 (green) measured at 20 °C and a concentration of 0.1 mg mL−1. | |
CD measurements were also performed for other peptide designs with interrupted polyQ sequences (Fig. S8, ESI†). A significant decrease of Trp–Trp cross-strand interactions was observed in general for variants with elongated termini. The elongation is accompanied with a larger Trp–Trp distance on each strand. We assume that this reduces also the cross-strand coupling, in particular for the terminal Trp–Trp pair which is not constrained by the turn, and we expect a disassembly of the hairpin termini. It has been shown that frayed hairpin termini reduce the cross-strand Trp–Trp exciton coupling50 suggesting that the hairpin termini get frayed the more elongated the polyQ sequences.
Concentration-dependent formation of oligomers
High peptide concentrations can favor oligomerization and aggregate formation, thus we evaluated concentration effects by NMR and FTIR approaches. One-dimensional 1H NMR dilution experiments were performed in a concentration range of 0.1–1 mM for the template Trpzip2, Trpzip-Q6 and Trpzip-Q10 (Fig. S3, ESI†). Only minor and similar changes in chemical shifts are monitored for each variant upon this concentration increase. The measurements of Trpzip2 are in good agreement with previous studies which suggested that the detected changes can be attributed to a limited self-association of the monomers occurring at concentrations of ∼1 mM.33 We suppose that we also observe transient interactions of monomers, and no oligomers at a concentration of 1 mM, but it should be noted that oligomers are not directly detectable in solution state NMR experiments when they precipitate. For Trpzip-Q10, precipitation was observed during lyophilisation and thus we assume a heterogenous mixture of monomers and oligomers where only the monomers in solution are detectable.
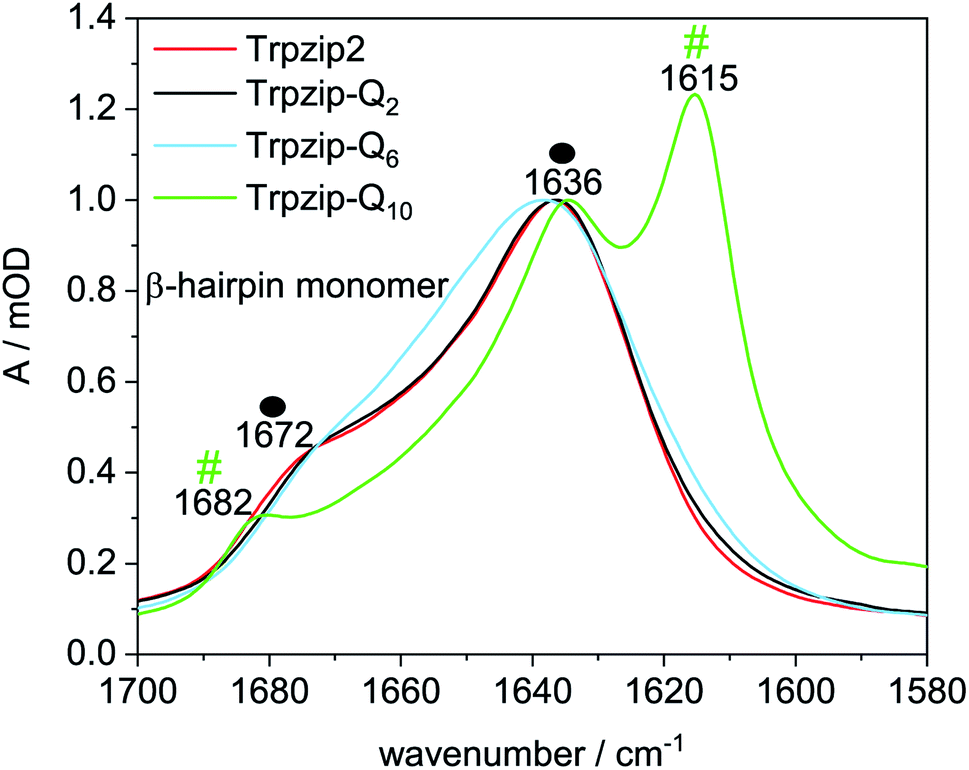
To further investigate the potential oligomer formation at concentrations in the mM range, we performed diffusion experiments by pulse-field gradient NMR (Fig. S4 and Table S2, ESI†) and FTIR measurements. Diffusion NMR spectroscopy focuses on the hydrodynamic dimension of the molecules under study (ESI for details†) whereas FTIR spectroscopy enables to distinguish hairpin monomers and oligomeric β-structures due to characteristic band components in the amide I′ spectrum (mainly CO stretching vibrations of the backbone). Fig. 6 shows amide I′ spectra of the template Trpzip2 and the Trpzip-Qn variants, all measured at a concentration of 10 mg mL−1 (3.7–6.2 mM, depending on the sequence). The bands at 1636 cm−1 and 1672 cm−1 reveal the monomeric β-hairpin structure of the template34,36,42,43,51–53 and Trpzip-Q2. The spectrum of Trpzip-Q6 becomes broader, but Trpzip-Q6 still forms a β-hairpin, as indicated by the maximum at 1636 cm−1 and the shoulder at 1672 cm−1. The broader band shape is assigned to a less structured β-hairpin what is supported by the NMR and CD measurements also revealing structural changes due to less Trp–Trp cross-strand interactions and frayed ends of the strands. Trpzip-Q10 forms oligomeric β-sheet structures, what is clearly indicated by the characteristic band shifts to 1615 cm−1 and 1682 cm−1.54,55 This result is not in contrast to the NMR measurements because an about 4 times higher concentration has been used in FTIR spectroscopy, favoring intermolecular interactions and oligomerization. The side chains of the glutamines also contain a carbonyl group which absorbs between 1635–1654 cm−1 (ref. 56) and overlaps the amide I′ region. The Trpzip-Q10 FTIR spectrum uncovers the carbonyl side chain at 1635 cm−1 since the backbone carbonyl vibration is shifted to 1615 cm−1 and does not overlay anymore.
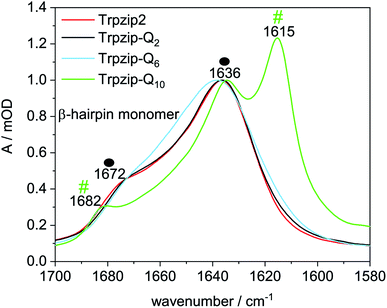 |
| | Fig. 6 Comparison of FTIR spectra in the amide I′ region of Trpzip2, Trpzip-Q2, Trpzip-Q6 and Trpzip-Q10 at 17.6 °C and a concentration of 10 mg mL−1. Spectra are normalized to the intensity observed at 1636 cm−1. The spectra of Trpzip2 and Trpzip-Q2 show two bands at 1636 cm−1 and 1672 cm−1 corresponding to monomeric β-hairpin structures (as indicated by the two black spheres). Trpzip-Q6 also reveals these two bands with a less pronounced band at 1672 cm−1 indicating a less structured hairpin. Oligomerization is observed for Trpzip-Q10 as revealed by the bands at 1615 cm−1 and 1682 cm−1 (two green hashes). The spectrum of Trpzip-Q10 also reveals the vibrational mode of the glutamine side-chain carbonyl at 1635 cm−1 (in D2O) which overlaps the amide I′ region. | |
Destabilization of β-hairpin conformation with glutamine content increase
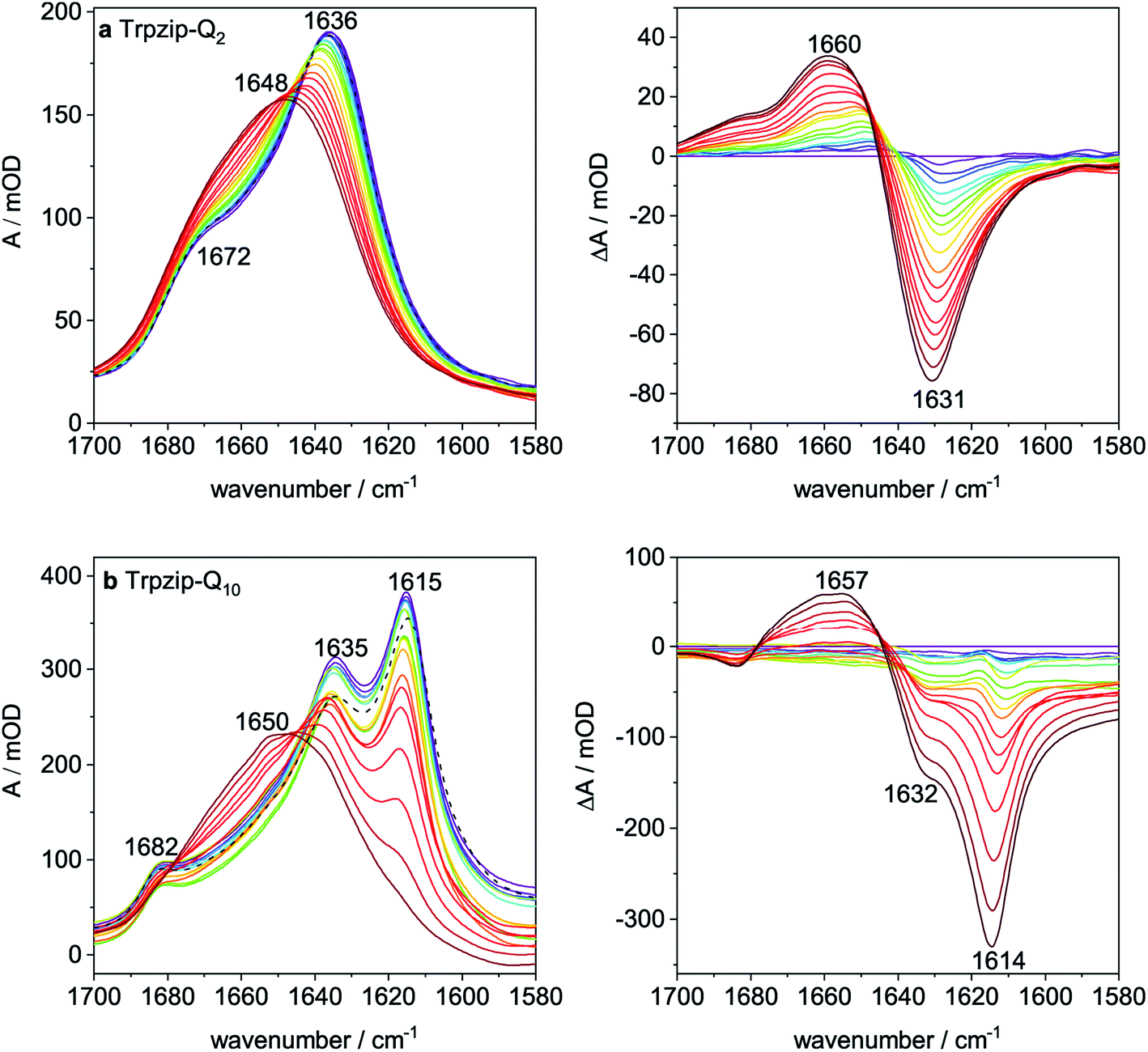
Thermal stability was studied by heating and recooling of the peptides and monitoring changes in the temperature-dependent amide I′ spectra (Fig. 7 and S9–S13, ESI†). The reversible thermal unfolding and refolding of Trpzip-Q2 from a hairpin structure (folded state) to a disordered structure (unfolded state) is observed by characteristic changes in the amide I′ spectrum. Fig. 7a reveals the typical split band components at 1636 cm−1 and 1672 cm−1 at low temperature, which are indicative for a monomeric β-hairpin structure. Thermal unfolding and transition to a disordered structure is shown by the change in the amide I′ band shape and a band maximum at 1648 cm−1. Difference spectra were calculated from the absorbance spectra with respect to the 5 °C (folded) state and reveal the maximum changes in absorbance.
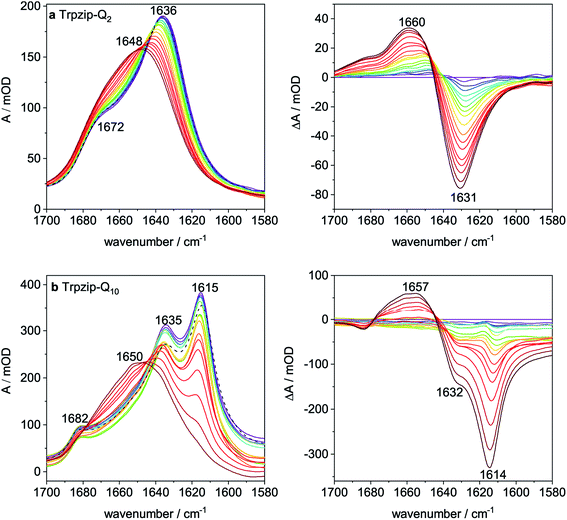 |
| | Fig. 7 Temperature-dependent FTIR absorption spectra (left) and corresponding difference spectra (right) in the amide I′ region (a) Trpzip-Q2 and (b) Trpzip-Q10. All spectra were recorded from 5 °C (blue colourish) to 95 °C (red colourish) in steps of 5 °C (in D2O; c = 10 mg mL−1). Absorption bands at 1636 cm−1 and 1672 cm−1 are indicative for the β-hairpin structure in the folded state of Trpzip-Q2. Upon heating, the band maximum shifts to 1648 cm−1 showing the unfolding of the peptide. Absorption bands at 1615 cm−1 and 1682 cm−1 show oligomeric sheet structures for Trpzip-Q10. Trpzip-Q2 could be reversibly unfolded and refolded as indicated by the black dashed spectrum in (a). Trpzip-Q10 can be converted into a disordered structure (1650 cm−1) upon temperature increase, however the oligomeric β-sheet structures are formed again after recooling (black dashed spectrum (b)). | |
Trpzip-Q10 forms an oligomeric sheet structure as indicated by the bands at 1615 cm−1 and 1682 cm−1 (Fig. 7b). The vibrational mode of the glutamine side-chain carbonyls can be assigned to the band component at 1635 cm−1. For some of the peptide variants, disaggregation of oligomeric sheet structures can be achieved by heating and recooling, but not for Trpzip-Q10 at this concentration. Although the oligomeric band disappears at high temperature (90 °C) and is shifted to higher wavenumbers (1650 cm−1), the oligomeric sheet structure (1615 cm−1) is reestablished after recooling (Fig. 7b, dashed black spectrum). The high number of glutamine side chains in the sequence probably facilitates the formation of intermolecular hydrogen bonds and thus the reestablishment of the oligomeric state. Some peptide variants could be transferred into a monomeric hairpin structure after heating and recooling (Fig. S11, S12 and S16, ESI†).
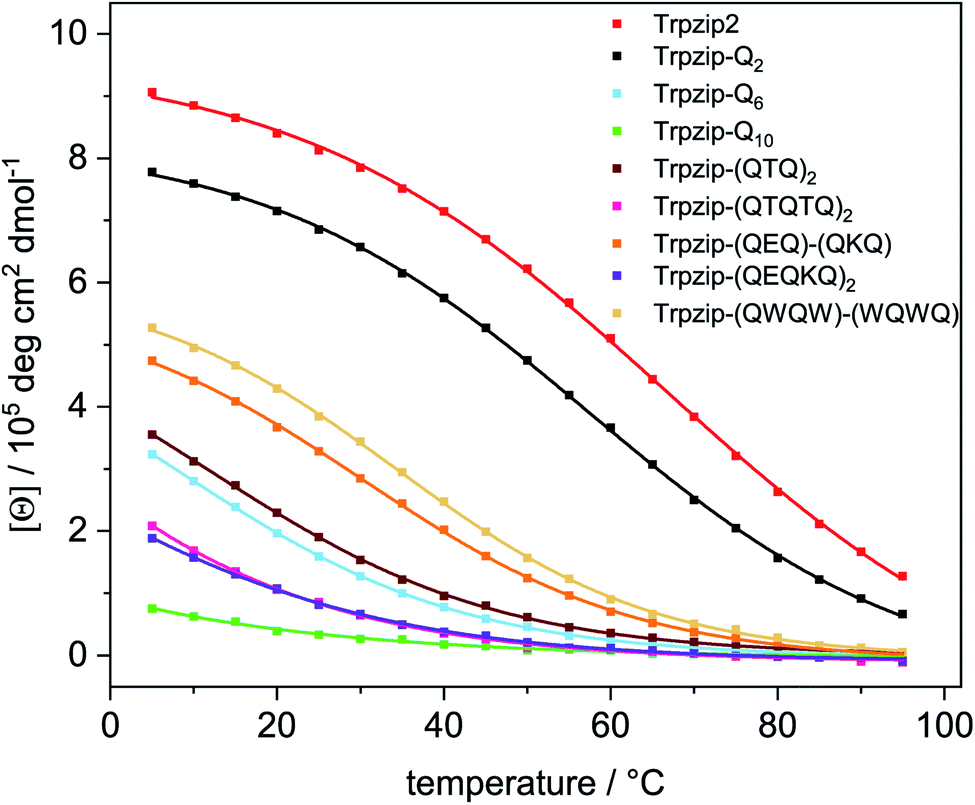
Transition temperatures were determined by fitting the second component of a SVD analysis of the amide I′ band (Tables S3, S4 and Fig. S14, 15, ESI†). For some of the peptides, the transition curves were too broad with no sigmoidal shape so that transition temperatures could not be determined for these cases but trends can be qualitatively evaluated. The template Trpzip2 has a transition temperature of about 70 °C.33,49 The melting temperature for Trpzip-Q2 is 68 °C and not significantly changed, as expected. No reliable transition temperature could be determined for Trpzip-Q6. For Trpzip-Q10, a higher transition temperature of 78 °C is obtained which can be explained by the oligomeric sheet structures establishing intermolecular hydrogen bonds between the sheets which inherently stabilize the structure. Trpzip-(QTQTQ)2 also forms oligomeric sheet structures, but the transition is only 65 °C. The decrease of the transition temperature might be caused by the reduction of two pairs of intramolecular glutamine cross-strand interactions in comparison to Trpzip-Q10. Exchanging threonines with charged amino acids (E, K) like for Trpzip-(QEQ)-(QKQ) shows a transition temperature of 45 °C which is significantly lower than for Trpzip2. Also Trpzip-(QWQW)-(WQWQ) has a comparable very low transition temperature of 39 °C. Both variants show a significant decrease of the hairpin stability. Transition temperatures were also determined from temperature-dependent CD measurements (Fig. S5 and S6, ESI†) by fitting the intensity loss of the aromatic exciton band at ∼228 nm of the tryptophans. The transition temperatures from CD (Table S4, ESI†) are lower than from FTIR, but show similar trends among the peptide variants. The main reason for the difference is that CD spectra – as analyzed here – monitor the tertiary contact of the hydrophobic Trp–Trp cross-strand interactions whereas FTIR spectra detect the polypeptide backbone vibrations and thus the secondary structure change. The Trp–Trp interactions are less stable than the hairpin structure.
A comparison of the transition curves (Fig. 8) reveals how sensitively the stability of the template hairpin is affected by the insertion of glutamines. We clearly observe a trend of decreasing hairpin stability as the number of glutamines increases indicating the ability of glutamines to destabilize β-hairpin conformations. Other studies have found that β-sheet formation in monomeric polyglutamine is thermodynamically unfavorable and propose that nucleation of β-sheet-rich aggregates requires an initial disorder to order transition.57 Our results are also in line with studies that used bottom-up designs of polyQ-rich peptides and observed a lack of β-structure.32
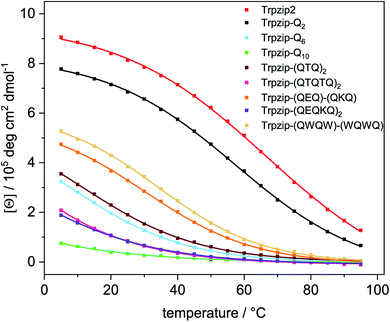 |
| | Fig. 8 Transition curves of Trpzip2, Trpzip-Qn models and other designs obtained from the intensity change at ∼228 nm from temperature-dependent CD spectra (Fig. S5, ESI†). Data were fitted using equation (eqn (S2), ESI†). The incorporation of glutamines or interrupted polyQ sequences results in a systematic weakening of the monitored Trp–Trp interactions and the hairpin structure lowering the intensity maxima and transition temperatures. The transition curves become less and even non-cooperative as the glutamine content increases. | |
Sub-millisecond structural dynamics
Laser-excited T-jump IR spectroscopy was applied to study the conformational dynamics of polyQ peptide models. We selected the maximum absorbance change at ∼1630 cm−1 in the temperature-dependent FTIR difference spectra (Fig. S9–S13, ESI†) representing the loss of the monomeric hairpin structure. The probe wavenumber varies slightly (±2 cm−1) among the variants. The final temperature after the T-jump was set to room temperature (∼25 °C) and the solvent dynamics were subtracted from the observed transient as described in Methods. Peptide transients are shown in Fig. S17, ESI† and were fitted with a mono-exponential decay function. The time constants of all models are in the μs time range (Table 2). T-jump measurements of Trpzip-Q10 and Trpzip-(QTQTQ)2 at ∼1630 cm−1 do not result in reliable transients since the absorbance changes were too small (as shown for Trpzip-Q10 in Fig. S18, ESI†). Both variants form oligomeric β-structures at 25 °C (Fig. S10 and S11, ESI†) which absorb at ∼1615 cm−1 thus we do not expect large absorbance changes at ∼1630 cm−1, the probe wavenumber of the monomeric hairpin. T-jump measurements at ∼1615 cm−1 were also performed for these two variants, but oligomer dynamics were not further analyzed. Since polyQ-rich peptides are prone for aggregation, T-jump measurements were repeated on the following day. If the time constant did not change (within the experimental error), we concluded that the peptide is still in a monomeric state. A significant change was observed for Trpzip-(QTQ)2, the relaxation time slows down from 3.22 μs to 3.88 μs after one day and the mono-exponential function does not fit the transient well. A bi-exponential fit function was better suited (Fig. S19a, ESI†) resulting in two time constants, τ1 = 3.24 μs and τ2 = 13.81 μs. The corresponding amplitudes provide a weighting factor, given here in percentages, with A1 = 88% and A2 = 12%. This shows that the first time constant remains the same (3.24 μs) as for the measurement the day before and yields a contribution of 88% to the total transient. In addition, a slower phase (13.81 μs) is observed with a significant contribution of 12%. We tentatively assign the slower phase to oligomeric structures which have formed to some extent after one day. For the peptide variants with charged amino acids (E, K), a better solubility is expected what should prevent aggregation. Trpzip-(QEQ)-(QKQ) has the same number of glutamines as Trpzip-(QTQ)2, and indeed the better solubility is supported by the mono-exponential fit of the transient and no change in time constant after one day indicating the monomeric structure. However, when the polyQ-rich sequence is elongated in Trpzip-(QEQKQ)2, two kinetic phases are observed even with a fresh sample. The transient is well described by a bi-exponential fit function resolving two time constants, 3.14 μs with a contribution of 64%, and 10.00 μs with a contribution of 36% which we assign to monomeric respectively oligomeric structures (Fig. S19b, ESI†). After one day, the time constants are the same (within the experimental error), only the amplitudes changed about 10% towards more monomer contribution. This peptide variant showed that heat application is capable to melt oligomeric structures into monomers (Fig. S12, ESI†) and the T-jump measurements of the first day might have shifted the monomer/oligomer distribution towards more monomers. The design Trpzip-(QWQW)-(WQWQ), which is closely related to the Trpzip2 template, reveals similar time constants as the template and stays in a monomeric state after one day.
Table 2 Relaxation times, τ, of peptide variants at ∼1630 cm−1, a final temperature of ∼25 °C and a concentration of 10 mg mL−1. Measurements were also performed on the following day
| Peptide |
τ
[μs] |
|
|
After 1 day |
|
Errors are statistical errors from the fitting procedure. Repeated measurements of independent samples result in an experimental error of ±200 ns.
When the transient was not well described with a mono-exponential fit function, a bi-exponential fit function was used providing two time constants (τ1, τ2) with corresponding amplitudes (A1, A2) given in percentages.
Precipitation of samples during lyophilisation is observed, oligomeric sheet structures are formed, no reliable data due to small absorbance changes at 1632 cm−1.
|
| Template Trpzip2 |
4.50 ± 0.14 |
4.58 ± 0.14 |
| Trpzip-Q2 |
4.83 ± 0.10 |
4.69 ± 0.09 |
| Trpzip-Q6 |
4.54 ± 0.06 |
4.49 ± 0.05 |
| Trpzip-Q10c |
— |
— |
| Trpzip-(QTQ)2b |
3.22 ± 0.02 |
3.88 ± 0.05 |
|
A
1
|
88% |
τ
1
|
3.24 ± 0.07 |
|
A
2
|
12% |
τ
2
|
13.81 ± 1.79 |
| Trpzip-(QTQTQ)2c |
— |
— |
| Trpzip-(QEQ)-(QKQ) |
5.17 ± 0.06 |
5.03 ± 0.05 |
| Trpzip-(QEQKQ)2b |
A
1
|
64% |
τ
1
|
3.14 ± 0.17 |
A
1
|
74% |
τ
1
|
3.14 ± 0.16 |
|
A
2
|
36% |
τ
2
|
10.00 ± 0.82 |
A
2
|
26% |
τ
2
|
10.73 ± 1.37 |
| Trpzip-(QWQW)-(WQWQ) |
4.84 ± 0.05 |
5.05 ± 0.05 |
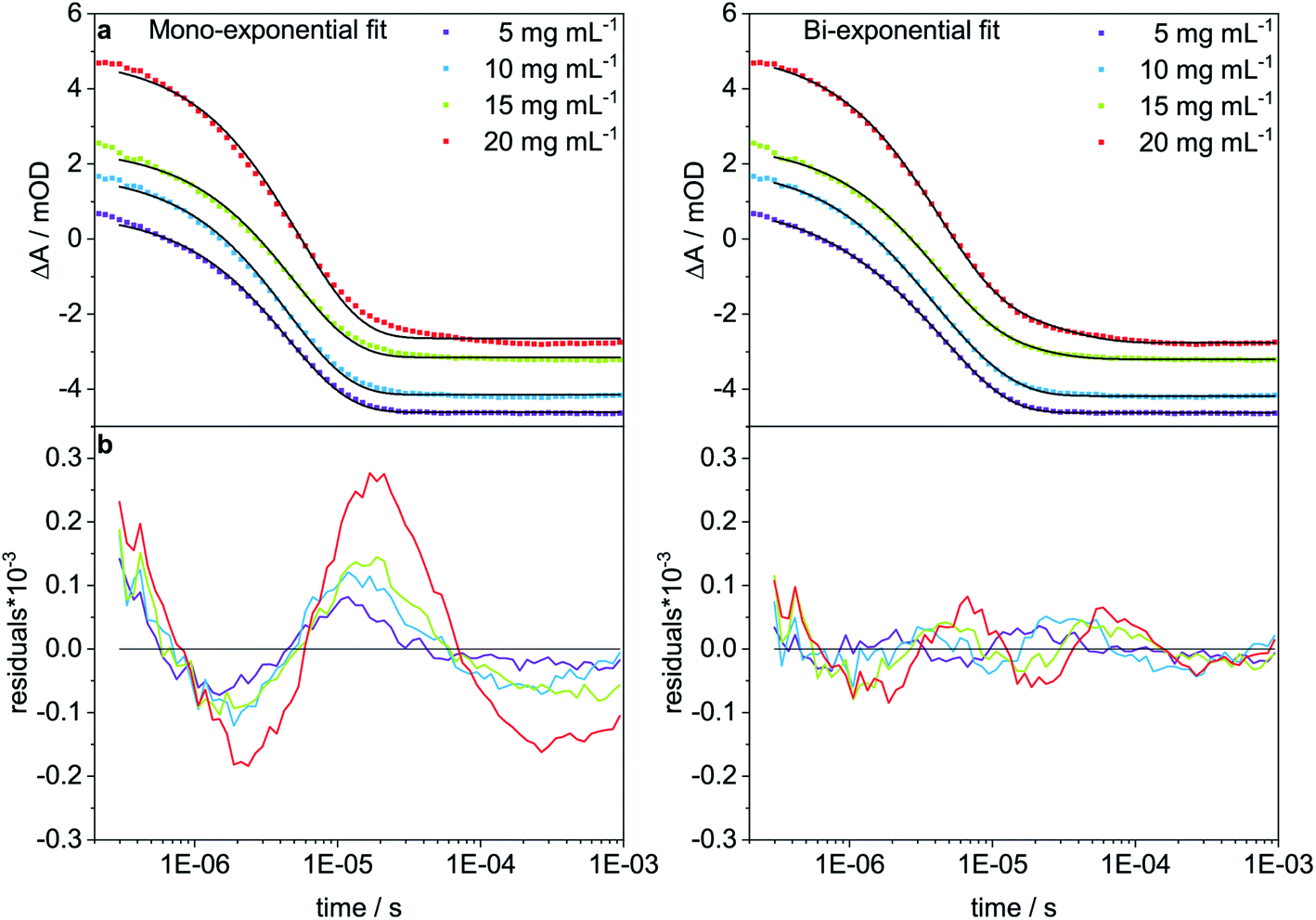
To analyze the concentration effect on the structural dynamics, Trpzip-Q6 measurements were performed at different concentrations (5–20 mg mL−1). Transients were fitted with mono- and bi-exponential fit functions and the quality of the fit was judged by the residuals. The residuals show the deviation of the fit-function from the experimental transient and clearly indicate that a mono-exponential fit is not adequate to describe the dynamics at high concentrations (Fig. 9). At a concentration of 5 mg mL−1, a contribution of 12% of a fast phase with 0.84 μs and a slower phase with a contribution of 88% of 4.95 μs are observed (Table 3). The main contribution is assigned to the hairpin dynamics, which is close to the time constant derived from the mono-exponential fit with 4.50 μs. Increasing the concentration leads to the increase of the time constants of both phases. At 10 mg mL−1, 4.54 μs is derived from the mono-exponential fit, lying in between the two time constants from the bi-exponential fit, both about a few μs with contributions of 36% and 64%. At 15 mg mL−1, a slower phase with 13.94 μs and a contribution of 20% and a faster phase with 3.77 μs and a contribution of 80% are observed. At 20 mg mL−1, one phase is similar with 4.04 μs (83%) and the other slowed down to 23.75 μs (17%). We conclude that the time constants of a few μs describe the hairpin dynamics, whereas time constants in the range of tens of μs are attributed to the presence of oligomeric structures. The measurements show that concentration is another important parameter that can favor oligomerization of polyQ model peptides.
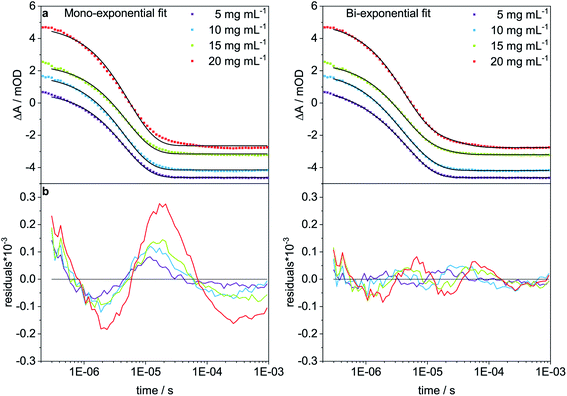 |
| | Fig. 9 T-jump measurements at 1632 cm−1 of Trpzip-Q6 at different concentrations (5–20 mg mL−1) and a final temperature of ∼25 °C. (a) Mono- and bi-exponential fits are shown in black for all transients and (b) residuals indicate the quality of the fit. | |
Table 3 Time constants, τ, of Trpzip-Q6 at a final temperature of ∼25 °C obtained from mono-and bi-exponential fits. For the bi-exponential fit the weighting factors A1 and A2 are given in percentage for the corresponding time constants τ1 and τ2
| Concentration [mg mL−1] |
Mono-exponential fit |
Bi-exponential fit |
|
τ [μs] |
A
1 [%] |
τ
1 [μs] |
A
2 [%] |
τ
2 [μs] |
|
Sample contains particles and is transparent.
Sample is turbid and not transparent.
|
| 5 |
4.50 ± 0.04 |
12 |
0.84 ± 0.10 |
88 |
4.95 ± 0.05 |
| 10 |
4.54 ± 0.06 |
36 |
2.20 ± 0.21 |
64 |
6.28 ± 0.28 |
| 15a |
4.94 ± 0.07 |
80 |
3.77 ± 0.13 |
20 |
13.94 ± 1.66 |
| 20b |
5.36 ± 0.11 |
83 |
4.04 ± 0.08 |
17 |
23.75 ± 1.98 |
Effect of individual glutamine side chains on structure and dynamics
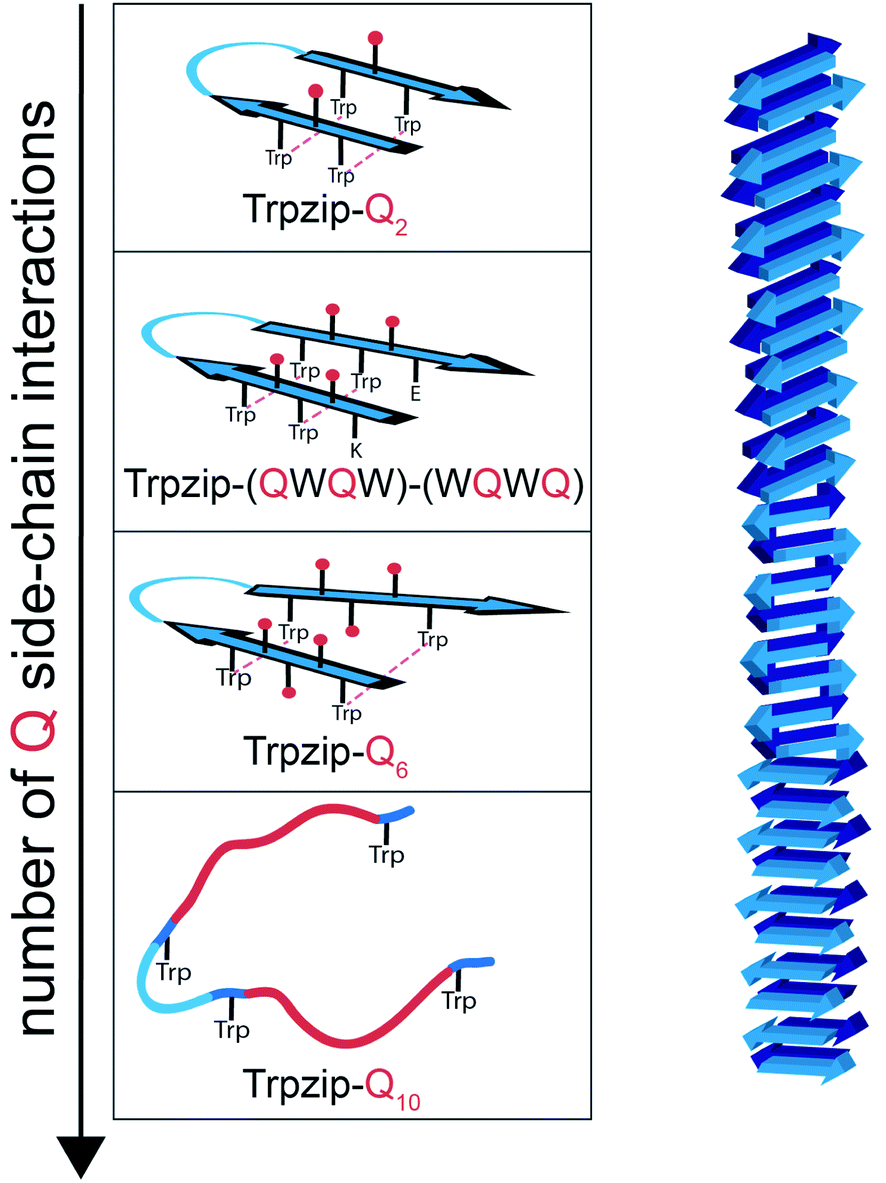
We have introduced various polyQ model peptides based on the template β-hairpin Trpzip2 and monitored the effect of the glutamines on structure and dynamics. By various spectroscopies, we could show that conformational changes depend primarily on two parameters: the number of glutamine side-chain interactions and the concentration. Fig. 10 sketches the structural changes for the different designs. Introduction of one Gln–Gln cross-strand pair in Trpzip-Q2 does not affect the soluble monomeric hairpin structure of the template which is stabilized by the hydrophobic cross-strand interactions of the two Trp–Trp pairs and the Asn–Gly turn. Increasing the number of glutamines up to three per strand weakens the interactions especially of the terminal Trp–Trp pair what is accompanied by more disordered, frayed termini. We can distinguish between more structured C-terminal ends in Trpzip-(QWQW)-(WQWQ) and less structured ones for Trpzip-Q6, but the template design maintains a monomeric hairpin structure, although somewhat distorted. Increasing the number of glutamines to five per strand in Trpzip-Q10 results in an almost complete loss of the hairpin-stabilizing Trp–Trp interactions and the peptide adopts a predominantly disordered structure with only weak hairpin features. It should be noted that besides the number of glutamine side-chain interactions, other amino acids might also distort the Trp–Trp interactions when they are part of the inserted polyQ-rich sequence. Among the different designs, the tendency for oligomerization is highest for Trpzip-Q10, as expected because of the relatively long and flexible glutamine tracts which favor the transient interaction of monomers. The designs have high potential to elucidate the impact of a varying number of Gln–Gln cross-strand interactions for the monomer conformation and furthermore for the oligomerization process. For Trpzip-Q6 we could show that the conformational dynamics change when the concentration is increased by the observation of two kinetic phases which we ascribe to the presence of both monomers and oligomers, respectively.
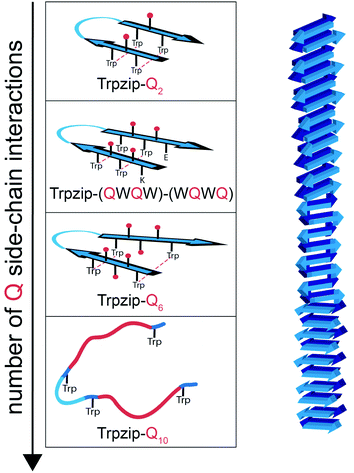 |
| | Fig. 10 Proposed conformational changes in designed polyQ peptide models (left) mimicking a hairpin structural motif of a polyQ fibril (right). All polyQ models are based on the hairpin template Trpzip2. The increase in glutamine side-chain interactions (spheres, colored in red) causes distortions in the monomeric hairpin structure and reduces the hairpin stabilizing Trp–Trp interactions (indicated by dashed lines), in particular at the termini. Trpzip-Q10 adopts a predominantly disordered structure with only weak hairpin features remaining from Trp–Trp interactions and the turn-promoting Asn–Gly motif (light blue). A high number of glutamines and high concentrations favor oligomer formation. The peptide models designed here allow to study the impact of individual glutamine side-chain interactions on structure and dynamics. | |
Conclusion
The combination of appropriately tailored peptide models and spectroscopic techniques has highest potential to analyze the unique interactions and dynamics of glutamine repeats. Laser-excited T-jump IR spectroscopy revealed one kinetic phase for the monomeric hairpins. Upon an increase in peptide concentration, we observe two time constants which we attribute to monomers and oligomeric β-sheets. The here studied sub-ms time domain provides access to the initial conformational states in polyQ fibril formation. Future NMR and IR studies will include site-specific 13CO labeling of both the glutamine backbone and side chain, respectively. This enables to distinguish even between backbone and side-chain dynamics of individual glutamines in the monomeric nucleus as well as in the oligomeric extended β-sheet.
Abbreviations
| CD | Circular dichroism |
| FMOC | Fluorenyl-methyloxycarbonyl |
| FTIR | Fourier-transform infrared |
| G | Glycine |
| Ho-YAG | Holmium yttrium aluminum garnet |
| HSQC | Heteronuclear single quantum coherence |
| MCT | Mercury cadmium telluride |
| NMR | Nuclear magnetic resonance |
| QCL | Quantum cascade laser |
| SVD | Singular value decomposition |
| TFA | Trifluoroacetate |
| T-jump | Temperature-jump |
| Trp | Tryptophan |
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (SFB 969, A2 to K. H.), the Young Scholar Fund of the University of Konstanz (to M. K.) and the Konstanz Research School Chemical Biology. We thank Timothy A. Keiderling for valuable discussions and Anke Friemel for technical assistance at the NMR Core Facility.
References
- C. de Chiara and A. Pastore, Kaleidoscopic protein–protein interactions in the life and death of ataxin-1: new strategies against protein aggregation, Trends Neurosci., 2014, 37(4), 211–218 CrossRef CAS.
- H. M. Saunders and S. P. Bottomley, Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins, Protein Eng., Des. Sel., 2009, 22(8), 447–451 CrossRef CAS.
- E. Scherzinger, A. Sittler, K. Schweiger, V. Heiser, R. Lurz, R. Hasenbank, G. P. Bates, H. Lehrach and E. E. Wanker, Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(8), 4604–4609 CrossRef CAS.
- S. Chen, F. A. Ferrone and R. Wetzel, Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(18), 11884–11889 CrossRef CAS.
- T. Yushchenko, E. Deuerling and K. Hauser, Insights into the Aggregation Mechanism of PolyQ Proteins with Different Glutamine Repeat Lengths, Biophys. J., 2018, 114(8), 1847–1857 CrossRef CAS.
- R. Schneider, M. C. Schumacher, H. Mueller, D. Nand, V. Klaukien, H. Heise, D. Riedel, G. Wolf, E. Behrmann, S. Raunser, R. Seidel, M. Engelhard and M. Baldus, Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy, J. Mol. Biol., 2011, 412(1), 121–136 CrossRef CAS.
- L. E. Buchanan, J. K. Carr, A. M. Fluitt, A. J. Hoganson, S. D. Moran, J. J. de Pablo, J. L. Skinner and M. T. Zanni, Structural motif of polyglutamine amyloid fibrils discerned with mixed-isotope infrared spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(16), 5796–5801 CrossRef CAS.
- D. Punihaole, R. S. Jakubek, R. J. Workman and S. A. Asher, Interaction Enthalpy of Side Chain and Backbone Amides in Polyglutamine Solution Monomers and Fibrils, J. Phys. Chem. Lett., 2018, 9(8), 1944–1950 CrossRef CAS.
- A. Natalello, A. M. Frana, A. Relini, A. Apicella, G. Invernizzi, C. Casari, A. Gliozzi, S. M. Doglia, P. Tortora and M. E. Regonesi, A major role for side-chain polyglutamine hydrogen bonding in irreversible ataxin-3 aggregation, PLoS One, 2011, 6(4), e18789 CrossRef CAS.
- R. Wetzel, Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence, J. Mol. Biol., 2012, 421(4–5), 466–490 CrossRef CAS.
- S. Chen, V. Berthelier, W. Yang and R. Wetzel, Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity, J. Mol. Biol., 2001, 311(1), 173–182 CrossRef CAS.
- S. Chen, V. Berthelier, J. B. Hamilton, B. O'Nuallain and R. Wetzel, Amyloid-like features of polyglutamine aggregates and their assembly kinetics, Biochemistry, 2002, 41(23), 7391–7399 CrossRef CAS.
- N. Slepko, A. M. Bhattacharyya, G. R. Jackson, J. S. Steffan, J. L. Marsh, L. M. Thompson and R. Wetzel, Normal-repeat-length polyglutamine peptides accelerate aggregation nucleation and cytotoxicity of expanded polyglutamine proteins, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(39), 14367–14372 CrossRef CAS.
- R. H. Walters and R. M. Murphy, Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation, J. Mol. Biol., 2009, 393(4), 978–992 CrossRef CAS.
- D. Punihaole, R. S. Jakubek, R. J. Workman, L. E. Marbella, P. Campbell, J. D. Madura and S. A. Asher, Monomeric Polyglutamine Structures That Evolve into Fibrils, J. Phys. Chem. B, 2017, 121(24), 5953–5967 CrossRef CAS.
- M. F. Perutz, J. T. Finch, J. Berriman and A. Lesk, Amyloid fibers are water-filled nanotubes, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(8), 5591–5595 CrossRef CAS.
- S. B. Priya and M. M. Gromiha, Structural insights into the aggregation mechanism of huntingtin exon 1 protein fragment with different polyQ-lengths, J. Cell. Biochem., 2019, 120(6), 10519–10529 CrossRef CAS.
- S. L. Crick, M. Jayaraman, C. Frieden, R. Wetzel and R. V. Pappu, Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(45), 16764–16769 CrossRef CAS.
- K. Kar, M. Jayaraman, B. Sahoo, R. Kodali and R. Wetzel, Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent, Nat. Struct. Mol. Biol., 2011, 18(3), 328–336 CrossRef CAS.
- Q. C. Zhang, T.-l. Yeh, A. Leyva, L. G. Frank, J. Miller, Y. E. Kim, R. Langen, S. Finkbeiner, M. L. Amzel, C. A. Ross and M. A. Poirier, A Compact β Model of Huntingtin Toxicity, J. Biol. Chem., 2011, 286(10), 8188–8196 CrossRef CAS.
- C. Peters-Libeu, J. Miller, E. Rutenber, Y. Newhouse, P. Krishnan, K. Cheung, D. Hatters, E. Brooks, K. Widjaja, T. Tran, S. Mitra, M. Arrasate, L. A. Mosquera, D. Taylor, K. H. Weisgraber and S. Finkbeiner, Disease-Associated Polyglutamine Stretches in Monomeric Huntingtin Adopt a Compact Structure, J. Mol. Biol., 2012, 421(4), 587–600 CrossRef CAS.
- M. F. Perutz, T. Johnson, M. Suzuki and J. T. Finch, Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(12), 5355–5358 CrossRef CAS.
- P. Sikorski and E. Atkins, New Model for Crystalline Polyglutamine Assemblies and Their Connection with Amyloid Fibrils, Biomacromolecules, 2005, 6(1), 425–432 CrossRef CAS.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. Ø. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Structure of the cross-β spine of amyloid-like fibrils, Nature, 2005, 435(7043), 773–778 CrossRef CAS.
- C. L. Hoop, H. K. Lin, K. Kar, G. Magyarfalvi, J. M. Lamley, J. C. Boatz, A. Mandal, J. R. Lewandowski, R. Wetzel and P. C. van der Wel, Huntingtin exon 1 fibrils feature an interdigitated beta-hairpin-based polyglutamine core, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(6), 1546–1551 CrossRef CAS.
- B. S. Heck, F. Doll and K. Hauser, Length-dependent conformational transitions of polyglutamine repeats as molecular origin of fibril initiation, Biophys. Chem., 2014, 185, 47–57 CrossRef CAS.
- S. Chen and R. Wetzel, Solubilization and disaggregation of polyglutamine peptides, Protein Sci., 2001, 10(4), 887–891 CrossRef CAS.
- D. Sharma, S. Sharma, S. Pasha and S. K. Brahmachari, Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions, FEBS Lett., 1999, 456(1), 181–185 CrossRef CAS.
- S. Sen, D. Dash, S. Pasha and S. K. Brahmachari, Role of histidine interruption in mitigating the pathological effects of long polyglutamine stretches in SCA1: a molecular approach, Protein Sci., 2003, 12(5), 953–962 CrossRef CAS.
- A. K. Thakur and R. Wetzel, Mutational analysis of the structural organization of polyglutamine aggregates, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(26), 17014–17019 CrossRef CAS.
- M. H. Smith, T. F. Miles, M. Sheehan, K. N. Alfieri, B. Kokona and R. Fairman, Polyglutamine fibrils are formed using a simple designed β-hairpin model, Proteins, 2010, 78(8), 1971–1979 CrossRef CAS.
- K. Kar, C. L. Hoop, K. W. Drombosky, M. A. Baker, R. Kodali, I. Arduini, P. C. van der Wel, W. S. Horne and R. Wetzel, Beta-hairpin-mediated nucleation of polyglutamine amyloid formation, J. Mol. Biol., 2013, 425(7), 1183–1197 CrossRef CAS.
- A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Tryptophan zippers: stable, monomeric β-hairpins, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(10), 5578–5583 CrossRef CAS.
- K. Hauser, C. Krejtschi, R. Huang, L. Wu and T. A. Keiderling, Site-Specific Relaxation Kinetics of a Tryptophan Zipper Hairpin Peptide Using Temperature-Jump IR Spectroscopy and Isotopic Labeling, J. Am. Chem. Soc., 2008, 130(10), 2984–2992 CrossRef CAS.
- W. Y. Yang and M. Gruebele, Detection-Dependent Kinetics as a Probe of Folding Landscape Microstructure, J. Am. Chem. Soc., 2004, 126(25), 7758–7759 CrossRef CAS.
- K. C. Jones, C. S. Peng and A. Tokmakoff, Folding of a heterogeneous β-hairpin peptide from temperature-jump 2D IR spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(8), 2828–2833 CrossRef CAS.
- C. D. Snow, L. Qiu, D. Du, F. Gai, S. J. Hagen and V. S. Pande, Trp zipper folding kinetics by molecular dynamics and temperature-jump spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(12), 4077–4082 CrossRef CAS.
- D. Scheerer, H. Chi, D. McElheny, T. A. Keiderling and K. Hauser, Enhanced Sensitivity to Local Dynamics in Peptides by Use of Temperature-Jump IR Spectroscopy and Isotope Labeling, Chem.–Eur. J., 2020, 26(16), 3524–3534 CrossRef CAS.
- D. Scheerer, H. Chi, D. McElheny, T. A. Keiderling and K. Hauser, Isotopically Site-Selected Dynamics of a Three-Stranded β-Sheet Peptide Detected with Temperature-Jump Infrared-Spectroscopy, J. Phys. Chem. B, 2018, 122(46), 10445–10454 CrossRef CAS.
- C. M. Davis and R. B. Dyer, Dynamics of an Ultrafast Folding Subdomain in the Context of a Larger Protein Fold, J. Am. Chem. Soc., 2013, 135(51), 19260–19267 CrossRef CAS.
- A. Popp, D. Scheerer, B. Heck and K. Hauser, Biomolecular dynamics studied with IR-spectroscopy using quantum cascade lasers combined with nanosecond perturbation techniques, Spectrochim. Acta, Part A, 2017, 181, 192–199 CrossRef CAS.
- D. Du, Y. Zhu, C.-Y. Huang and F. Gai, Understanding the key factors that control the rate of β-hairpin folding, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(45), 15915 CrossRef CAS.
- A. Popp, L. Wu, T. A. Keiderling and K. Hauser, Effect of Hydrophobic Interactions on the Folding Mechanism of β-Hairpins, J. Phys. Chem. B, 2014, 118(49), 14234–14242 CrossRef CAS.
- A. Popp, D. Scheerer, H. Chi, T. A. Keiderling and K. Hauser, Site-Specific Dynamics of β-Sheet Peptides with DPro–Gly Turns Probed by Laser-Excited Temperature-Jump Infrared Spectroscopy, ChemPhysChem, 2016, 17(9), 1273–1280 CrossRef CAS.
- D. K. Wilkins, S. B. Grimshaw, V. Receveur, C. M. Dobson, J. A. Jones and L. J. Smith, Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques, Biochemistry, 1999, 38(50), 16424–16431 CrossRef CAS.
- D. Scheerer, H. Chi, D. McElheny, A. Samer, T. A. Keiderling and K. Hauser, Role of Aromatic Cross-Links in Structure and Dynamics of Model Three-Stranded β-Sheet Peptides, J. Phys. Chem. A, 2018, 122(2), 543–553 CrossRef CAS.
- W. O. Wray, T. Aida and R. B. Dyer, Photoacoustic Cavitation and Heat Transfer Effects in the Laser-Induced Temperature Jump in Water, Appl. Phys. B, 2002, 74(1), 57–66 CrossRef CAS.
- R. Huang, L. Wu, D. McElheny, P. Bouř, A. Roy and T. A. Keiderling, Cross-Strand Coupling and Site-Specific Unfolding Thermodynamics of a Trpzip β-Hairpin Peptide Using 13C Isotopic Labeling and IR Spectroscopy, J. Phys. Chem. B, 2009, 113(16), 5661–5674 CrossRef CAS.
- L. Wu, D. McElheny, R. Huang and T. A. Keiderling, Role of tryptophan-tryptophan interactions in Trpzip β-hairpin formation, structure, and stability, Biochemistry, 2009, 48(43), 10362–10371 CrossRef CAS.
- V. Setnicka, R. Huang, C. L. Thomas, M. A. Etienne, J. Kubelka, R. P. Hammer and T. A. Keiderling, IR study of cross-strand coupling in a β-hairpin peptide using isotopic labels, J. Am. Chem. Soc., 2005, 127(14), 4992–4993 CrossRef CAS.
- T. Wang, Y. Xu, D. Du and F. Gai, Determining β-sheet stability by Fourier transform infrared difference spectra, Biopolymers, 2004, 75(2), 163–172 CrossRef CAS.
- A. W. Smith, H. S. Chung, Z. Ganim and A. Tokmakoff, Residual Native Structure in a Thermally Denatured β-Hairpin, J. Phys. Chem. B, 2005, 109(36), 17025–17027 CrossRef CAS.
- A. W. Smith and A. Tokmakoff, Amide I two-dimensional infrared spectroscopy of β-hairpin peptides, J. Chem. Phys., 2007, 126(4), 045109 CrossRef.
- R. Sarroukh, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, ATR-FTIR: a “rejuvenated” tool to investigate amyloid proteins, Biochim. Biophys. Acta, 2013, 1828(10), 2328–2338 CrossRef CAS.
- S. D. Moran and M. T. Zanni, How to Get Insight into Amyloid Structure and Formation from Infrared Spectroscopy, J. Phys. Chem. Lett., 2014, 5(11), 1984–1993 CrossRef CAS.
- A. Barth, The infrared absorption of amino acid side chains, Prog. Biophys. Mol. Biol., 2000, 74(3), 141–173 CrossRef CAS.
- X. Wang, A. Vitalis, M. A. Wyczalkowski and R. V. Pappu, Characterizing the conformational ensemble of monomeric polyglutamine, Proteins, 2006, 63(2), 297–311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05299j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Karin
Hauser
* and
Karin
Hauser