
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal endogenous antibody recruiting nanobodies capable of triggering immune effectors for targeted cancer immunotherapy†
Haofei
Hong
 a,
Chen
Li
a,
Liang
Gong
a,
Jinfeng
Wang
a,
Dan
Li
a,
Jie
Shi
a,
Zhifang
Zhou
a,
Zhaohui
Huang
*bc and
Zhimeng
Wu
*a
a,
Chen
Li
a,
Liang
Gong
a,
Jinfeng
Wang
a,
Dan
Li
a,
Jie
Shi
a,
Zhifang
Zhou
a,
Zhaohui
Huang
*bc and
Zhimeng
Wu
*a
aKey Laboratory of Carbohydrate Chemistry & Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi, 214122, China. E-mail: zwu@jiangnan.edu.cn
bWuxi Cancer Institute, Affiliated Hospital of Jiangnan University, Wuxi, 214062, China. E-mail: Zhaohuihuang@jiangnan.edu.cn
cLaboratory of Cancer Epigenetics, School of Medicine, Jiangnan University, Wuxi, 214122, China
First published on 11th February 2021
Abstract
Developing monoclonal antibodies (mAbs) for cancer immunotherapy is expensive and complicated. Nanobodies are small antibodies possessing favorable pharmacological properties compared with mAbs, but have limited anticancer efficacy due to the lack of an Fc region and poor pharmacokinetics. In this context, engineered universal endogenous antibody-recruiting nanobodies (UEAR Nbs), as a general and cost-effective approach, were developed to generate functional antibody-like nanobodies that could recapitulate the Fc biological functions for cancer immunotherapy. The UEAR Nbs, composed of the IgG binding domain and nanobody, were recombinantly expressed in E. coli and could recruit endogenous IgGs onto the cancer cell surface and trigger potent immune responses to kill cancer cells in vitro. Moreover, it was proved that UEAR Nbs displayed significantly improved half-lives in vivo. The in vivo antitumor efficacy of UEAR Nbs was demonstrated in a murine model using EGFR positive triple-negative breast cancer (TNBC).
Introduction
Nanobodies, also known as single-domain antibodies, are heavy-chain-only antibodies with a single variable antigen-binding domain derived from camelids.1 Compared to conventional monoclonal antibodies (mAbs), nanobodies have unique characteristics including small size, high specific affinity, outstanding stability and solubility, easy production and manipulation and low immunogenicity.2,3 Now, it is well recognized that nanobodies are a promising alternative to conventional mAbs from basic research to clinical application, where nanobodies or chemical functionalized nanobodies are extensively explored as imaging, diagnostic and therapeutic agents in diverse areas.4,5 For example, the first nanobody, caplacizumab, was approved by the EU and FDA for acquired thrombotic thrombocytopenic purpura treatment recently,6,7 and more than dozens of nanobody-based medicines are in different stages of clinical trials. However, for cancer immunotherapy, the capability of nanobodies was largely limited because of the lack of an Fc region and the poor pharmacokinetic profile. Detailed mechanism studies have established that the Fc region in mAbs could trigger a potent innate immunity to kill cancer cells and prolong the serum half-life of mAbs, through antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and Fc receptor-mediated recycling mechanism, respectively.8 Therefore, a general and cost-effective technology that is able to generate antibody-like nanobodies that could recapitulate the Fc biological functions for cancer immunotherapy is required.Currently, nanobody fusion with anti-HSA nanobody and modification with a half-life extension moiety are two effective technologies to improve the pharmacokinetics of nanobodies, but these are unable to reinstate the Fc-mediated biological functions.9,10 Engineered nanobody fusion with the Fc portion is a popular technology, whose product has advanced into clinical trials for cancer immunotherapy.11 However, Fc-fused nanobodies suffer from similar shortcomings to the conventional mAbs, such as complicated and expensive production,12 potential immunogenicity caused by human allotype of the Fc portion,13 and reduced ADCC or CDC activity due to the low affinity to FcγRIIIa on immune cells or the complement.14,15 Antibody-recruiting molecules (ARMs) are bifunctional molecules composed of a cell-targeting moiety and an antibody-binding component, which could bridge the target cells and immune system and induce downstream immunity to eliminate the target cells.16,17 Many rationally designed ARMs, where preferred haptens that could be recognized by natural occurring endogenous antibodies, such as dinitrophenyl (DNP), galactose-α-1,3-galactose (αGal) and rhamnose (Rha), as an antibody-binding component, have been successfully achieved for cancer,18–22 viruses,23,24 bacteria25,26 and others.27 For example, we and others demonstrated that nanobody-DNP conjugates could form an in situ immune-complex with specific endogenous anti-DNP antibody existing in human serum and subsequently provoke potent ADCC and CDC cytotoxicity to target destructing cancer cells in vitro and exhibit in vivo antitumor activity in mouse xenograft models.28,29 Notably, it is observed that the pharmacokinetic profile of nanobody-DNP conjugates was improved more than 20-fold in the presence of anti-DNP antibodies.28 This strategy provides a novel and effective solution to reconstitute the missing Fc-mediated anticancer biological function and extend the half-life of the nanobody simultaneously, which could potentially avoid the adverse immunogenicity issues associated with nanobody-Fc fusion protein.
However, the success of the specific endogenous antibody-recruiting strategy for cancer immunotherapy, including ARMs, nanobody-DNP conjugates and others, was heavily dependent on the amount of anti-hapten antibodies in the human system and their affinity to ARMs, given the fact that only approximately 1–3% of hapten-specific antibodies pre-exist in human blood.30–32 Although pre-vaccination could generate high titers of the hapten-specific antibody for this purpose, the complicated treatment procedure as well as the unknown side effects may attenuate the possibility of clinical transformation of this technology. To address the short supply of specific endogenous antibody, relative abundant human IgGs (10–20% in total serum protein),33 a product of delayed immune response to infections in life, attract our attention. We speculated that an engineered nanobody that could bind with the target cancer cells and recruit the universal endogenous IgGs in human serum without compromising the capability of Fc biological function may solve the above challenge. In this case, a synthetic IgG-binding domain derived from domain B in Staphylococcus aureus protein A,34,35 also designated as the ZZ domain, which has been successfully applied in IgG purification,36,37 detection38,39 and immunological therapy,40 seems to fit these requirements. Importantly, crystal structure studies demonstrated that this domain and the human Fc receptor on immune cells could bind at different regions of an IgG Fc portion,35,41 respectively, indicating that the fusion of this domain with the nanobody may not affect the required Fc biological functions.
We report here the design and construction of an engineered universal endogenous antibody-recruiting nanobody (termed UEAR Nbs to describe this technology) that was recombinantly expressed in E. coli by fusing the IgG binding domain with the nanobody through a GS linker. As shown in Fig. 1, the UEAR Nbs could recognize the cancer cells and recruit general endogenous IgGs onto the surface of cancer cells, followed by triggering immune responses, such as ADCC, CDC and ADCP, to eliminate tumor cells in a precise manner by the engagement of IgG Fc-mediated mechanisms. Furthermore, the pharmacokinetics of the UEAR Nbs as well as the anti-cancer efficacy in triple-negative breast cancer (TNBC) xenograft mice was evaluated in vivo.
 | ||
| Fig. 1 UEAR Nb-mediated cell lysis via triggering ADCC, CDC and ADCP; and half-life extension via forming an immunocomplex. | ||
Results and discussion
Design and expression of UEAR Nbs 7D12-ZZ and ZZ-7D12 in E. coli
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein belonging to a family of four receptor tyrosine kinases. Ligand-induced activation of the EGFR and its downstream signaling pathways are involved in many cellular processes, such as cell proliferation, mobility and differentiation.42 Overexpression of the EGFR has been observed in a variety of cancer types, including breast cancer (especially in TNBC), lung cancer, metastatic colorectal cancer, and head and neck cancer.43 Consequently, the EGFR is a validated therapeutic target as well as a diagnostic biomarker in clinic. In this work, an EGFR targeting nanobody 7D12 was selected as the tumor-targeting molecule. Previous studies have confirmed that 7D12 binds to domain III of human EGFR with high affinity44 and has the potential to overcome EGFR ecto-domain mutant-mediated primary and secondary resistance to clinically approved full mAbs.45 For a universal antibody-recruiting moiety, the ZZ domain was selected as the Fc-binding molecule because of its high binding affinity and specificity.Two UEAR Nbs, 7D12-ZZ and ZZ-7D12, where the ZZ domain was placed at the N- and C-terminus of nanobody 7D12 via a GS linker (GGGGSGGGGS), respectively, were designed (Fig. 2A). In addition, a His6 tag was genetically engineered to the C-terminus for affinity purification purpose. After the recombinant plasmids of pET22b-7D12-ZZ and pET22b-ZZ-7D12 were constructed, they were transformed into E. coli BL21 (DE3) cells, which were cultured at 37 °C in Terrific-Broth (TB) medium and induced with isopropyl-β-D-thiogalactopyranoside (IPTG) to express proteins. The recombinant UEAR Nbs were harvested and purified by Ni-resin affinity chromatography to give 7D12-ZZ and ZZ-7D12 in a yield of 50 and 100 mg L−1, respectively. Nanobody 7D12 and the ZZ domain were recombinantly expressed similarly in E. coli BL21 (DE3) with a yield of 40 and 90 mg L−1. All proteins were then characterized by SDS-PAGE. 7D12 or ZZ domain protein appeared as a single band around 16 kDa (Fig. 2B, lane 1 and 2), while 7D12-ZZ and ZZ-7D12 appeared as a single band with a molecular weight of approximately 32 kDa (Fig. 2B, lane 3 and 4). These proteins were unambiguously confirmed by MS, which was consistent with the calculated molecular weight (Fig. S1†).
 | ||
| Fig. 2 Design and recombinant expression of UEAR. (A) Illustration of 7D12, ZZ domain, 7D12-ZZ and ZZ-7D12. (B) SDS-PAGE analysis of purified proteins: lanes 1–4, purified 7D12, ZZ domain, 7D12-ZZ and ZZ-7D12, respectively; lane M, marker. | ||
Characterization of UEAR Nb binding with IgGs and cancer cell overexpressing EGFR
With UEAR Nbs, 7D12-ZZ and ZZ-7D12, in hand, we first evaluated their binding affinity to IgGs from different mammalian species using an ELISA method. The experiments were performed by coating the plates with PBS buffer, 7D12, ZZ domain, 7D12-ZZ and ZZ-7D12 proteins, respectively, and then incubated with IgGs derived from mouse, rabbit and human, followed by HRP-modified secondary antibodies. The results were determined by using a TMB kit. As shown in Fig. 3A, no appreciable signal was observed in PBS and 7D12 coated wells. By contrast, strong signals were observed in the ZZ domain, 7D12-ZZ and ZZ-7D12 coated wells incubated with three IgG samples. The intensities of absorption at 450 nm from 7D12-ZZ and ZZ-7D12 groups were comparable to those of the ZZ domain group. This result suggested that placing the ZZ domain at the C- or N-terminus of nanobody 7D12 did not alter its structure significantly; both formats retained their binding capability to IgGs after fusion with nanobody 7D12. | ||
| Fig. 3 IgG and EGFR binding assays. (A) ELISA evaluation of the binding affinity of 7D12, ZZ, 7D12-ZZ and ZZ-7D12 to IgGs from mouse, rabbit and human. (B) Competitive ELISA evaluation of the IC50 of 7D12, 7D12-ZZ and ZZ-7D12 to the EGFR. Error bars represent the SD of three parallel experiments. ****: P < 0.0001. | ||
Then, to evaluate the binding affinity of various Nbs to the EGFR, a standard competitive cell-based ELISA was performed. After being fixed with EGFR positive A431 cells, the wells were treated with 7D12, 7D12-ZZ or ZZ-7D12 in the presence of recombinant human EGFR with different concentrations (0.016 to 500 nmol L−1). Thereafter, wells were incubated with anti-His6 tag antibodies and HRP-modified anti-mouse IgG antibodies. The results were finally determined using a TMB kit. As shown in Fig. 3B, 7D12, 7D12-ZZ and ZZ-7D12 all presented an EGFR dose-dependent binding efficiency, with an IC50 of 6.3, 3.8 and 9.7 nmol L−1, respectively. This result indicated that both of UEAR Nbs remained at a nanomolar level of binding affinity to the EGFR. However, compared with 7D12, the binding affinity of 7D12-ZZ was improved approximately 1.7 fold while ZZ-7D12 exhibited slightly decreased binding affinity.
Next, we need to prove whether UEAR Nbs could bind specifically to EGFR positive cancer cells and recruit endogenous IgGs onto cell surfaces. Accordingly, three cell lines, namely human TNBC MDA-MB-468 cells, human squamous carcinoma A431 cells with high EGFR expression, and MCF7 cells with low EGFR expression, were used for this study. The cells were seeded in 24-well plates and treated with 7D12, ZZ domain, 7D12-ZZ and ZZ-7D12, respectively, followed by incubation with mouse IgGs and Dylight 488-conjugated anti-mouse IgG antibodies. The cells were then imaged using a fluorescence microscope. As presented in Fig. 4A and S2–S4,† only strong fluorescence signals were observed on EGFR positive A431 and MDA-MB-468 cells treated with UEAR Nbs. No obvious fluorescence was observed in 7D12 and ZZ domain groups as well as the EGFR negative MCF7 group. This result demonstrated that UEAR Nbs not only retained their EGFR binding activity and specificity but were capable of recruiting universal IgGs onto EGFR positive cell surfaces.
 | ||
| Fig. 4 Cell-binding and IgG-recruiting assays. (A) Immunofluorescence image of cells treated with nanobody 7D12, ZZ domain, UEAR Nbs 7D12-ZZ or ZZ-7D12 (scale bar = 20 μm). (B) Flow cytometry assays and (C) the corresponding MFI of cells treated with 7D12, ZZ domain, UEAR Nbs 7D12-ZZ or ZZ-7D12. Error bars show the SD of three parallel experiments. ****: P < 0.0001. | ||
Furthermore, we conducted a quantitative evaluation of cell-binding and IgG-recruiting ability of UEAR Nbs using flow cytometry. As shown in Fig. 4B and C, only EGFR positive cells (A431 and MDA-MB-468), but not EGFR negative cells (MCF7), displayed significantly IgG anchoring when treated with UEAR Nbs. The mean fluorescence intensity (MFI) detected on A431 after incubation with 7D12-ZZ or ZZ-7D12 was 6.8- and 7.3-fold higher than that of 7D12, respectively. Notably, the MFI detected from the MDA-MB-468 cell group treated with 7D12-ZZ or ZZ-7D12 was 20.4- and 20.9-fold higher as compared with the result of the 7D12 control group. To further reveal the effect of ZZ domain location on binding affinity in UEAR Nbs, A431 and MDA-MB-468 cells were incubated with different concentrations (5, 20 and 50 nmol L−1) of 7D12-ZZ or ZZ-7D12 and then analyzed using flow cytometry. Interestingly, the two UEAR Nbs displayed similar binding affinities to EGFR positive cancer cells as well as the capabilities to recruit IgGs (Fig. S5†).
In vitro cytotoxicity evaluation induced by UEAR Nbs
ADCC, CDC and ADCP are three known crucial anti-cancer mechanisms engaged by the Fc-portion of therapeutic mAbs. ADCC critically relies on the binding of Fc with the specific FcγRIIIa receptor on innate immune cells, such as NK cells and δT cells existing in human peripheral blood mononuclear cells (PBMCs). Based on the different binding profiles of the ZZ domain and FcγRIIIa receptor to Fc-portion,46,47 it was expected that UEAR Nbs may bridge the cancer cells and immune cells through the Fc-terminus of the recruited universal endogenous IgGs to trigger ADCC cytotoxicity. To verify this hypothesis, we first utilized A431 cells and MDA-MB-468 cells to assess ADCC. Following incubation with different concentrations (0.2, 2, 20 and 50 nmol L−1) of UEAR Nbs and mouse serum (as the source of endogenous IgGs), cells were co-cultured with freshly isolated PBMCs (as the source of immune cells). Then, cell lysis was determined using a lactate dehydrogenase (LDH) cytotoxicity kit. As presented in Fig. 5A, a dose-dependent ADCC activity in the experiment group was observed, where cell lysis increased with the increase of UEAR Nb concentrations. By using a dosage of 50 nmol L−1 of UEAR Nbs, over 25 and 44% of A431 and MDA-MB-468 cells were killed, respectively. However, the viabilities of A431 and MDA-MB-468 cells were not influenced (<10%) when treated with 7D12. To induce an effective ADCC in this system, an immune complex, formed by cancer cells, UEAR Nbs, endogenous IgGs and FcγRIIIa receptor on immune cells, is required. Immunofluorescence image and flow cytometry experiments have demonstrated that a ternary complex by cancer cells, UEAR Nbs and endogenous IgGs was formed successfully. The ADCC results further demonstrated that immune cells were involved in this system. Although we were unable to isolate the active immune complex, the result clearly indicated that UEAR Nbs, both 7D12-ZZ and ZZ-7D12, could bridge the cancer cells and immune cells by the engagement of endogenous IgGs effectively, thereby triggering ADCC to destruct target cells. | ||
| Fig. 5 In vitro cytotoxicity assays. (A) ADCC mediated by different concentrations of 7D12, 7D12-ZZ or ZZ-7D12. (B) CDC mediated by 50 nmol L−1 of 7D12, 7D12-ZZ or ZZ-7D12 in the presence of RC or HIRC. Error bars represent the SD of three parallel experiments. *: P < 0.05, **: P < 0.01, ***: P < 0.001. | ||
CDC is a classic complement pathway initiated by the formation of the antibody-complement component 1q (C1q) complex and a series of subsequent cascade reactions.48 We next evaluated the CDC cytotoxicity mediated by UEAR Nbs. To this end, cells were incubated with 50 nmol L−1 of UEAR Nbs in the presence of mouse serum and rabbit complement (RC); then, the cell viability was measured using a CCK8 kit. As shown in Fig. 5B, distinct cytotoxicity was observed when cancer cells were treated with 7D12-ZZ or ZZ-7D12. For example, approximately 53.2 and 61.2% of MDA-MB-468 cell lysis were observed when incubated with 7D12-ZZ and ZZ-7D12, respectively. The potency was 10.5- and 12.2-fold higher as compared with the results of the 7D12 treatment group. Additionally, no obvious cell lysis was observed when the heat inactivated rabbit complement (HIRC) was applied in UEAR Nb groups, further demonstrating that the observed cell-killing effect was indeed induced by CDC. The similar potency induced by UEAR Nbs in the two cancer cell lines proved that the fusion sites of the ZZ domain had no significant impact on the CDC activities.
ADCP is another important mechanism to induce target cell phagocytosis through the activation of FcγRs on macrophages by the Fc-moiety of mAbs.49 THP-1 is a human monocyte cell line that has been successfully used in phagocytosis assays.50 In this regard, we employed THP-1 as effector cells to investigate the ADCP level induced by UEAR Nbs. To do this, target cells and THP-1 cells were first stained with DiO (green cell membrane probe) and DiI (orange red cell membrane probe), respectively. Then, target cells were treated with UEAR Nbs and human serum (as the source of endogenous IgGs), followed by incubation with THP-1 cells. As directly observed by confocal fluorescence imaging (Fig. S6 and S7†), both cancer cell A431 and MDA-MB-468 cells were clearly phagocytosed by THP-1 cells in the presence of UEAR Nbs and human serum, indicating that UEAR Nbs were capable of evoking ADCP. We further quantitatively analysed the phagocytosis efficiency by counting the cell numbers in R1 (double-positive cells) and R2 (remaining target cells) in flow cytometry experiments. As shown in Fig. 6A, considerable amounts of double-positive cells were only observed in the groups of 7D12-ZZ and ZZ-7D12, whereas nearly no double-positive cells were observed in the groups of PBS and 7D12. Interestingly, the potency of 7D12-ZZ- and ZZ-7D12-mediated ADCP displayed discriminated profiles. The phagocytoses of A431 and MDA-MB-468 cells induced by 7D12-ZZ could reach 45.3 and 57.6%, respectively, which were significantly higher than those induced by ZZ-7D12 (A431, 21.5%; MDA-MB-468, 22.2%) (Fig. 6B). The underlying mechanism is unknown at this stage. It is speculated that the difference may be caused by the different interactions of the UEAR Nb-IgG immune complex with multiple FcγRs involved ADCP activation.
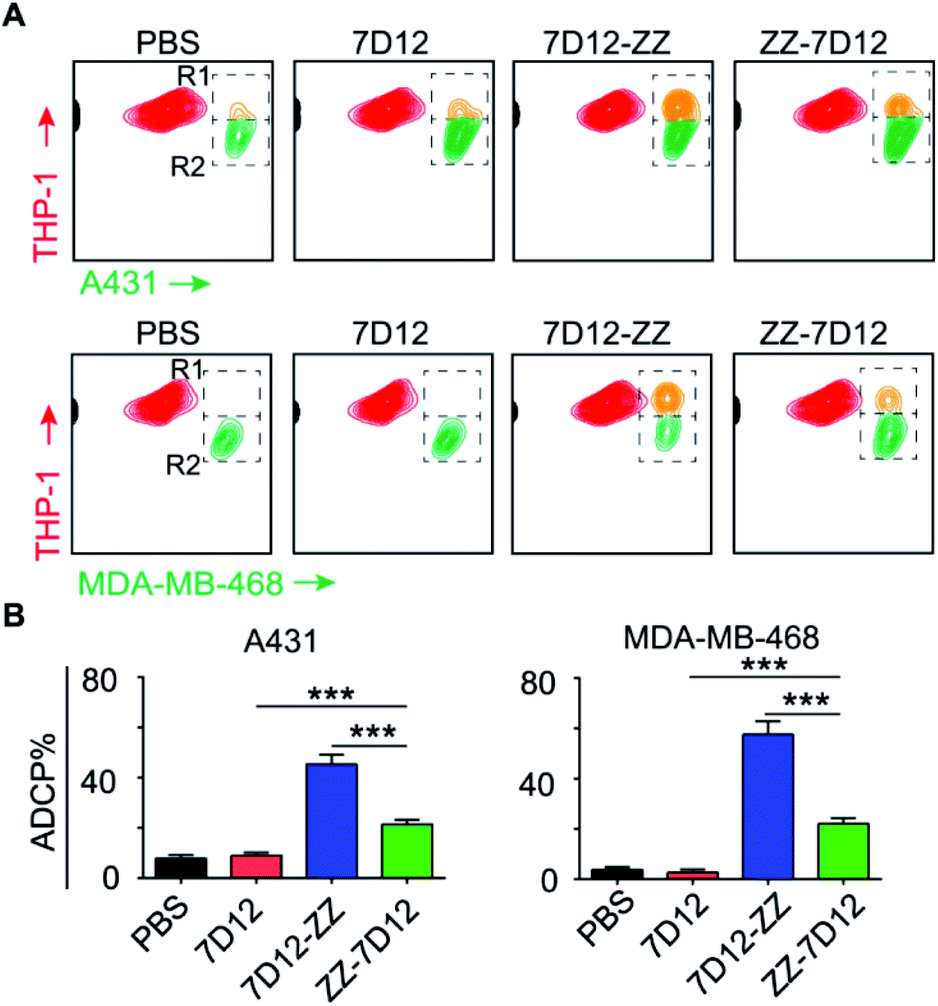 | ||
| Fig. 6 In vitro ADCP assays. (A) Flow cytometry assays and (B) the corresponding phagocytosis of target cells treated with PBS, 7D12, or UEAR Nbs 7D12-ZZ or ZZ-7D12. Error bars show the SD of three parallel experiments. ***: P < 0.001. | ||
Pharmacokinetics evaluation of UEAR Nbs
One of the main challenges for nanobody-based therapeutics is their relatively short half-life in vivo. For example, the half-life of 7D12 in mice was less than 10 min. To investigate whether ZZ domain fusion could prolong the serum half-life of UEAR Nbs, we performed in vivo experiments to determine their half-lives. Thus, 7D12-ZZ and ZZ-7D12 were intravenously (i.v.) injected into the healthy mice and the blood samples were collected at different time points. The corresponding plasma concentrations of 7D12-ZZ and ZZ-7D12 were determined according to a standard curve obtained by ELISA assay (Fig. S8†). As shown in Fig. 7, the half-life of 7D12-ZZ and ZZ-7D12 could reach 25.9 and 29.5 h, respectively, which were approximately 160.9- and 183.4-fold improved compared to the previously reported 0.16 h half-life of 7D12.28 Additionally, no significant difference in half-life was observed between 7D12-ZZ and ZZ-7D12. The significant improvement of the pharmacokinetics of UEAR Nbs could be attributed to the in situ formed immune complex of the ZZ domain with endogenous IgGs, whose sizes were increased to above the renal threshold, thereby avoiding waste-pass clearance.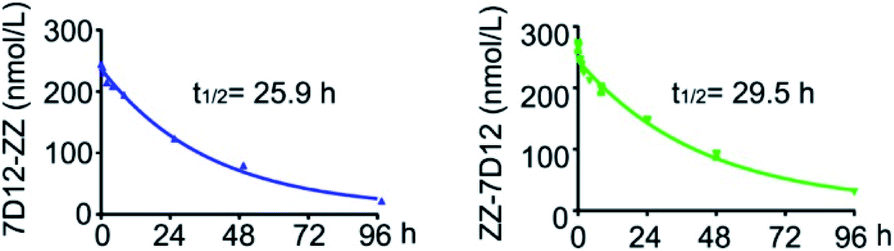 | ||
| Fig. 7 The plasma concentrations and half-life of 7D12-ZZ and ZZ-7D12 in Balb/c mice. Blood samples collected from mice in the same group at the same time point were pooled and then analyzed using ELISA. Error bars represent the SD of three parallel experiments. | ||
In vivo antitumor efficacy evaluation of UEAR Nbs
TNBC is a notorious breast cancer subtype that lacks the overexpression of the estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor-2 (HER2), which comprises 10–24% of all breast cancers.51 However, previous studies using human TNBC tissue demonstrated that EGFR overexpression frequently occurred in TNBC. MDA-MB-468 cells are validated triple-negative basal-A mammary carcinoma with positive EGFR overexpression. Thus, MDA-MB-468 cells were s.c. injected into Balb/c nude mice to create a TNBC xenograft mice model; then, these mice were randomly divided into four groups which were treated by i.p. injection of PBS, 7D12, 7D12-ZZ, or ZZ-7D12 (Fig. 8A), respectively. Treatments were given every two days in 10 days using 50 μL of nanobodies in PBS (30 μmol L−1) and 50 μL of pooled normal mouse serum as the source of endogenous IgGs. During the course of experiments (18 days), we monitored the tumor size in mice every two days. As described in Fig. 8B, the tumor volume in groups treated with 7D12-ZZ or ZZ-7D12 was significantly reduced and persistent tumor regression was observed in the treatment period as well as after treatment cessation. To further ascertain the antitumor efficacy of UEAR Nbs, we excised tumors from each mouse and conducted tumor weight measurement at the experimental endpoint. As shown in Fig. 8C–E, for PBS and 7D12 groups, the mean tumor weight was 419.2 ± 122.7 mg, 255 ± 62.5 mg, indicating that nanobody 7D12 exhibited a limited tumor growth inhibition effect (TGI, 39.2%). By contrast, for 7D12-ZZ and ZZ-7D12 treatment groups, the mean tumor weights were 27.8 ± 9.0 and 13.4 ± 2.2 mg. The TGI was 89.1 and 94.7% (7D12-ZZ and ZZ-7D12 vs. 7D12), respectively. This result clearly suggested that UEAR Nbs could significantly enhance the antitumor efficacy of nanobodies by reconstituting the Fc functions in the presence of endogenous IgGs. The improved pharmacokinetics of UEAR Nbs may also contribute to this outstanding antitumor activity. | ||
| Fig. 8 Evaluation the in vivo antitumor efficacy of UEAR Nbs. (A) Schematic illustration of the experimental plan for immunotherapy of xenograft MDA-MB-468 tumors in Balb/c nude mice. (B) Tumor volumes of mice treated with PBS, nanobody 7D12, 7D12-ZZ or ZZ-7D12. Each arrow represents a treatment. Error bars represent the SEM (n = 5). (C) Tumors excised from the treated mice at the experimental endpoint. (D) Tumor weights of the treated mice at the experimental endpoint. (E) Tumor growth inhibition (TGI) of the treated mice at the experimental endpoint. Tumor growth inhibition was calculated according to the tumor weights. Error bars represent the SEM (n = 5). **: P < 0.01. | ||
To evaluate the toxicity of UEAR Nbs, the mouse weight was monitored and no obvious weight loss was observed in mice treated with either nanobody 7D12 or UEAR Nbs (Fig. S9†). In addition, hematoxylin and eosin (H&E) staining results presented in Fig. S10† revealed that none of the collected tissues showed acute or chronic inflammation or necrotic regions. Moreover, to evaluate the anaphylactic shock risk of UEAR Nbs, C57BL/6J mice were i.p. injected with 100 μL of 7D12-ZZ or ZZ-7D12 (1 mg mL−1 in PBS) at day 0, followed by day 7 to 15 with a frequency of every two days. During the course of experiment, no anaphylactic shock was observed. In addition, the nanobody-specific IgE level of serum samples at day 15 was as low as that at day 0 (Fig. S11†), indicating the low anaphylactic shock risk of UEAR Nbs. All the above results suggested that UAER Nbs were essentially nontoxic.
Conclusions
mAb-based cancer immunotherapy has achieved great success in the past few decades.52,53 However, it is widely recognized that developing therapeutic mAbs as drugs is complicated and expensive. Nanobodies, as a new type of antibody fragment derived from camelids, offer competitive advantages over full length mAbs and other antibody fragments, including physiochemical properties, discovery, optimization and production. The ARM strategy is a promising modality for cancer immunotherapy. However, currently, only hapten-specific naturally occurring antibodies were harnessed to induce humoral and cellular immune responses to fight cancer cells. Recently, Fc-ARMs, composed of folic acid and a Fc binding peptide, were nicely constructed and could redirect endogenous antibodies to trigger an anti-cancer immune response.54 However, this molecule exhibited marginal in vivo anti-tumor efficacy. In this work, to make a functional antibody-like nanobody for cancer immunotherapy, a simple and robust UEAR Nb was constructed based on the ARM concept and our previous studies, to reinstate the missing Fc biological functions. We demonstrated that the UEAR Nbs could recruit abundant universal endogenous IgGs, independent of hapten-specific antibodies in human serum, onto cancer cell surfaces and exert the Fc-mediated anticancer biological mechanisms to eliminate cancer cells in vitro and in vivo. A beneficial pharmacokinetic improvement of UEAR Nbs was achieved simultaneously with this strategy. Considering the superior targeting capability of nanobodies, this strategy will provide a general and economic approach to generate antibody-like nanobodies for cancer and other disease applications.Ethical statement
All animal experiments were performed according to the guidelines and protocols approved by the Institutional Animal Care and Use Committee of the Jiangnan University (JN. No. 20190315b0650625 and 20201130c0650220).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21472070, 32000904, 21602084, 21907038), the Natural Science Foundation of Jiangsu Province (BK20200601), the National Postdoctoral Program for Innovative Talents of China (BX20200153), the China Postdoctoral Science Foundation (2018M632227), and the Social Development Key Project of Jiangsu Province (BE2019632). The project was partly funded by the 111 Project (No. 111-2-06), the Open Foundation of Key Laboratory of Carbohydrate Chemistry & Biotechnology Ministry of Education (No. KLCCB-KF202006) and the National First-class Discipline Program of Food Science and Technology [JUFSTR20180101]. We appreciate the help of Professor Xiaodong Gao and Professor Morihisa Fujita at Jiangnan University for the flow cytometry analysis and Professor Wei Huang at the Shanghai Institute of Materia Medica for the MS spectrum of the nanobody.Notes and references
- C. Hamerscasterman, T. Atarhouch, S. Muyldermans, G. Robinson, C. Hamers, E. B. Songa, N. Bendahman and R. Hamers, Nature, 1993, 363, 446–448 CrossRef CAS.
- J. P. Salvador, L. Vilaplana and M. P. Marco, Anal. Bioanal. Chem., 2019, 411, 1703–1713 CrossRef CAS.
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS.
- D. Schumacher, J. Helma, A. F. L. Schneider, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2018, 57, 2314–2333 CrossRef CAS.
- S. Steeland, R. E. Vandenbroucke and C. Libert, Drug Discov. Today, 2016, 21, 1076–1113 CrossRef CAS.
- S. Duggan, Drugs, 2018, 78, 1639–1642 CrossRef CAS.
- C. Morrison, Nat. Rev. Drug Discovery, 2019, 18, 485–487 CrossRef CAS.
- G. J. Weiner, Immunol. Res., 2007, 39, 271–278 CrossRef CAS.
- B. M. Tijink, T. Laeremans, M. Budde, M. S. V. Walsum, T. Dreier, H. J. de Haard, C. R. Leemans and G. A. M. S. van Dongen, Mol. Cancer Ther., 2008, 7, 2288–2297 CrossRef CAS.
- H. T. Pan, J. Y. Liu, W. T. Deng, J. Y. Xing, Q. Li and Z. Wang, Int. J. Nanomed., 2018, 13, 3189–3201 CrossRef CAS.
- X. Sun, X. Yan, W. Zhuo, J. K. Gu, K. Zuo, W. Liu, L. Liang, Y. Gan, G. He, H. Wan, X. J. Gou, H. B. Shi and J. P. Hu, Int. J. Mol. Sci., 2018, 19, 1984 CrossRef.
- A. Beck, T. Wurch, C. Bailly and N. Corvaia, Nat. Rev. Immunol., 2010, 10, 345–352 CrossRef CAS.
- D. Levin, B. Golding, S. E. Strome and Z. E. Sauna, Trends Biotechnol., 2015, 33, 27–34 CrossRef CAS.
- D. J. Falconer, G. P. Subedi, A. M. Marcella and A. W. Barb, ACS Chem. Biol., 2018, 13, 2179–2189 CrossRef CAS.
- M. Dechant, W. Weisner, S. Berger, M. Peipp, T. Beyer, T. Schneider-Merck, J. J. L. van Bueren, W. K. Bleeker, P. W. H. I. Parren, J. G. J. van de Winkel and T. Valerius, Cancer Res., 2008, 68, 4998–5003 CrossRef CAS.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS.
- P. J. McEnaney, C. G. Parker and A. X. Zhang, Annu. Rep. Med. Chem., 2017, 50, 481–518 CAS.
- A. F. Rullo, K. J. Fitzgerald, V. Muthusamy, M. Liu, C. Yuan, M. Huang, M. Kim, A. E. Cho and D. A. Spiegel, Angew. Chem., Int. Ed., 2016, 55, 3642–3646 CrossRef CAS.
- A. Uvyn, R. De Coen, M. Gruijs, C. Tuk, J. De Vrieze, M. van Egmond and B. de Geest, Angew. Chem., Int. Ed., 2019, 58, 12988–12993 CrossRef CAS.
- J. Wehr, E. L. Sikorski, E. Bloch, M. S. Feigman, N. J. Ferraro, T. R. Baybutt, A. E. Snook, M. M. Pires and D. Thevenin, J. Med. Chem., 2020, 63, 3713–3722 CrossRef CAS.
- S. S. Li, B. C. Yu, J. J. Wang, Y. Q. Zheng, H. J. Zhang, M. J. Walker, Z. N. Yuan, H. Zhu, J. Zhang, P. G. Wang and B. H. Wang, ACS Chem. Biol., 2018, 13, 1686–1694 CrossRef CAS.
- X. X. Li, X. J. Rao, L. Cai, X. L. Liu, H. X. Wang, W. N. Wu, C. G. Zhu, M. Chen, P. G. Wang and W. Yi, ACS Chem. Biol., 2016, 11, 1205–1209 CrossRef CAS.
- K. P. Naicker, H. G. Li, A. Heredia, H. J. Song and L. X. Wang, Org. Biomol. Chem., 2004, 2, 660–664 RSC.
- C. G. Parker, R. A. Domaoal, K. S. Anderson and D. A. Spiegel, J. Am. Chem. Soc., 2009, 131, 16392–16394 CrossRef CAS.
- J. M. Fura, S. Sarkar, S. E. Pidgeon and M. M. Pires, Curr. Top. Med. Chem., 2017, 17, 290–304 CrossRef CAS.
- M. N. Idso, A. S. Akhade, M. L. Arrieta-Ortiz, B. T. Lai, V. Srinivas, J. P. Hopkins, A. O. Gomes, N. Subramanian, N. Baliga and J. R. Heath, Chem. Sci., 2020, 11, 3054–3067 RSC.
- E. Chirkin, V. Muthusamy, P. Mann, T. Roemer, P. G. Nantermet and D. A. Spiegel, Angew. Chem., Int. Ed., 2017, 56, 13036–13040 CrossRef CAS.
- H. F. Hong, Z. F. Zhou, K. Zhou, S. Z. Liu, Z. W. Guo and Z. M. Wu, Chem. Sci., 2019, 10, 9331–9338 RSC.
- M. A. Gray, R. N. Tao, S. M. DePorter, D. A. Spiegel and B. R. McNaughton, ChemBioChem, 2016, 17, 155–158 CrossRef CAS.
- R. T. C. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393–1398 CrossRef CAS.
- W. L. Chen, L. Gu, W. P. Zhang, E. Motari, L. Cai, T. J. Styslinger and P. G. Wang, ACS Chem. Biol., 2011, 6, 185–191 CrossRef CAS.
- C. B. Carlson, P. Mowery, R. M. Owen, E. C. Dykhuizen and L. L. Kiessling, ACS Chem. Biol., 2007, 2, 119–127 CrossRef CAS.
- G. Vidarsson, G. Dekkers and T. Rispens, Front. Immunol., 2014, 5, 520 Search PubMed.
- B. Nilsson, T. Moks, B. Jansson, L. Abrahmsen, A. Elmblad, E. Holmgren, C. Henrichson, T. A. Jones and M. Uhlen, Protein Eng., 1987, 1, 107–113 CrossRef CAS.
- A. C. Braisted and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5688–5692 CrossRef CAS.
- S. Inouye and Y. Sahara-Miura, Protein Expression Purif., 2017, 137, 58–63 CrossRef CAS.
- J. A. Brockelbank, V. Peters and B. H. A. Rehm, Appl. Environ. Microbiol., 2006, 72, 7394–7397 CrossRef CAS.
- J. Z. Hui, A. Al Zaki, Z. L. Cheng, V. Popik, H. T. Zhang, E. T. Luning Prak and A. Tsourkas, Small, 2014, 10, 3354–3363 CrossRef CAS.
- M. Iijima, H. Kadoya, S. Hatahira, S. Hiramatsu, G. Jung, A. Martin, J. Quinn, J. Jung, S. Y. Jeong, E. K. Choi, T. Arakawa, F. Hinako, M. Kusunoki, N. Yoshimoto, T. Niimi, K. Tanizawa and S. Kuroda, Biomaterials, 2011, 32, 1455–1464 CrossRef CAS.
- Z. Cai, T. Fu, Y. Nagai, L. Lam, M. Yee, Z. Q. Zhu and H. T. Zhang, Cancer Res., 2013, 73, 2619–2627 CrossRef CAS.
- L. Jendeberg, M. Tashiro, R. Tejero, B. A. Lyons, M. Uhlen, G. T. Montelione and B. Nilsson, Biochemistry, 1996, 35, 22–31 CrossRef CAS.
- Y. Yarden and M. X. Sliwkowski, Nat. Rev. Mol. Cell Biol., 2001, 2, 127–137 CrossRef CAS.
- I. Uberall, Z. Kolar, R. Trojanec, J. Berkovcova and M. Hajduch, Exp. Mol. Pathol., 2008, 84, 79–89 CrossRef CAS.
- K. R. Schmitz, A. Bagchi, R. C. Roovers, P. M. van Bergen en Henegouwen and K. M. Ferguson, Structure, 2013, 21, 1214–1224 CrossRef CAS.
- J. Tintelnot, N. Baum, C. Schultheiss, F. Braig, M. Trentmann, J. Finter, W. Fumey, P. Bannas, B. Fehse, K. Riecken, K. Schuetze, C. Bokemeyer, T. Rosner, T. Valerius, M. Peipp, F. Koch-Nolte and M. Binder, Mol. Cancer Ther., 2019, 18, 823–833 CrossRef CAS.
- S. Radaev, S. Motyka, W. H. Fridman, C. Sautes-Fridman and P. D. Sun, J. Biol. Chem., 2001, 276, 16469–16477 CrossRef CAS.
- G. L. Moore, H. Chen, S. Karki and G. A. Lazar, Mabs, 2010, 2, 181–189 CrossRef.
- N. S. Merle, S. E. Church, V. Fremeaux-Bacchi and L. T. Roumenina, Front. Immunol., 2015, 6, 262 Search PubMed.
- K. R. VanDerMeid, M. R. Elliott, A. M. Baran, P. M. Barr, C. C. Chu and C. S. Zent, Cancer Immunol. Res., 2018, 6, 1150–1160 CrossRef CAS.
- M. E. Ackerman, B. Moldt, R. T. Wyatt, A. S. Dugast, E. McAndrew, S. Tsoukas, S. Jost, C. T. Berger, G. Sciaranghella, Q. Liu, D. J. Irvine, D. R. Burton and G. Alter, J. Immunol. Methods, 2011, 366, 8–19 CrossRef CAS.
- B. G. Haffty, Q. F. Yang, M. Reiss, T. Kearney, S. A. Higgins, J. Weidhaas, L. Harris, W. Hait and D. Toppmeyer, J. Clin. Oncol., 2006, 24, 5652–5657 CrossRef.
- C. Zhang and K. Y. Pu, Chem. Soc. Rev., 2020, 49, 4234–4253 RSC.
- Z. L. Zeng and K. Y. Pu, Adv. Funct. Mater., 2020, 30, 2004397 CrossRef CAS.
- K. Sasaki, M. Harada, Y. Miyashita, H. Tagawa, A. Kishimura, T. Mori and Y. Katayama, Chem. Sci., 2020, 11, 3208–3214 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General information and supplementary figures. See DOI: 10.1039/d0sc05332e |
| This journal is © The Royal Society of Chemistry 2021 |